

Abstract
Bruton's tyrosine kinase (Btk), belonging to the Tec family of tyrosine kinases (TFKs), is essential for B-lymphocyte development. Abrogation of Btk signaling causes human X-linked agammaglobulinemia (XLA) and murine X-linked immunodeficiency (Xid). We employed affinity purification of Flag-tagged Btk, combined with tandem mass spectrometry, to capture and identify novel interacting proteins. We here characterize the interaction with ankryin repeat domain 54 protein (ANKRD54), also known as Lyn-interacting ankyrin repeat protein (Liar). While Btk is a nucleocytoplasmic protein, the Liar pool was found to shuttle at a higher rate than Btk. Importantly, our results suggest that Liar mediates nuclear export of both Btk and another TFK, Txk/Rlk. Liar-mediated Btk shuttling was enriched for activation loop, nonphosphorylated Btk and entirely dependent on Btk's SH3 domain. Liar also showed reduced binding to an aspartic acid phosphomimetic SH3 mutant. Three other investigated nucleus-located proteins, Abl, estrogen receptor β (ERβ), and transcription factor T-bet, were all unaffected by Liar. We mapped the interaction site to the C terminus of the Btk SH3 domain. A biotinylated, synthetic Btk peptide, ARDKNGQEGYIPSNYVTEAEDS, was sufficient for this interaction. Liar is the first protein identified that specifically influences the nucleocytoplasmic shuttling of Btk and Txk and belongs to a rare group of known proteins carrying out this activity in a Crm1-dependent manner.
INTRODUCTION
Cytoplasmic (nonreceptor) protein tyrosine kinases (cPTKs or nPTKs) are essential cellular components in multicellular organisms (12, 21), orchestrating signal transduction in diverse cell types, including hematopoietic cells (16, 32). Btk is a cytoplasmic PTK belonging to the Tec family of tyrosine kinases (TFKs), the second largest family of cPTKs. It is expressed in the B-cell lineage and plays a pivotal role in signaling and development (7, 26, 41). Mutations in the corresponding gene give rise to X-linked agammaglobulinemia (XLA) in humans and X-linked immunodeficiency (Xid) in mice, severely impairing B-lymphocyte development (34, 43–45). Activation of Btk, through various receptors at the plasma membrane, ignites a cascade of signaling events, resulting in the formation of a multiprotein complex, the “signalosome.” Despite the fact that a broad spectrum of signaling partners has been identified, both in vitro and in vivo (19, 26), still little is known about the composition of signaling components downstream of Btk.
The Btk protein has a conserved multidomain architecture comprised of PH, Tec homology (TH), SH3, SH2, and kinase (SH1) domains (26, 41). Each of the Btk domains has the capacity to interact with various signaling molecules, enabling Btk to carry out diverse biological processes (26, 41). Membrane tethering of Btk is mediated by its PH domain through interaction with phosphatidylinositol 3-kinase (PI3K)-generated phophatidylinositol 3,4,5-triphosphate (PIP3), which results in the full activation of the kinase and also locates Btk close to its downstream substrates (13, 18, 29). The significance of this interaction is manifested by the phenotype caused by the Xid missense mutation, R28C (34, 43), which impairs membrane tethering of the PH domain. An identical mutation found in humans causes XLA (46), demonstrating that there are species-specific phenotypes. Two phosphorylated tyrosine residues have been identified in Btk, tyrosine-551 (35) and tyrosine-223 (31) within the kinase and SH3 domains, respectively. Y551 represents the conserved regulatory tyrosine in the activation loop of the kinase domain and is phosphorylated by Src family tyrosine kinases (1, 35). Y223 is located in the SH3 domain, found to be regulatory and modulated by trans- or autophosphorylation (31). In addition to having a cytoplasmic-membrane location, Btk (25, 47, 38) and other TFKs, Itk (33) and Txk (6), are also found in the nucleus. Regulation of the nuclear translocation and nucleocytoplasmic shuttling of Txk, Btk, and Itk remains to be elucidated.
Here, we identified a novel Btk-interacting protein, ankyrin repeat domain 54 (ANKRD54)/Liar. During our studies on ANKRD54, Liar was reported as a novel Lyn-binding protein influencing erythropoietin-induced differentiation of erythrocytes (39). Our data demonstrate an SH3-dependent, functional interaction between Liar and Btk in the nuclear compartment, resulting in shuttling to the cytoplasm.
MATERIALS AND METHODS
Cell culture.
Namalwa and Ramos (human Burkitt B-cell lymphoma), Nalm-6 (pre-B-cell leukemia), K562 (human erythromyeloblastoid leukemia), A20 (mouse B-lymphoma), Jurkat (human T-lymphocyte), Cos7 (African green monkey kidney), Phoenix GP and PG13 (retrovirus producer lines based on human embryonic kidney HEK293T cells), and NIH 3T3 (mouse embryonic fibroblast) cell lines were obtained from the American Type Culture Collection (ATCC). The cell lines Phoenix GP, Cos7, and NIH 3T3 were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Invitrogen). All other hematopoietic cells were cultured in RPMI 1640 medium with supplements. All cells were cultivated at 37°C in a humidified 5% CO2 incubator.
Reagents.
Transient transfections were performed in six-well plates using polyethylenimine (PEI) 25K (Polyscience, Inc.) or FuGENE 6 (Roche) according to the manufacturer's instructions. Monoclonal mouse and polyclonal rabbit antibodies against Btk have been described earlier (29). Other antibodies used were as follows: anti-Flag M2 affinity gel and anti-Flag M2 (Sigma), anti-c-Myc 9E10 (Roche), anti-c-Myc (C-33) (Santa Cruz Biotechnology), antiphosphotyrosine 4G10 (Upstate Biotechnology), Cy3-conjugated anti-IgG (Jackson ImmunoResearch), Cy5-conjugated anti-IgG (Chemicon Temecula), anti-human Liar (ProSci Inc.), anti-green fluorescent protein (anti-GFP) (Santa Cruz), and anti-DsRed (Clontech). The secondary antibodies used, goat anti-mouse 800CW, goat anti-rabbit 800CW, goat anti-mouse 680LT, and goat anti-rabbit 680 antibodies, were from LI-COR Biosciences GmbH.
We generated affinity-purified polyclonal antibody (C54) against Liar using a synthetic peptide epitope (QMTSTKEQVDEVTDLLAC), and immunization was performed by GenScript (Piscataway, NJ). The SDS-PAGE (4 to 20% Tris-glycine) gels and nitrocellulose membranes of the iBlott dry-blotting system were purchased from Invitrogen. Statistical analysis in Table 1 was performed using a chi-square test of association using 2 × 2 contingency tables with Yates's correction, and results with P values of less than 0.05 were considered statistically significant observations.
Table 1.
Distribution pattern of expressed protein in nuclear (N) and cytoplasmic (C) compartmentsa
| Protein(s) expressed | Plasmid amt (μg) or ratiob | % of cells expressing indicated protein(s) |
|||||
|---|---|---|---|---|---|---|---|
| GFP (combined with Btk, Btk-NLS, or Btk-ΔSH3-NLS) |
DsRed-Liar |
||||||
| N only | C only | Both N and C | N only | C only | Both N and C | ||
| DsRed-Liar | 0.8 | 18 | 0 | 82 | |||
| Btk-GFP | 0.8 | 0 | 93 | 7 | |||
| GFP-Btk | 0.8 | 0 | 94 | 6 | |||
| GFP-Btk-NLS | 0.8 | 100c | 0 | 0 | |||
| GFP–Btk-ΔSH3-NLS | 0.8 | 100c | 0 | 0 | |||
| DsRed-Liar + GFP-Btk | 1:1d | 0 | 100 | 0 | 23 | 0 | 77 |
| DsRed-Liar + GFP-Btk-NLS | 1:1d | 0 | 100 | 0 | 10 | 0 | 90 |
| DsRed-Liar + GFP–Btk-ΔSH3-NLS | 1:1d | 100 | 0 | 0 | 22 | 0 | 78 |
| DsRed-Liar | 0.3 | 56 | 0 | 44 | |||
| DsRed-Liar + GFP-Btk-NLS + Btk-SH3 | 1:1:8e | 18 | 56 | 26 | 0 | 62 | 38 |
| DsRed-Liar + GFP–Btk-ΔSH3-NLS + Btk-SH3 | 1:1:8e | 100 | 0 | 0 | 0 | 37 | 63 |
| Btk-SH3 | DsRed-Liar | ||||||
| Btk-SH3 | 0.8 | 0 | 0 | 100 | |||
| DsRed-Liar + Btk-SH3 | 1:1 | 0 | 95 | 5 | 0 | 0 | 100 |
Distribution of DsRed-Liar protein alone or in the presence of Btk-GFP, GFP-Btk, GFP-Btk-NLS, or GFP–Btk-ΔSH3-NLS is shown. In some experiments, the Btk SH3 domain was utilized as a competitor for the Btk-Liar interaction. Mean values were calculated for 100 to 300 cells from three separate experiments.
Plasmid amounts and cotransfection ratios are indicated.
Almost entirely nuclear-resident Btk, but with a small fraction of overexpressed Btk being localized in the cytoplasm and around the nucleus.
1 = 0.8 μg of plasmid.
1 = 0.3 μg of plasmid (an amount used to reach similar total levels of transfected plasmid).
Protease inhibitor, complete mini EDTA-free tablets (Roche), phosphatase inhibitor cocktail (Sigma), protein A- and G-Sepharose and HiTrap streptavidin HP columns (GE Healthcare), PCI-32765 (Pharmacyclics, Inc.), dasatinib, and leptomycin B (LMB; LC Laboratories) were used in this study. All other high-grade laboratory chemicals and reagents were obtained from Sigma.
Plasmid constructs, PCR, and cloning.
cDNAs encoding wild-type (wt) and mutant Btk constructs (R28C, K430E, Y223A, Y551F, Y223A/Y551F, ΔSH3, ΔPH, E41K) with or without GFP were generated by subcloning into pEGFP or pSGT vectors as described previously (3, 19, 29). The cDNA of human Btk was cloned into the KpnI and XhoI sites of the eukaryotic expression vector pSVK3. Double-stranded synthetic oligonucleotides corresponding to three glycine linkers followed by the Flag tag (GGG-DYKDDDDK) were inserted into the XbaI and XhoI sites of the plasmid pSVK3-Btk to generate a C terminus epitope-tagged protein. Primers were as follows: sense, 5′-CTA GAT GTC ATG GAT GAA GAA TCC GGT GGC GGT GAC TAC AAG GAC GAC GAT GAC AAG TGA TCA C; antisense, 5′-TA CAG TAC CTA CTT CTT AGG CCA CCG CCA CTG ATG TTC CTG CTG CTA CTG TTC ACT AGT GAG CT.
Finally, the entire Btk–3×G-Flag fragment was transferred from the pSVK3 plasmid and cloned into the EcoRI/XhoI sites of the retroviral vector pMSCV-IRES-GFP (where IRES represents the internal ribosome entry site). To create a nucleus-resident Btk (pEGFP–Btk-NLS), two complementary synthetic oligonucleotides corresponding to the classical simian virus 40 (SV40) nuclear localization signal (NLS) heptapeptide were annealed and subsequently introduced into the C terminus of plasmid pGFP-Btk by standard DNA cloning. To obtain Btk-ΔSH3 deletion mutants lacking the entire SH3 domain (L97 to E280), a site-directed deletion was generated, using a multiple overlapping-PCR strategy (Mutagenex, Inc.). The deletion mutant vector pSGT–Btk-ΔSH3 was also used to construct two fusions, pSGT–Btk-ΔSH3-NLS and pEGFP–Btk-ΔSH3-NLS. Full-length expression of the Txk/Rlk constructs was achieved using pCEFL-Rlk-GFP and pCEFL-Rlk-myc (FL), kindly provided by Pamela Schwartzberg (NIH). The Liar gene was synthesized and subcloned into the pMSCV-IRES-GFP vector, with Flag and Myc tags at the N and C termini, respectively. Later, the Liar gene was subcloned into the pEGFP-N1 vector at EcoRI/SalI sites. A fusion was generated by in-frame cloning into the expression vector pDsRed-monomer-C1 (Clontech) (Kpn21 and BamHI sites). For PCR amplifications, the following primers were used: sense, 5′-GCT AGC CGT TGC CAT GGC AGC C; antisense, 3′-AAG CTT GCT ACC TCT TCT CCA T. The inserts were verified by sequencing.
For tissue expression analysis of Liar transcripts, human multiple-tissue cDNA, panel 1 (Clontech), was used. All other total RNA preparations from various cell lines were extracted with an RNeasy minikit (Qiagen). cDNA synthesis from cell lines was performed using a first-strand cDNA synthesis kit (Roche). For semiquantitative PCR, DNA amplifications were performed using the primers 5′-GCT AGC CGT TGC CAT GGC AGC C (sense) and 3′-AAG CTT GCT ACC TCT TCT CCA T (antisense), generating a single, approximately 1-kb band corresponding to the full-length Liar gene that was compared with an ACTB housekeeping gene (40).
Affinity purification of Btk-Flag for mass spectrometric analysis.
A total of 1.2 liters (≈109 cells each) of Namalwa cells (wt and stably expressed Btk-Flag cell lines) was grown to an optimal density. Cells were collected and resuspended in 30 ml serum-free RPMI medium. Then, cells were starved for 3 h at 37°C. The cells were pelleted and washed once with phosphate-buffered saline (PBS) buffer at room temperature. Resuspended cells in 30 ml PBS were subdivided into three test tubes (≈10 ml each). The first tube was left unstimulated, the second was treated with 100 μM pervanadate with 1% FBS for 15 min at 37°C, and the third tube was treated with 100 nM dasatinib for 15 min at 37°C. Finally, the cells were pelleted and kept at −80°C until use.
A 30-ml volume of lysis buffer containing 50 mM HEPES-KOH (pH 7.6), 120 mM KCl, 2 mM EDTA, 0.5% NP-40, 10 mM β-glycerophosphate, and 20 mM NaF was prepared, and the following reagents were freshly added: 0.5 mM dithiothreitol (DTT), 1 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride (PMSF), 3 tablets of protease inhibitor, and 300 μl phosphatase inhibitor. Each pellet of cells was lysed with 5 ml lysis buffer and then incubated on ice for 20 min, with interval vortex mixing. For preclearing, 500 μl of a 50% slurry mixture of proteins A and G and agarose anti-IgG was added to each tube. The samples were incubated by rocking for 60 min at 4°C, followed by centrifugation at 4°C for 10 min at 3,000 rpm. The supernatants were passed through 0.8-μm filters to new tubes containing 50 μl of a 50% slurry of Flag M2 agarose beads. The samples were incubated under rocking conditions for 5 h at 4°C. The beads were washed six times via centrifugation and supernatant aspiration cycles with washing buffer (lysis buffer with lower NP-40 content, 0.1%). Then the beads were washed three times with freshly made cold Flag-rinsing buffer (50 mM NH4HCO3, pH 8.0, 75 mM KCl, and 2 mM EDTA). The purified Btk-Flag complexes on the anti-Flag M2 agarose beads were eluted gently with 2% NH4OH and then lyophilized and prepared for gel-free mass spectrometry analysis according to the protocol described by Chen and Gingras (5). Residual bound proteins from beads were denatured with 25 μl SDS-PAGE sample buffer. These samples were run on an SDS-PAGE gel and stained with GelCode blue stain (Pierce) to check for the elution yield. Samples were digested in solution and analyzed by liquid chromatography-mass spectrometry using a nano-1100 high-performance liquid chromatography (HPLC) system (Agilent) coupled to an LTQ mass spectrometer (MS; Thermo Fisher) or a capillary 1100 HPLC system (Agilent) coupled to an LTQ-Orbitrap mass spectrometer (Thermo Fisher). The gradient was 2 to 60% acetonitrile over 105 min with a total run time of 150 min. The resulting MS/MS spectra were searched using the MASCOT program (version 2.02.01) against the human Ensembl Database release (9).
Retroviral transduction of hematopoietic cell lines.
Phoenix GP and PG13 adherent cell lines were used for production of viral particles. The preparation of viral particles from the Btk–3×G-Flag construct pMSCV and the transduction procedure for lymphocyte cell lines were performed essentially as described previously (2).
Immunofluorescence and confocal microscopy.
Cells were seeded overnight at 50% confluence in six-well dishes. The following day, cells were transfected with plasmids and grown for an additional 48 h, followed by live-cell confocal microscopy to identify colored fusion proteins. For immunostaining, cells were washed several times with PBS and then fixed in 2.5% formaldehyde for 15 min. The fixed cells were permeabilized in 0.1% Triton X-100 in PBS for 15 min, blocked in 0.1% bovine serum albumin (BSA)-c (Aurion), and diluted in a 1% PBS-Tween solution for 1 h. Without washing, the cells were incubated overnight at 4°C with primary antibodies, anti-c-Myc, and 9E10 monoclonal antibody (1:1,000). The cells were washed and stained further with a Cy3-conjugated goat anti-mouse IgG (1:500) or with a fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (1:200). All antibodies, primary and secondary, were diluted in blocking solution. In all cases, the cell nuclei were stained with 5 μg/ml DAPI (4′6-diamidino-2-phenylindole, dihydrochloride) (Molecular Probes), diluted in PBS. Confocal microscopy analysis was performed as described previously (29).
Immunoprecipitation and biotin-streptavidin pulldown assay.
All immunoprecipitations were performed as described previously (3, 25, 29). For endogenous Liar pulldown, we synthesized a Btk SH3 peptide “bait” conjugated with biotin (produced by GenScript). The primary sequence of the peptide (biotin-Btk SH3 domain) was biotin-CGGG linked to 254ARDKNGQEGYIPSNYVTEAEDS275, corresponding to the C-terminal 22 amino acids of the Btk SH3 domain from alanine-254 to serine-275. The pulldown assay of biotin-Btk SH3 peptide was conducted using HiTrap streptavidin HP beads (GE Healthcare, Sweden) according to the manufacturer's instructions. For each sample, 50 μl of streptavidin gel slurry beads was incubated with 3 μg of biotin-Btk SH3 peptide for 20 min, followed by the addition of 300 μl lysate (4 × 106 cells) of mouse A20 B cells, under rotation at 4°C for 60 min. After incubation, samples were centrifuged for 3 min at 500 × g and the supernatant was removed. The beads were washed twice with 40× bead volumes (1 ml) of lysate buffer. Samples were prepared for SDS-PAGE and Western blotting as described previously (25, 47). The Western blot signals (except those shown in Fig. 3B) were quantified by using LI-COR and Odyssey software.
Fig 3.

Validation of the Btk-Liar interaction. (A) Coimmunoprecipitation validation of ectopic expression of Liar (DsRed-Liar-Myc) with pSGT-Btk in Cos-7 cells; (B) endogenous interaction of Btk and Liar in Namalwa B cells; (C) influence of Btk phosphorylation status on Liar interaction using the Btk inhibitor PCI-32765. Cells were activated by pervanadate; (D) influence of phosphomimetic mutations Y223D and Y551D on Liar interaction. Lane 1, untransfected control (other controls were mock transfected); lane 2, cells transfected with pEGFP plasmid only; lane 3, cells transfected with pSGT empty vector only.
RESULTS
Btk-Flag-stable cell lines and coimmunopurification of protein complexes.
To identify novel Btk complexes, we expressed Flag-tagged Btk in two hematopoietic cell lines, Namalwa and K562. Since the cell lines endogenously express Btk, we expect a complete set of Btk signaling pathways to operate, although cell lineage-specific patterns may exist. Obtaining stable Btk-Flag expression in hematopoietic cell lines was considered critical for the proteomics approach to purify and identify Btk interaction complexes in vivo.
Btk-Flag was cloned into retroviral vector pMSCV containing IRES-driven GFP (Fig. 1A). Phoenix GP cells were transiently transfected with pMSCV-Btk-Flag-IRES-GFP (Fig. 1A), and viral particles were prepared from supernatant (2) and used to transduce Namalwa (Fig. 1B) and K562 (not shown) cells. Following four rounds of fluorescence-activated cell sorting (FACS), GFP-positive cells were enriched to 98% purity and the selected population was gated for cells with low expression of Btk-Flag from Namalwa and K562 cells (not shown). In both cell lines, Western blot analysis confirmed GFP and Btk-Flag expression together with endogenous Btk (Fig. 1C).
Fig 1.

Strategy used to create the biological model for proteomics analysis. (A) Schematic representation of the pMSCV vector cloned with Btk-Flag and live-cell imaging of Phoenix GP cells expressing the Btk-Flag-IRES-EGFP for production of viral particles. (B) Live-cell imaging of Namalwa cells after retroviral transduction to establish stable expression of Btk-Flag and EGFP with the same transcript. (C) Western blotting of the stable cell lines Namalwa and K562. (D) Typical experiment of immunopurification of Btk-Flag protein complexes. Protein staining shows residual bound proteins on anti-Flag M2-agarose beads after the elution procedure (see Materials and Methods). The eluted samples were prepared for gel-free mass spectrometry analysis. The eluted proteins were much less contaminated than the gel-stained proteins (D), with respect to heavy (IgG Hc)- and light (IgG Lc)-chain protein bands. (E) ANKRD54 is identified in Btk immunoprecipitates. Flag-tagged Btk was captured with anti-Flag M2 agarose from Namalwa cells under the indicated conditions. Mass spectrometry analysis was employed to identify interacting partners. The number of unique peptides and Mascot search algorithm scores are shown.
Namalwa cells were expanded for large-scale affinity purification of Btk-Flag. Prior to Btk isolation, Namalwa cells were subjected to three conditions: (i) pervanadate activation (phosphatase inhibition), (ii) dasatinib treatment (Btk inhibition), or (iii) no treatment. Purified Btk-Flag complexes, captured with anti-Flag M2-agarose beads, were eluted, lyophilized, and prepared for gel-free MS/MS analysis. The residual bound proteins on the M2-agarose beads, after the elution step, were run on an SDS-PAGE gel and stained with GelCode blue stain (Fig. 1D). Raw, mass spectrometry data were processed with the Mascot database search tool to score and match all sequenced peptides against the Ensembl protein database (data available on request). One of the proteins that copurified with Btk, but was absent in control samples lacking Btk, was ANKRD54, recently designated Liar in mouse (39). Liar was isolated from both untreated and dasatinib-treated samples (Fig. 1E).
The primary sequence of human Liar protein corresponds to a 34-kDa polypeptide of exactly 300 amino acids, consisting of unique N- and C-terminal regions with four ankyrin repeat domains in the center of the molecule (Fig. 2A), similar to murine Liar (39). Ankyrin repeat domains (ANKRDs) are functional structural motifs, consisting of 30 to 40 amino acids shaped into a β-hairpin–helix-loop-helix structure. Several repeating units of these domains cooperate to form an extended recognition fold that exhibits specificity for protein-protein interactions (4, 17, 27, 28). The structure and function of ANK repeat-interacting proteins are very diverse. Often, a set of ankyrin repeat domains has a very specific protein-binding partner. For example, the 7-ANK repeats in IκB interact with the p65 subunit of NF-κB, preventing p65's entry into the nucleus, thereby blocking the activation of the NF-κB signaling cascade (11, 14).
Fig 2.

Schematic structure of Liar, homology alignments, and expression patterns. (A) Domain structure of human Liar, indicating the putative sequences of the NLS and NES motifs. (B) Sequence alignments between human, mouse, and rat proteins using Clustal X 2.0 software. The shaded spots indicate variations in amino acid sequences. The GLPGRA box indicates the P loop region missing in human Liar, while the box in the C terminus shows the peptide used for immunization resulting in the C54 Liar antibody. (C and D) Expression patterns of ANKRD54/Liar transcripts in human tissues (C) and haematopoietic cell lines (D), including primary peripheral blood mononuclear cells (PBMCs). Sk., skeletal.
Characterization and tissue expression of Liar.
Using the Gene-Card database and Motif-Finder searches, the Liar domains and motif structure were identified and predicted (Fig. 2A). The human and mouse Liar proteins have almost identical domain and motif features, containing 4 ankyrin repeats in the core of the protein surrounded by N-terminal bipartite NLS and C-terminal nuclear export signal (NES) motifs (39). In addition, the mouse Liar protein has a putative ATP-binding P loop (51GLPGRS56) (39), while the human protein lacks this motif (Fig. 2B, the GLPGRA box). Sequence alignment of Liar proteins shows remarkably high sequence identity and conservation between the human, murine, and rat proteins (Fig. 2B), and a highly conserved C-terminal epitope was selected and used as the antigen for generation of anti-Liar (C54) antibodies (Fig. 2B, C-terminal box). Sequence similarity was also found to be high in more distantly related eukaryotes, such as Drosophila (not shown).
Next, we investigated transcript expression of Liar using cDNAs from human tissues, hematopoietic cell lines, and primary peripheral blood mononuclear cells (PBMCs) (Fig. 2C and D). Amplicons were equivalent to the full-size Liar mRNA (900 bp). As shown in Fig. 2C, Liar has a wide expression pattern but was not readily detected in lung tissue. Pancreas and brain tissues expressed very high levels of Liar, which has also been reported for ciliated cells (22). Human control PBMCs from a healthy individual expressed Liar; the cell lines Namalwa, Ramos (B cells), Nalm-6 (pre-B cell), K562 (myeloid cell), and Jurkat (T cell) were also positive (Fig. 2D).
To investigate the function of the Liar protein, we generated a cDNA with an N-terminal Flag tag and a C-terminal Myc tag, subsequently cloned into the pMSCV-IRES-GFP vector for mammalian gene expression. Later, the Liar-myc fusion gene was cloned into the pDsRed plasmid at the N terminus (pDsRed-Liar-myc) and into the pEGFP plasmid at the C terminus without myc (pLiar-EGFP). To verify the MS/MS interaction, following ectopic expression of pSGT-Btk and pDsRed-Liar-myc in Cos7-cells, reciprocal coimmunoprecipitations of both proteins were performed (Fig. 3A).
Importantly, endogenous interaction was confirmed by coimmunoprecipitation of human B cells (Fig. 3B). Thus, consistent with the mass spectrometry data, coimmunoprecipitation of endogenous Liar verified the interaction (Fig. 1D).
Liar interacts with nonphosphorylated Btk but less so with the phosphomimetic Y223D mutant.
We next sought to determine the phosphorylation status of Btk during the interaction with Liar. To study this, we transfected the wt Btk (pSGT-Btk) alone or together with the Liar (pLiar-EGFP) construct into Cos7 cells. After 48 h, cells were harvested, starved for 3 h, and activated with pervanadate in the presence or absence of the Btk inhibitor PCI-32765 (Fig. 3C). The Btk-Liar interaction was independent of Btk kinase activity (Fig. 3C), while the phosphomimetic Y223D mutant showed reduced binding (Fig. 3D).
Subcellular localization of Liar and Btk.
To further investigate the role of Liar's interaction with Btk, in terms of subcellular localization, we conducted immunofluorescence analysis using two different approaches. First, antibody staining of myc-tagged Liar (pMSCV-Flag-Liar-myc) alone identified Liar in the nucleus, with a small fraction being localized in the cytoplasm (Fig. 4A, middle panel). The murine stem cell virus (MSCV) plasmid expresses GFP protein from the same transcript driven by an IRES, which is used as a positive marker of transfection (Fig. 4A, left panel). Second, using a fusion of Liar with monomeric DsRed fluorescent protein (pDsRed-Liar-myc) alone, we verified that the Liar protein was localized mainly in the nucleus, independent of the size of the myc or DsRed tag (Fig. 4A and B; Table 1, top section). Thus, two different tagged versions of Liar, in fixed (myc-tagged) or live (red fluorescent protein-tagged) cells, showed that the protein resides predominantly in the nucleus. These results are in accordance with observations presented by Samuels et al. (39) on mouse Liar.
Fig 4.

Liar is a nucleocytoplasmic protein colocalizing with Btk in the cytoplasm, inner membrane, and perinuclear regions. (A) Liar localization with anti-Myc staining enhanced with Cy3. GFP was expressed together with Liar using the pMSCV-Liar-IRES-GFP plasmid. (B) DsRed-Liar expressed alone. (C) Btk-GFP expressed alone. (D) Btk-GFP coexpressed with DsRed-Liar. Equal amounts of plasmids were transiently transfected in Cos7, and colocalization was monitored 48 h posttransfection.
In a steady-state situation, about 82% of the cells had DsRed-Liar protein in both the nucleus and cytoplasm, while in the remaining 18% of the cells, when 0.8 μg plasmid DNA was used for transfection, the protein was confined to the nucleus (Table 1, top section). In order to compare localization dynamics of Liar, we employed statistical scoring of our microscopy observations. In some competition experiments, in order to use similar amounts of plasmids, the amount of the Liar construct was reduced because the Btk SH3 construct was added in an 8-fold excess (Table 1, top and middle sections). When Liar was expressed at lower levels (0.3-μg plasmid transfection), we observed that Liar resided predominantly in the nucleus, i.e., in 56% of cells, with only 44% of cells showing Liar in both the nucleus and the cytoplasm simultaneously (Table 1, middle section). The corresponding values for 0.8 μg plasmid DNA were 18% and 82%, respectively (Table 1, top section).
We (25) and others (38, 47) have previously demonstrated that Btk is found mainly in the cytoplasm but also shuttles between the cytoplasm and the nucleus. In accordance with this, two different GFP-tagged versions of Btk (Btk-GFP and GFP-Btk) were examined and found to show similar localizations, with >90% of all cells demonstrating cytoplasmic distribution, while in 6 to 7% of cells the fusion proteins were consistently found in both the nucleus and the cytoplasm (Table 1, top section).
We next investigated the subcellular localization of Btk-GFP (wt) coexpressed with DsRed-Liar (Fig. 4D). Importantly, when the proteins were expressed simultaneously, Btk (wt) and Liar colocalized entirely in the cytoplasm (Fig. 4D; Table 1, top section, row 6). Thus, in the presence of increased amounts of Liar, Btk (wt) is no longer resident in the nucleus at all (Table 1, top section, P < 0.02). There was also a dramatic decrease in the number of cells expressing Btk at the cell membrane (not shown). Conversely, in the presence of Btk (wt), about the same percentage of cells demonstrated nuclear residency of Liar (Table 1, top section, compare rows 1 and 6). However, a significant portion of Liar protein colocalized with Btk at an intense rim surrounding the nucleus (compare Fig. 4D to Fig. 4B and C).
Overexpression of Liar downregulates tyrosine phosphorylation of Btk.
To further assess the possible functional role of Liar, tyrosine phosphorylation of Btk was investigated. To test this, we overexpressed Liar and Btk in different ratios (Fig. 5A). The transient-transfection amount of pSGT-Btk was kept constant (0.5 μg plasmid), while the amount of DsRed-Liar-myc plasmid was increased (0.5, 1.0, and 2.0 μg) and then the level of Btk phosphorylation at pY551 was quantified under steady-state conditions. Y551 is located in the activation loop of Btk, and its phosphorylation is a measure of Btk kinase activity (19). Indeed, the phosphorylation level of Btk at pY551 was gradually diminished upon higher-level Liar expression (Fig. 5A and B), suggesting that Liar downregulates Btk phosphorylation. This is a selective effect, since overexpression of Liar did not affect the phosphorylation level of ERK. Thus, while the interaction per se is independent of Y551 phosphorylation (Fig. 3C), upon interaction, phosphorylation is reduced.
Fig 5.
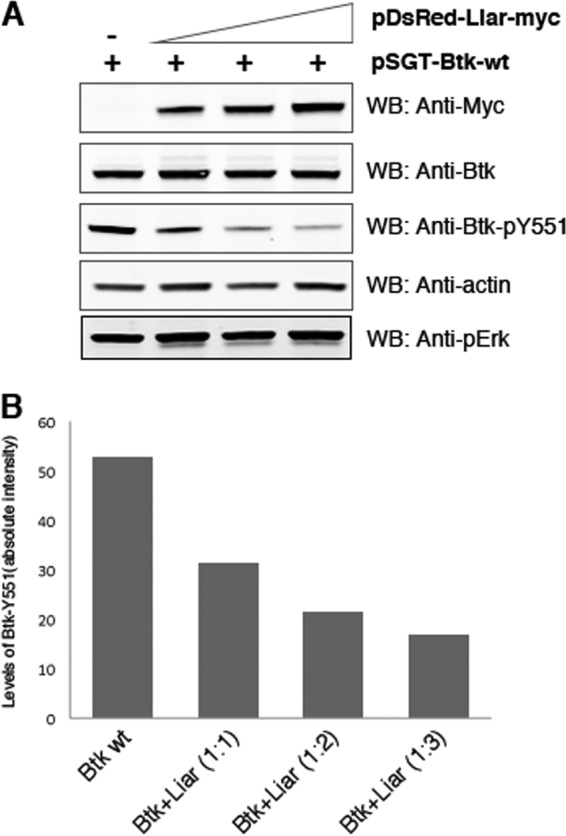
Liar interacts with nonphosphorylated Btk and overexpression of Liar severely downregulates tyrosine phosphorylation of active Btk in vivo. (A) Coimmunoprecipitation analysis of Btk and phosphorylated Btk and pulldown with anti-Btk antiserum, with the respective anti-GFP directed against the fusion protein. (B) The pDSRed-Liar-myc plasmid was transfected at ratios of 1:1, 2:1, and 4:1 to the pSGT-Btk plasmid in Cos7 cells. The phosphorylation state of Btk was monitored using the phosphotyrosine-specific pY551 antibody. Equal loading of Btk and actin expression are indicated. Quantification of the Btk pY551 level, using a densitometric intensity graph, is shown.
Liar modulates Btk shuttling and exclusion of nucleus-resident Btk.
In order to further study shuttling, we generated a nucleus-targeted GFP-Btk fusion protein containing a synthetic NLS signal located at the C terminus of Btk. This form of Btk (GFP–Btk-NLS) resided entirely in the nucleus (Fig. 6A; Table 1, top section), over a wide range of expression levels (not shown). Unexpectedly, despite the nuclear distribution of GFP–Btk-NLS, a complete nuclear exclusion of the protein was observed in the presence of Liar (Fig. 6C; Table 1, top section, P ≪ 10−10). Under these circumstances, in 90% of the cells, Liar was still detected in the nucleus (Fig. 6C; Table 1, top section), but there was also what seems to be an essentially complete colocalization of Btk-NLS with Liar in the cytoplasm. Thus, these observations, together with the effect on wt Btk, strongly suggest the existence of a functional link between Liar and Btk in terms of shuttling and subcellular localization. To characterize this phenomenon in detail, we generated Btk mutants to study the efficacy of nucleocytoplasmic shuttling.
Fig 6.
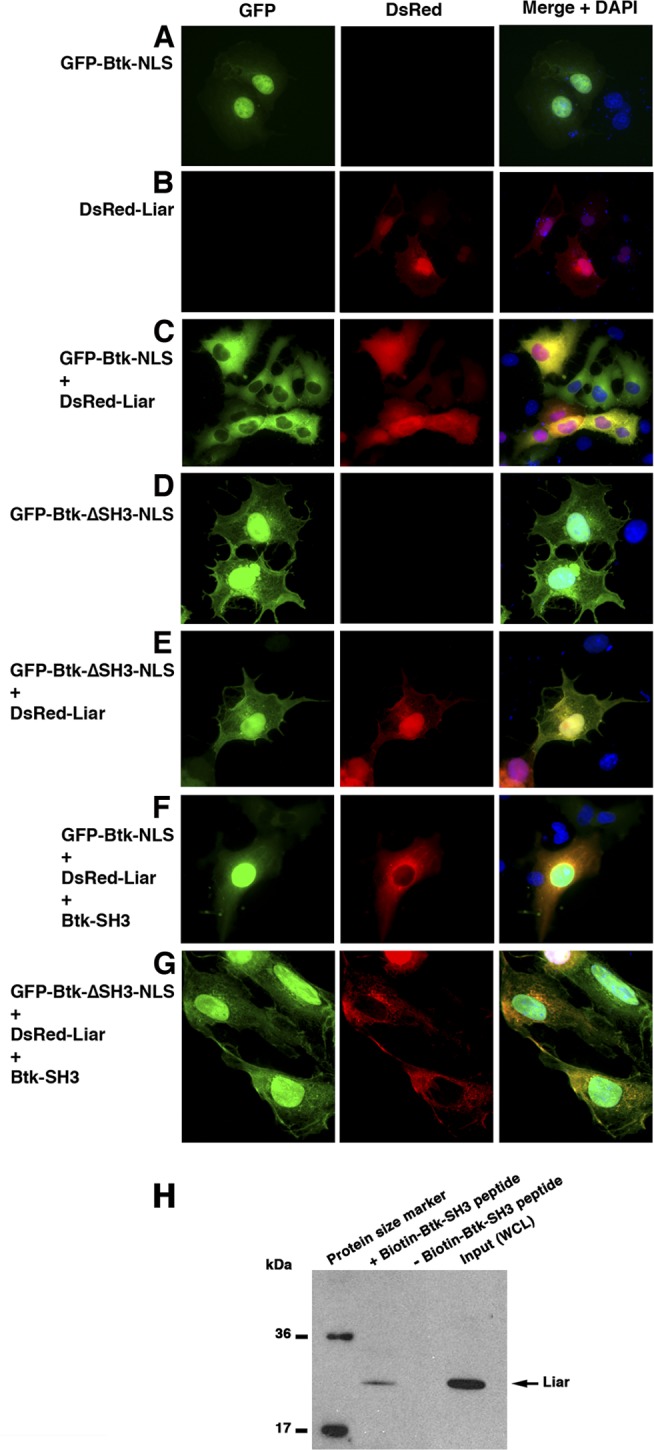
Liar interacts with the Btk SH3 domain and expels nucleus-targeted Btk (Btk-NLS) from the nucleus. (A to E) Ectopic expression of GFP–Btk-NLS (A), DsRed-Liar (B), GFP–Btk-NLS plus DsRed-Liar (C), GFP–Btk-ΔSH3-NLS (D), and GFP–Btk-ΔSH3-NLS with DsRed-Liar (E). (F and G) Competitive assay with the Btk SH3 domain alone coexpressed with GFP–Btk-NLS and DsRed-Liar (F) and together with GFP–Btk-ΔSH3-NLS mutant and DsRed-Liar (G). (H) Pulldown assay of endogenous Liar by biotinylated Btk SH3 peptide and tyrosine phosphorylation dependent on the Liar-Btk interaction. The biotinylated interaction peptide has the following sequence: biotin-CGGG-ARDKNGQEGYIPSNYVTEAEDS. For a negative control, the streptavidin beads were incubated without the Btk SH3 peptide (middle lane). The right lane shows Liar from a whole-cell lysate (WCL). The assay is representative of four different experiments using various B-cell lines, namely, Namalwa, K562, Nalm6, and RBL2H3.
Apart from showing that Btk shuttles between the cytoplasm and the nucleus, we have also reported previously that a Btk mutant lacking the SH3 domain (Btk-ΔSH3–GFP) localizes in the nucleus in the absence of the export signal blocker leptomycin B (LMB) (25). In fact, our conclusion from this work was that the SH3 domain is needed to retain Btk in the cytoplasm (25). Based on these observations, we constructed a new GFP–Btk-NLS mutant lacking the SH3 domain (GFP–Btk-ΔSH3-NLS) to verify the above-described findings. As expected, the GFP–Btk-ΔSH3-NLS mutant was predominantly a nucleus-resident protein (Fig. 6D; Table 1, top section), resembling the Btk-ΔSH3–GFP mutant (25). The result from cotransfection of GFP–Btk-ΔSH3-NLS with DsRed-Liar-myc is shown in Fig. 6E. In fact, Liar failed to influence the localization of the Btk-ΔSH3-NLS mutant (Fig. 6E; Table 1, top section). This strongly suggests that this process (interaction and colocalization) is an SH3-dependent phenomenon (Table 1; compare Fig. 6C and 6E).
Interestingly, the ankyrin repeats of murine Liar have previously been identified as novel SH3 binding regions for Lyn in erythrocytes and also for some other SH3-containing proteins, such as HS1 and Vav1 (39). To test the hypothesis that Liar interacts with the Btk SH3 domain, we performed a competition experiment to investigate whether the expression of the SH3 domain of Btk alone could reverse the cytoplasmic retention of Btk-NLS imposed by Liar. Consistent with our hypothesis, in a significant fraction of cells, the GFP–Btk-NLS protein kept its nuclear residency in the presence of Liar when the Btk SH3 domain was overexpressed in a ratio to the other components of 8:1:1 (Fig. 6F; Table 1, middle section). Thus, the Btk SH3 domain alone appears to be sufficient to bind stably with Liar in the cytoplasm, allowing unbound, full-length Btk-NLS protein to maintain nuclear residency (Fig. 6F; Table 1, middle section). As expected, expression of Btk SH3 failed to compete with the nuclear residency of Btk-ΔSH3-NLS, presumably since Liar does not interact with Btk-ΔSH3-NLS (Fig. 6E and G; Table 1, middle section), while Liar appears in the cytoplasm. We next considered what influence Liar could have on the localization of the Btk SH3 domain alone. Surprisingly, the Btk SH3 domain was confined to the cytoplasm in the presence of Liar (95%), in sharp contrast to its 100% nucleocytoplasmic distribution when expressed alone (Table 1, bottom section; P < 0.0001). Collectively, we observed that in the presence of Liar, the three proteins GFP-Btk, GFP–Btk-NLS, and Btk SH3 were essentially entirely localized to the cytoplasm. In conclusion, we show that the Liar protein profoundly influences Btk's localization in an SH3 domain-dependent manner.
We next sought to confirm that the physical interaction between Liar and Btk was dependent on the SH3 domain of Btk. Therefore, we designed a pulldown assay using a biotinylated Btk SH3 peptide as “bait” for the A20 B-cell line extracts. The SH3 peptide contains the C-terminal 22 amino acid residues of the Btk SH3 domain, with the sequence biotin-CGGG-ARDKNGQEGYIPSNYVTEAEDS. The design of this peptide is based on our preliminary experiments, in which we used various deletion mutants made for other purposes and observed that an internal N-terminal SH3 domain deletion mutant still was able to bind Liar similar to the way wt Btk did. Therefore, we decided to design a peptide corresponding to the remaining C-terminal portion of the SH3 domain of Btk. Indeed, we could capture endogenous Liar as a binding partner to this short portion of the Btk SH3 domain (Fig. 6H, left lane). To eliminate the possibility of an unspecific interaction of Liar with the streptavidin beads, we treated a parallel sample under identical pulldown assay conditions in the absence of bait (Fig. 6H, middle lane). In this control sample, we found no trace of Liar, even with prolonged enhanced chemiluminescence (ECL) exposure (not shown). Thus, an SH3-derived peptide devoid of the proline-binding region specifically captures endogenous Liar.
Liar does not affect localization of other nucleus-resident proteins.
To study the specificity of the Btk-NLS nuclear exclusion events in the presence of Liar (Fig. 6C), we investigated the outcome of cotransfection of other nucleus-resident proteins. The distribution of GFP, a 27-kDa protein that freely shuttles between the nucleus and cytoplasm, was not modulated by the presence of Liar (Fig. 7A). Similarly, the localization of the nucleus-resident proteins estrogen receptor β (pYFP-ERβ) (Fig. 7B) and transcription factor T-bet (8, 24) (DsRed-T-bet) (Fig. 7C) was unaffected when they were cotranfected with pMSCV-Liar-myc. Since Liar is not influencing the subcellular distribution of three different nucleus-resident proteins, we conclude that the effect of Liar on nuclear proteins is selective.
Fig 7.
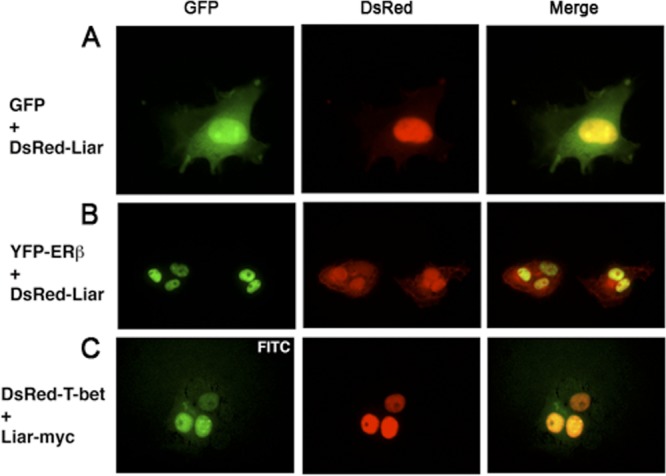
Localization of other nucleus-resident proteins is not affected by Liar. GFP (A), YFP-ERβ (B), and DsRed–T-bet (C) were not excluded from the nucleus by DsRed-Liar. Fluorescent imaging of Myc-tagged Liar was obtained with anti-Myc followed by the addition of anti-mouse Ig–FITC.
LMB inhibits nuclear export of Btk and Liar.
Due to the fact that both Liar and Btk proteins shuttle to the nucleus, we wanted to directly examine the nuclear export pathway. To test this, we employed LMB, a specific inhibitor of the Crm-1-dependent nuclear export machinery. We designed an experiment using ectopic expression of GFP-Btk, DsRed-Liar, GFP-Btk plus DsRed-Liar, and GFP–Btk-NLS plus DsRed-Liar. Twenty-four hours posttransfection, cells were treated with LMB for 12 h and compared to an untreated control (Fig. 8A to D). In the absence of LMB (Fig. 8A and B, left panels), GFP-Btk localized in the cytoplasm, while Liar localized predominantly in the nucleus. However, nuclear Liar relocates to the cytoplasm in the presence of GFP-Btk and GFP–Btk-NLS (Fig. 8C and D, left panels). Nevertheless, after 12 h of LMB treatment, both Btk and Liar accumulate in the nucleus (Fig. 8A and D, right panels), suggesting that they both utilize a Crm-1-dependent export pathway, in accordance with earlier findings (25, 39). In this context, we sought to modulate the export pathway using LMB treatment for shorter incubation times, 3 and 6 h, in order to monitor the rate of accumulation, in particular colocalization of Liar with Btk (Fig. 8E to G). Here we observed that Liar accumulates rapidly in the nucleus, even after only 3 h of LMB treatment, while Btk at this point is almost entirely cytoplasmic. This is consistent with the fact that Liar is mainly a nucleus-resident protein, while wt Btk enters more slowly into the nucleus (Fig. 8E and F, left panels). To this end, we observed that Btk-NLS migrates to the nucleus even more readily than Liar (Fig. 6A; Table 1, top section). Therefore, this difference implies that Liar and Btk do not meet or interact in the cytoplasm. In fact, a prolonged treatment for 6 h with LMB is also compatible with the above idea (Fig. 8G, left and right panels). It should be kept in mind that Btk does not harbor a classical NES, while Liar does (amino acid positions 282 to 292) (Fig. 2). It is not known how nucleus-trapped Btk (LMB treated) and nucleus-resident Btk-NLS are expelled from nucleus. We believe that Liar plays a crucial role in mediating nuclear export of Btk. We already show that Btk interacts with Liar through an SH3-dependent interaction, since the ΔSH3 mutant of Btk stays in the nucleus and does not interact with Liar (Fig. 6E). To summarize the above functional findings, we propose a model (Fig. 8H) for how Liar mediates nuclear export of Btk. In section 1 of the model, Liar and Btk interact in the nucleus via the SH3 domain. Upon interaction, the NES of Liar gets exposed (Fig. 8H, section 1) and both proteins translocate to the cytoplasm as a complex (Fig. 4D and 8C). In a similar fashion, in section 2 of the model, expression of the Btk SH3 domain alone results in the export of Liar from the nucleus (Fig. 6G). Supporting the proposed role of Liar, Liar is not able to interact (Fig. 6H) or induce export of the Btk-ΔSH3-NLS mutant (Fig. 6E and 8H, section 3). Conversely, full-length Btk-NLS functionally interacts with Liar in the nucleus and is readily expelled (Fig. 8H, section 4, and 6C).
Fig 8.

Leptomycin B (LMB) treatment influences nuclear accumulation of Btk and Liar differentially. (A to D) Localization in the absence of LMB (left panels) of Btk-GFP (A), DsRed-Liar (B), GFP-Btk and DsRed-Liar (C), and GFP–Btk-NLS and dDsRed-Liar (D), in comparison to localization with 12 h of LMB pretreatment (right panels). (E to G) Lower panels show LMB treatment for 3 h (left) and 6 h (right) for Btk-GFP (E), DsRed-Liar (F), and GFP-Btk and DsRed-Liar (G). (H) Model of the nucleocytoplasmic shuttling of Btk regulated the novel SH3-dependent interaction with Liar. In this model, we propose that the NES of Liar becomes unprotected (exposed) upon interaction with Btk's SH3 domains and exports Btk from the nucleus effectively, even if the nucleus-resident form of Btk, Btk-NLS, is coexpressed. Owing to the fact that nucleus-resident Btk is hypophosphorylated (25), the model suggests that hypophosphorylated Btk, upon binding to Liar, is exported to the cytoplasm (the degree of Btk phosphorylation is not indicated in the Figure). Our data show clearly that coexpression of the SH3 domain alone is sufficient for competing with the nuclear export sequestration of Btk by Liar.
Liar prevents Txk/Rlk, another member of the Tec kinase family, from entering the nucleus.
After making the observations described above, we wanted to study the behavior of another member of the TFKs, namely, the Txk/Rlk. Unlike Btk, Txk lacks the pleckstrin homology (PH) domain and instead contains a cysteine string motif, critical for palmitoylation and required for cytoplasmic membrane translocation (6). When full-length Txk is depalmitoylated upon activation, it translocates to the nucleus (6, 23, 36). We confirmed that Txk readily migrates into the nucleus of Cos7-cells (Fig. 9A). We next tested the ability of Liar to modulate Txk translocation and observed that nuclear exclusion of Txk was detected in the presence of Liar (Fig. 9B). The colocalization in the cytoplasm was diffuse and not perinuclear, as seen for Btk/Liar. These results suggest that Liar is a common denominator for the shuttling of Txk and Btk. We subsequently investigated the ability of Liar to modulate another tyrosine kinase, c-Abl, which is well known for its ability to shuttle into the nucleus (42, 48). Coexpression of Liar with the nucleus-resident c-Abl–NES− mutant did not alter the subcellular distribution of this protein (Fig. 9C and D). Taken together, these results strengthen the idea that the interaction between Liar and Btk or Txk is selective and potentially controls the elusive nuclear function of the kinases.
Fig 9.

Liar removes nucleus-resident Txk/Rlk-GFP but not the nucleus-trapped c-Abl–NES− tyrosine kinase. (A and B) Native Txk localized to the nucleus and to the cytoplasm in the same cell (A), while in the presence of Liar, it was found predominantly in the cytoplasm (B). (C) Expression of nuclear trapped c-Abl—NES−. (D) Liar did not modulate nucleus-trapped c-Abl–NES− localization. Abl was stained with polyclonal anti-Abl antibody and decorated with a FITC-labeled secondary antibody. Txk, c-Abl, and Liar were expressed in Cos7 cells.
DISCUSSION
In this work, we have identified and characterized a new functional interaction partner for Btk, the ankyrin repeat protein Liar, using a proteomics approach based on affinity-purified Btk-Flag as bait in Namalwa B cells. We validated the interaction using biochemical analysis and also by subcellular localization with confocal microscopy. The full physiological relevance of the Liar-Btk interaction remains to be elucidated, but Liar causes nuclear exclusion of Btk and also suppresses its tyrosine phosphorylation.
Human and murine Liar proteins show 92% sequence identity and 95% similarity. Liar consists of four ankyrin repeat domains in the core of the molecule, flanked by unique and conserved regions in both the N and C termini. The N-terminal region of Liar contains a bipartite nuclear localization signal, while the C-terminal region harbors a typical nuclear export signal. Sequence alignments of Liar from several eukaryotic species demonstrate remarkable conservation (not shown). Interestingly, the ortholog of the ANKRD54 gene is not present in species where the Btk gene is absent (unpublished data), such as nematodes or yeasts, a finding compatible with the idea that an evolutionary relationship exists between Liar and Btk. This implies that Liar has a unique conserved feature with a distinct functional role in Btk nucleocytoplasmic shuttling. This is reminiscent of the situation with SH3BP5, a known negative regulator of Btk (30).
First, we validated the observed mass spectrometry interaction of Btk and Liar, using anti-Liar immunoprecipitates (c-Myc, DsRed) and GFP and our polyclonal antibody, C54 (data not shown). Importantly, endogenous interactions between Liar and Btk in B cells suggest that the proteins are functionally linked. Mass spectrometry data were validated by using endogenous as well as ectopic expression of both proteins in a coimmunoprecipitation assay, demonstrating stable interactions between Btk and Liar. We extended the analysis and showed that Liar displays a unique interaction with the Btk SH3 domain that is similar to its interaction with a selected set of SH3-containing proteins (39), since the ankyrin repeats do not contain classical polyproline-rich (PPR) PXXP motifs (20). This suggests an entirely new binding mode. Indeed, we have demonstrated that the Liar protein binds to a peptide derived from the C-terminal portion of the SH3 domain lacking the proline-rich binding core, implying that Liar binds the SH3 domain partner in a polyproline-independent manner. Overexpression of Liar led to inhibition of phosphorylation of the regulatory phospho-tyrosine Y551 of Btk in a dose-dependent manner.
Interestingly, the Y223D, but not the Y551D, Btk mutant showed reduced binding to Liar. Our previous nuclear magnetic resonance (NMR) structure of the Btk SH3 domain (10) showed Y223 in close proximity to, e.g., Y268. Since Y268 is located in the putative Liar-binding region of the Btk SH3 domain, this finding further supports the suggested mode of interaction.
We found that Liar resides mainly in the nucleus but that cytoplasmic localization is also detected. However, when DsRed-Liar is expressed in the presence of Btk-GFP, the two proteins seem to recolocalize in the cytoplasm. Thus, wt Btk and Btk-NLS were excluded from the nucleus when Liar was coexpressed. The extent of nuclear exclusion for the Btk-NLS fusion was surprisingly prominent, changing from 100% nuclear residency to 0% in the presence of the DsRed-Liar protein.
We (25), as well as others (47), have reported the nucleocytoplasmic shuttling nature of Btk. In a previous study, we also observed that the SH3 domain is a negative regulator of the nuclear shuttling of Btk (20). In this work, we observed that GFP–Btk-ΔSH3-NLS, similar to the GFP–Btk-NLS fusion protein, is exclusively nuclear. However, importantly, Liar fails to remove GFP–Btk-ΔSH3-NLS protein from nucleus. Also, we show that the efficacy of nuclear export of GFP–Btk-NLS, but not that of the GFP–Btk-ΔSH3-NLS fusion protein, by Liar is compromised in the presence of the Btk SH3 domain. We found that the expression of the Btk SH3 domain alone is sufficient to act as a competitive binder for Liar. Thus, collectively, we identify a new partner for Liar in B lymphocytes and demonstrate that Liar relocalizes Btk into the cytoplasm in an SH3-dependent manner.
The recent report showing that the ankyrin repeats of Liar specifically interact with the SH3 domains of Lyn, HS1, ESE2L, Vav1, Hip55, and LASP1 (39) is compatible with the idea that the ankyrin repeats of Liar also control its interaction with Btk. The conserved ankyrin repeats often have diverse protein-protein partners, the specificity also being determined by the number of repeats (17, 27, 28).
We also investigated the localization of three unrelated nucleus-resident proteins, namely, GFP, ERβ, and T-bet, the last two of which are transcription factors residing in the nucleus. We found that Liar does not modulate the localization of these proteins. We also tested the possible effect on nucleus-resident Txk/Rlk (6), and we observed that Liar exports nucleus-resident Txk/Rlk into the cytoplasm. In contrast, c-Abl tyrosine kinase with an established nucleocytoplasmic shuttling behavior (42, 48) was not affected by Liar in spite of the fact that it contains a bona fide SH3 domain.
Importantly, we found differences in relocation of Liar and Btk to the nucleus in the presence of LMB. Drug treatment induced more rapid accumulation of Liar in the nucleus than of Btk, but when coexpressed, both proteins were ultimately trapped in the nucleus. Taken together, these observations suggest that Btk and Liar move to the nucleus separately and that they interact with each other in the nucleus in an SH3-dependent manner. Thus, for Btk, we hypothesize that the Liar NES is utilized for Btk export and that the Liar NES is automasked in the unbound state, while upon Btk interaction, the NES is exposed, thereby driving both proteins out of the nucleus. In contrast, it is well known that another ankyrin repeat protein, IκBα, masks the NF-κB NLS sequence, thereby sequestering NF-κB in the cytoplasm (37). Recently it was observed that this process is even more complex, since A kinase-interacting protein 1 (AKIP1) and protein kinase A regulate the rate of NF-κB translocation (15).
Alternatively, it is possible that Liar's NLS is blocked by Btk. However, in the case of the Btk-NLS fusion, there would also be a need for Liar to interfere with Btk's NLS for Btk to become enriched in the cytoplasm. Even if Liar is shuttling in the absence of Btk, its normal NES function could be weak, since in the presence of Btk, Liar becomes more cytoplasmic. However, the precise shuttling mechanism of Liar remains elusive, and it is possible that shielding of the Liar NLS is the underlying cause.
Based on our findings, we propose a model for how Liar regulates the nucleocytoplasmic shuttling and concomitantly suppresses the phosphorylation of Btk. One possibility is that Liar is critical for terminating Btk-mediated signaling in the nucleus, although we do not yet have a clear picture about the nuclear targets for Btk since, in the case of Btk, this interaction is SH3 domain dependent. It will be interesting to find out whether this is also true for Txk, and, moreover, to get further insights into the physiological effects of these interactions.
ACKNOWLEDGMENTS
We thank Pamela Schwartzberg for the Txk/Rlk plasmids, Laurie H. Glimcher for the T-bet plasmid, Giulio Superti-Furga for the c-Abl constructs, Anders Ström for the estrogen receptor β plasmid, and Joseph J. Buggy (Pharmacyclics, Inc.) for the PCI-32765 inhibitor. We thank Adrian Pasculescu and Lorne Taylor for discussions and technical issue assistance on mass spectrometry.
B.F.N. thanks Erik Edith Fernström Stiftelse for providing funding travel and cooperation with the Mount Sinai Hospital, Toronto, Canada. Additional support to B.F.N was from the Karolinska Institutet Research Foundation, the Åke Wiberg Foundation, the Lars Hierta Foundation, the Magnus Bergvall Foundation, and Genome Canada through the Ontario Genomics Institute (T.P.). M.O.G. maintains a Ph.D. scholarship from Södertörn University-College, A.H. received a Ph.D. fellowship from COMSATS, Institute of Information-Technology, Islamabad, Pakistan, and D.K.M. maintains a Ph.D. fellowship from MoHE in the Kurdistan Regional Government, Iraq. This work was further supported by the Swedish Cancer Society, the Swedish Research Council, the Stockholm County Council (research grant ALF), and the European Council FP7 grant EURO-PADnet (C.I.E.S.)
M.O.G. performed most of the clonings, carried out the bulk of experiments related to Liar, and contributed to the writing; B.F.N. designed and performed most of the initial experiments and wrote the major part of the paper; A.H., D.K.M., and A.J.M. performed some of the clonings, designed some experiments, and performed statistical analysis. V.N. and P.M. performed proteomics analysis; K.C. helped with proteomics data analysis and editing of the manuscript; T.P. provided knowledge and advice as well as funding for the proteomics facility at SLRI; C.I.E.S. conceived the project, designed some experiments, contributed to writing and editing, and obtained research funding.
We declare no competing financial interests.
Footnotes
Published ahead of print 23 April 2012
REFERENCES
- 1. Afar DE, et al. 1996. Regulation of Btk by Src family tyrosine kinases. Mol. Cell. Biol. 16: 3465–3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aints A, Belusa R, Andersson RM, Guven H, Dilber MS. 2002. Enhanced ouabain resistance gene as a eukaryotic selection marker. Hum. Gene Ther. 13: 969–977 [DOI] [PubMed] [Google Scholar]
- 3. Bäckesjö CM, Vargas L, Superti-Furga G, Smith CIE. 2002. Phosphorylation of Bruton's tyrosine kinase by c-Abl. Biochem. Biophys. Res. Commun. 299: 510–515 [DOI] [PubMed] [Google Scholar]
- 4. Barrick D, Ferreiro DU, Komives EA. 2008. Folding landscapes of ankyrin repeat proteins: experiments meet theory. Curr. Opin. Struct. Biol. 18: 27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen GI, Gingras AC. 2007. Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases. Methods 42: 298–305 [DOI] [PubMed] [Google Scholar]
- 6. Debnath J, et al. 1999. rlk/TXK encodes two forms of a novel cysteine string tyrosine kinase activated by Src family kinases. Mol. Cell. Biol. 19: 1498–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gomez-Rodriguez J, et al. 2007. Tec kinases, actin, and cell adhesion. Immunol. Rev. 218: 45–64 [DOI] [PubMed] [Google Scholar]
- 8. Halcomb KE, Musuka S, Gutierrez T, Wright HL, Satterthwaite AB. 2008. Btk regulates localization, in vivo activation, and class switching of anti-DNA B cells. Mol. Immunol. 46: 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hamada N, Backesjo CM, Smith CI, Yamamoto D. 2005. Functional replacement of Drosophila Btk29A with human Btk in male genital development and survival. FEBS Lett. 579: 4131–4137 [DOI] [PubMed] [Google Scholar]
- 10. Hansson H, et al. 1998. Solution structure of the SH3 domain from Bruton's tyrosine kinase. Biochemistry 37: 2912–2924 [DOI] [PubMed] [Google Scholar]
- 11. Haskill S, et al. 1991. Characterization of an immediate-early gene induced in adherent monocytes that encodes I kappa B-like activity. Cell 65: 1281–1289 [DOI] [PubMed] [Google Scholar]
- 12. Hunter T. 2000. Signaling—2000 and beyond. Cell 100: 113–127 [DOI] [PubMed] [Google Scholar]
- 13. Hyvonen M, Saraste M. 1997. Structure of the PH domain and Btk motif from Bruton's tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia. EMBO J. 16: 3396–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Israel A. 2000. The IKK complex: an integrator of all signals that activate NF-kappaB? Trends Cell Biol. 10: 129–133 [DOI] [PubMed] [Google Scholar]
- 15. King CC, Sastri M, Chang P, Pennypacker J, Taylor SS. 2011. The rate of NF-kappaB nuclear translocation is regulated by PKA and A kinase interacting protein 1. PLoS One 6: e18713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kurosaki T, Hikida M. 2009. Tyrosine kinases and their substrates in B lymphocytes. Immunol. Rev. 228: 132–148 [DOI] [PubMed] [Google Scholar]
- 17. Li J, Mahajan A, Tsai MD. 2006. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45: 15168–15178 [DOI] [PubMed] [Google Scholar]
- 18. Li T, et al. 1995. Activation of Bruton's tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity 2: 451–460 [DOI] [PubMed] [Google Scholar]
- 19. Lindvall JM, et al. 2005. Bruton's tyrosine kinase: cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol. Rev. 203: 200–215 [DOI] [PubMed] [Google Scholar]
- 20. Liu X, Pawson T. 1994. Biochemistry of the Src protein-tyrosine kinase: regulation by SH2 and SH3 domains. Recent Prog. Horm. Res. 49: 149–160 [DOI] [PubMed] [Google Scholar]
- 21. Manning G, Plowman GD, Hunter T, Sudarsanam S. 2002. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 27: 514–520 [DOI] [PubMed] [Google Scholar]
- 22. McClintock TS, Glasser CE, Bose SC, Bergman DA. 2008. Tissue expression patterns identify mouse cilia genes. Physiol. Genomics 32: 198–206 [DOI] [PubMed] [Google Scholar]
- 23. Mihara S, Suzuki N. 2007. Role of Txk, a member of the Tec family of tyrosine kinases, in immune-inflammatory diseases. Int. Rev. Immunol. 26: 333–348 [DOI] [PubMed] [Google Scholar]
- 24. Miller AT, Wilcox HM, Lai Z, Berg LJ. 2004. Signaling through Itk promotes T helper 2 differentiation via negative regulation of T-bet. Immunity 21: 67–80 [DOI] [PubMed] [Google Scholar]
- 25. Mohamed AJ, et al. 2000. Nucleocytoplasmic shuttling of Bruton's tyrosine kinase. J. Biol. Chem. 275: 40614–40619 [DOI] [PubMed] [Google Scholar]
- 26. Mohamed AJ, et al. 2009. Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 228: 58–73 [DOI] [PubMed] [Google Scholar]
- 27. Mosavi LK, Cammett TJ, Desrosiers DC, Peng ZY. 2004. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 13: 1435–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mosavi LK, Minor DL, Jr, Peng ZY. 2002. Consensus-derived structural determinants of the ankyrin repeat motif. Proc. Natl. Acad. Sci. U. S. A. 99: 16029–16034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nore BF, et al. 2000. Redistribution of Bruton's tyrosine kinase by activation of phosphatidylinositol 3-kinase and Rho-family GTPases. Eur. J. Immunol. 30: 145–154 [DOI] [PubMed] [Google Scholar]
- 30. Ortutay C, Nore BF, Vihinen M, Smith CI. 2008. Phylogeny of Tec family kinases identification of a premetazoan origin of Btk, Bmx, Itk, Tec, Txk, and the Btk regulator SH3BP5. Adv. Genet. 64: 51–80 [DOI] [PubMed] [Google Scholar]
- 31. Park H, et al. 1996. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity 4: 515–525 [DOI] [PubMed] [Google Scholar]
- 32. Pawson T, Scott JD. 2005. Protein phosphorylation in signaling–50 years and counting. Trends Biochem. Sci. 30: 286–290 [DOI] [PubMed] [Google Scholar]
- 33. Perez-Villar JJ, O'Day K, Hewgill DH, Nadler SG, Kanner SB. 2001. Nuclear localization of the tyrosine kinase Itk and interaction of its SH3 domain with karyopherin alpha (Rch1alpha). Int. Immunol. 13: 1265–1274 [DOI] [PubMed] [Google Scholar]
- 34. Rawlings DJ, et al. 1993. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science 261: 358–361 [DOI] [PubMed] [Google Scholar]
- 35. Rawlings DJ, et al. 1996. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science 271: 822–825 [DOI] [PubMed] [Google Scholar]
- 36. Readinger JA, Mueller KL, Venegas AM, Horai R, Schwartzberg PL. 2009. Tec kinases regulate T-lymphocyte development and function: new insights into the roles of Itk and Rlk/Txk. Immunol. Rev. 228: 93–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sachdev S, Hoffmann A, Hannink M. 1998. Nuclear localization of IkappaB alpha is mediated by the second ankyrin repeat: the IkappaB alpha ankyrin repeats define a novel class of cis-acting nuclear import sequences. Mol. Cell. Biol. 18: 2524–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saito K, et al. 2003. BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity 19: 669–678 [DOI] [PubMed] [Google Scholar]
- 39. Samuels AL, Klinken SP, Ingley E. 2009. Liar, a novel Lyn-binding nuclear/cytoplasmic shuttling protein that influences erythropoietin-induced differentiation. Blood 113: 3845–3856 [DOI] [PubMed] [Google Scholar]
- 40. Schmidt EE, Ichimura K, Messerle KR, Goike HM, Collins VP. 1997. Infrequent methylation of CDKN2A(MTS1/p16) and rare mutation of both CDKN2A and CDKN2B(MTS2/p15) in primary astrocytic tumours. Br. J. Cancer 75: 2–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smith CI, et al. 2001. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays 23: 436–446 [DOI] [PubMed] [Google Scholar]
- 42. Taagepera S, et al. 1998. Nuclear-cytoplasmic shuttling of C-ABL tyrosine kinase. Proc. Natl. Acad. Sci. U. S. A. 95: 7457–7462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thomas JD, et al. 1993. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science 261: 355–358 [DOI] [PubMed] [Google Scholar]
- 44. Tsukada S, et al. 1993. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 72: 279–290 [DOI] [PubMed] [Google Scholar]
- 45. Vetrie D, et al. 1993. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 361: 226–233 [DOI] [PubMed] [Google Scholar]
- 46. Vihinen M, et al. 1997. BTKbase, mutation database for X-linked agammaglobulinemia (XLA). Nucleic Acids Res. 25: 166–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Webb CF, et al. 2000. The transcription factor Bright associates with Bruton's tyrosine kinase, the defective protein in immunodeficiency disease. J. Immunol. 165: 6956–6965 [DOI] [PubMed] [Google Scholar]
- 48. Yoshida K. 2008. Nuclear trafficking of proapoptotic kinases in response to DNA damage. Trends Mol. Med. 14: 305–313 [DOI] [PubMed] [Google Scholar]