

Abstract
The largest transcription factor IID (TFIID) subunit, TBP-associated factor 1 (TAF1), possesses protein kinase and histone acetyltransferase (HAT) activities. Both enzymatic activities are essential for transcription from a subset of genes and G1 progression in mammalian cells. TAF7, another TFIID subunit, binds TAF1 and inhibits TAF1 HAT activity. Here we present data demonstrating that disruption of the TAF1/TAF7 interaction within TFIID by protein phosphorylation leads to activation of TAF1 HAT activity and stimulation of cyclin D1 and cyclin A gene transcription. Overexpression and small interfering RNA knockdown experiments confirmed that TAF7 functions as a transcriptional repressor at these promoters. Release of TAF7 from TFIID by TAF1 phosphorylation of TAF7 increased TAF1 HAT activity and elevated histone H3 acetylation levels at the cyclin D1 and cyclin A promoters. Serine-264 of TAF7 was identified as a substrate for TAF1 kinase activity. Using TAF7 S264A and S264D phosphomutants, we determined that the phosphorylation state of TAF7 at S264 influences the levels of cyclin D1 and cyclin A gene transcription and promoter histone H3 acetylation. Our studies have uncovered a novel function for the TFIID subunit TAF7 as a phosphorylation-dependent regulator of TAF1-catalyzed histone H3 acetylation at the cyclin D1 and cyclin A promoters.
INTRODUCTION
Cell proliferation involves the coordinated expression of protein encoding genes that control progression through the cell cycle. These regulators include cyclin D1, a growth factor sensor that integrates extracellular signals with the core cell cycle machinery (35). Overexpression of cyclin D1, which accelerates entry into S phase, is frequently found in human cancers and is often associated with a poor prognosis (34). Common mechanisms for cyclin D1 overexpression in cancer cells are gene amplification and gene rearrangements, causing abnormally elevated levels of transcript and protein. Such genomic aberrations are not a feature of all cancer cells that overexpress cyclin D1, suggesting the involvement of alternative transcriptional upregulation mechanisms.
In eukaryotes, expression of protein-encoding genes is carried out by the RNA polymerase II (Pol II)-dependent transcription machinery. Initiation of transcription is mediated by members of either the TFIID or the SAGA (TFTC, PCAF, SAGA) family of coactivator complexes (8, 12, 27, 30, 38, 40). TFIID complexes contain the TATA-binding protein (TBP) and a set of TBP-associated factors (TAFs) (8, 38). The SAGA family of complexes does not contain TBP and instead is composed of the histone acetyltransferase Gcn5 and a subset of TAFs present in TFIID. Members of the SAGA family are essential for transcription of only 10% of yeast genes, which suggests that TFIID complexes are responsible for the majority of RNA Pol II-dependent transcription (17). Within TFIID, TBP and the ∼14 TAFs interact to form a trilobed structure, as determined by immunoelectron microscopy (20). TAF1, the largest subunit of TFIID, makes extensive contacts with TBP and many other TAFs, including TAF7.
Human TAF1 is a unique molecule in that it possesses intrinsic protein kinase, histone acetyltransferase (HAT), and ubiquitin-activating and -conjugating activities (6, 24, 29). We previously reported that TAF1 HAT activity is required for efficient transcription of cyclin D1 and cyclin A genes in mammalian cells (7, 16). The HAT domain of TAF1 is located in the central region of the protein and is highly conserved in all eukaryotes. In vitro, histones H3 and H4 and the basal transcription factors TFIIEβ and TFIIF have been identified as the substrates for TAF1 HAT activity (18, 24). Recent work has suggested that TAF7, another TFIID subunit, may play a pivotal role in the regulation of TAF1 HAT activity. Gegonne et al. identified TAF7 as a protein capable of directly binding to TAF1 and inhibiting TAF1 HAT activity (10). TAF1 also contains two independent serine/threonine kinase domains, one in the N terminus (NTK) and one in the C terminus (CTK) of the protein. Kinase activity has been demonstrated in vitro for human, yeast, and Drosophila TAF1 (6, 22, 23). Both domains are classified as atypical kinases and share little amino acid homology with each other; however, the NTK and CTK domains both are capable of autophosphorylation and transphosphorylation of substrates such as the TFIID subunit TAF7 (11).
In the ts13 mutant hamster cell line, a temperature-sensitive missense mutation in the HAT domain of TAF1 causes a G1/S phase cell cycle arrest at the nonpermissive temperature of 39.5°C (7, 14). These cells exhibit transcriptional downregulation of cyclins A, D1, and E but not c-fos or c-myc (32, 37, 39). Thus, TAF1 HAT inactivation does not induce a global defect in gene transcription but rather has an effect at only a subset of promoters. Transfection of TAF1 kinase mutants into ts13 cells failed to rescue the G1/S phase cell cycle arrest (26, 31). Thus, TAF1 HAT activity alone cannot promote G1/S phase progression, which suggests that the kinase activity of TAF1 also is required.
Here we present data in support of a model in which TAF7, when associated with the TFIID complex and bound to TAF1, serves as a negative regulator of TAF1 HAT activity and cyclin D1 and cyclin A gene transcription. Overexpression of TAF7 in HeLa cells inhibited cyclin D1 and cyclin A gene transcription and caused the cells to accumulate in early S phase. In contrast, depletion of TAF7 from TFIID complexes by small interfering RNA (siRNA) knockdown increased histone H3 acetylation at both cyclin promoters and stimulated cyclin D1 and cyclin A gene transcription. We found that TAF1 phosphorylation of TAF7 on serine-264 disrupted TAF7 binding to TAF1. Release of TAF7 from the TFIID complex via a phosphorylation-dependent mechanism activated TAF1 HAT activity and H3 histone acetylation at the cyclin D1 and cyclin A promoters. Expression of a TAF7 mutant, S264A, refractory to TAF1 phosphorylation, was even more effective at reducing H3 acetylation and transcription at target promoters than comparable levels of wild-type (WT) TAF7. Our studies have uncovered a novel function for the TAF7 subunit of TFIID as a phosphorylation-dependent transcriptional regulator and demonstrate that altering the subunit composition of TFIID can have profound consequences on TFIID function and gene transcription.
MATERIALS AND METHODS
Tissue culture cell lines.
HeLa cells were grown in Dulbecco's modified Eagle medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (HyClone), 2 mM l-glutamine, and penicillin-streptomycin in a humidified incubator containing 5% CO2 at 37°C. Sf9 insect cells were propagated in Hink's TNM-FH insect medium containing 10% FBS, l-glutamine, and penicillin-streptomycin. Cultures were grown at 27°C in the absence of CO2.
siRNA knockdown.
HeLa cells were seeded at 3.3 × 104 cells/well in a 24-well plate and maintained for 16 h in antibiotic-free DMEM containing 10% FBS. Cells were transfected with 50 nM siGENOME TAF7 siRNA (ThermoScientific) or nontargeting siRNA (Dharmacon) and 1 μl of DharmaFECT 3 reagent (Dharmacon). After 72 h of incubation at 37°C in antibiotic-free DMEM containing 10% FBS, cells were harvested for subsequent analysis.
RNA isolation and two-step qRT-PCR analysis.
RNA was harvested from cells using TRIzol reagent according to the manufacturer's protocol (Gibco BRL). Transcript levels were determined by two-step quantitative reverse transcription PCR (qRT-PCR). First, cDNA was synthesized from 1 μg of RNA using the iScript cDNA synthesis kit (Bio-Rad). One microliter of cDNA was analyzed by quantitative-PCR (q-PCR) on a MX3000 platform (Stratagene) using SsoFast EvaGreen Supermix (Bio-Rad) and primers for the TAF7, cyciln D1, cyclin E, cyclin A, c-fos, GAPDH (glyceraldehyde-3-phosphate dehydrogenase), and ARBP genes (primer sequences available upon request).
Transient transfections.
HeLa cells were seeded at 1 × 105 cells/well in a 24-well plate. Cells were transfected with 0.5 μg TAF7 expression plasmid using FuGENE HD transfection reagent (Roche) according to the manufacturer's protocol. Between 48 and 72 h posttransfection, cells were harvested and subjected to RNA expression, protein expression, or cell cycle analysis.
HeLa nuclear extracts and immunoprecipitation of TFIID.
For preparation of nuclear extracts, HeLa cells were resuspended in buffer A (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT]) and incubated on ice for 15 min. Cells were lysed by pushing through a 25-gauge needle 5 times. The crude nuclear pellet was isolated by centrifugation for 20 s at 12,000 × g, resuspended in buffer C (20 mM HEPES [pH 7.9], 25% [vol/vol] glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride [PMSF], 0.5 mM DTT), and incubated for 30 min at 4°C. After centrifugation for 5 min at 12,000 × g, the supernatant/nuclear extract was incubated with 0.5 μl anti-TBP polyclonal antibody (gift from R. Tjian) overnight at 4°C. Ten microliters of protein A–Sepharose CL-4B (GE Healthcare) was added, and the samples were incubated for 2 h at 4°C. The precipitated proteins were washed 5 times with buffer C and analyzed by Western blot or equilibrated with kinase buffer and used in kinase assays.
Cell cycle analysis.
HeLa cells were resuspended at 3 × 106 cells/ml, and cold 70% ethanol was added dropwise while vortexing. Cells were fixed for 1 h at 4°C, washed 2 times with phosphate-buffered saline (PBS), resuspended in propidium iodide (PI) staining buffer (1× PBS, 2% FBS, 50 μg/ml propidium iodide, 200 μg/ml RNase A, 0.1% Igepal), and incubated for 3 h at 4°C. Data were collected on a FACScan machine using CellQuest Pro software (Becton, Dickinson and Company) and analyzed using FlowJo software (Tree Star, Inc.).
Cell synchronization.
HeLa cells were synchronized using a thymidine/nocodazole block protocol. Cells were incubated for 24 h in DMEM with 10% FBS containing 2 mM thymidine (USB). Thymidine was removed by washing once with PBS, and the cells were released from the block by incubation in DMEM containing 10% FBS for 3 h. Nocodazole was added to the media at 100 ng/ml for 12 h to arrest the cells at G2/M. Cells were released into the cell cycle by washing with PBS, and fresh DMEM containing 10% FBS was added. Cells were collected by centrifugation at different time points for subsequent analysis.
Chromatin immunoprecipitations.
Synchronized HeLa cells, seeded on 10-cm plates, were cross-linked by the addition of 400 μl of 37% formaldehyde to 10 ml medium. After 15 min, cells were washed twice with PBS and lysed in 0.5 ml immunoprecipitation (IP) buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, and 0.5% Nonidet P-40) containing protease inhibitors. The lysed cells were centrifuged, and the nuclear pellet was collected, resuspended in 1.0 ml IP buffer, and sonicated 4 times for 20 s (Branson Sonifier 450, output 2, duty cycle 60). Half of the nuclear extract was incubated overnight at 4°C with either 1 μl of anti-TAF1 (gift from X. Liu), anti-TBP (gift from R. Tjian), anti-histone H3 K9K14Ac (Upstate), anti-histone H3 K9Ac (Upstate), or anti-TAF7 (gift from I. Davidson) antibody. The other half of the nuclear extract was incubated with mouse IgG (Abcam). Ten microliters protein A–Sepharose CL-4B (GE Healthcare) was added, and the samples were incubated for 2 h at 4°C. Immunoprecipitated complexes were washed five times with IP buffer, and bound DNA-protein complexes were eluted with 2× 250 μl elution buffer (1% SDS, 0.1 M NaHCO3). Cross-links were reversed by the addition of 20 μl 5 M NaCl and incubation overnight at 65°C. Samples were ethanol precipitated with glycogen as a carrier, and pellets were resuspended in 100 μl proteinase K buffer (10 mM Tris-HCl [pH 7.8], 5 mM EDTA, 0.5% SDS) and digested with 200 μg/ml proteinase K for 30 min at 50°C. DNA was purified by two phenol-chloroform extractions and one chloroform-isoamyl alcohol extraction, ethanol precipitated, and resuspended in 25 μl TE (10 mM Tris-HCl [pH 8.0], 1 mM EDTA [pH 8.0]). Twenty-five nanograms of purified chromatin immunoprecipitation (ChIP) DNA was amplified with SsoFast EvaGreen Supermix (Bio-Rad) using primers spanning the promoter of cyclin D1 and other genes. qPCRs were performed on the MX3000 platform (Stratagene).
In vitro kinase assays.
TFIID complexes immunoprecipitated from synchronized HeLa cell nuclear extracts were immobilized on anti-TBP antibody bound to protein A–Sepharose CL-4B (GE Healthcare). The immobilized proteins were incubated for 30 min at 30°C in 25 μl of phosphorylation buffer (25 mM HEPES [pH 7.9], 12.5 mM MgCl2, 100 mM KCl, 0.1 mM EDTA) with 1 μCi [γ-32P]ATP and 200 ng His-TAF7ΔN (comprising amino acids [aa] 103 to 349) as exogenous kinase substrate. Reaction products were subjected to SDS-PAGE and transferred to nitrocellulose, and the membrane was exposed to film to detect phosphorylated His-TAF7ΔN. The same membrane was probed overnight at 4°C with anti-TAF1 antibody (gift from X. Liu), diluted 1:10,000 in PBST (PBS containing 0.1% Tween 20), or with anti-His antibody (Bethyl), diluted 1:5,000 in PBST, to determine the total amount of TAF1 and His-TAF7ΔN in each reaction. The appropriate horseradish peroxidase (HRP)-conjugated secondary antibody was used at 1:5,000 to 1:10,000 dilutions. For kinase reactions that did not involve the addition of recombinant His-TAF7ΔN as the substrate, TFIID was immunoprecipitated from HeLa nuclear extracts that were fractionated on phosphocellulose as described previously (5). The 1.0 M KCl elution from the phosphocellulose column (P1.0) was incubated with anti-TBP antibody and protein A–Sepharose CL-4B and extensively washed. The immobilized proteins, equilibrated with the appropriate reaction buffer, were used in in vitro kinase assays. The identity of phosphorylated TFIID subunits was determined by immunoblotting with different TAF antibodies. Anti-TAF7 antibody (a gift from I. Davidson) was used at a 1:5,000 dilution and the anti-TAF5 monoclonal antibody (MAb 6B1) at a 1:2,500 dilution. To confirm the TAF7 phosphosites, 150 ng of baculovirus-expressed and purified glutathione S-transferase (GST)–TAF1 CTK was incubated with 250 ng of purified, bacterially expressed recombinant WT or mutant His-TAF7 under kinase conditions as described above. Reaction products were separated on SDS-polyacrylamide, and the TAF7 proteins were visualized by silver staining. Silver-stained SDS-polyacrylamide gels were dried between cellophane and subjected to autoradiography to detect phosphorylated His-TAF7.
Expression and purification of recombinant TAF1 and TAF7 proteins.
Full-length hemagglutinin (HA)-tagged TAF1, GST-tagged TAF1 CTK, and GST-tagged TAF1 C.RAP were expressed and purified from 15-cm plates of Sf9 cells infected with 1 ml of recombinant baculovirus as described previously (7). Proteins were affinity purified using an anti-HA antibody column or glutathione-Sepharose beads (GE Healthcare). For expression of His-tagged TAF7 proteins, cDNA of TAF7 was PCR amplified and subcloned into the BamHI and SalI sites of pET28a. TAF7 S264A and S264D mutants were created using the QuikChange site-directed mutagenesis kit (Stratagene). TAF7 expression constructs were transformed into BL21* cells for protein expression. Starter cultures of 2 ml were diluted into 100 ml LB containing 30 μg/ml kanamycin, and cells were grown to an optical density (OD) of 1.0 at 600 nm. TAF7 expression was induced by the addition of 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). After 16 h, cells were harvested and lysed in 0.4 M HEMG buffer (400 mM KCl, 25 mM HEPES [pH 7.9], 12.5 mM MgCl2, 0.5 mM EDTA, 0.1% NP-40, 10% glycerol). His-TAF7 was purified using Ni-nitrilotriacetic acid (NTA)- agarose (Qiagen) and competitively eluted from the beads with 200 mM imidazole.
In vitro histone acetyltransferase assay.
TFIID immunoprecipitated from the P1.0 phosphocellulose fraction of HeLa nuclear extracts and immobilized on protein A–Sepharose was washed 2 times with assay buffer (50 mM Tris-HCl [pH 8.0], 10% glycerol, 0.1 mM EDTA, 1 mM DTT, 0.2 mM PMSF) and resuspended in 30 μl assay buffer containing 300 ng histone H3 peptide (aa 1 to 20) and 0.25 μCi [3H]acetyl coenzyme A ([3H]acetyl-CoA) (Amersham). Reaction mixtures were incubated for 60 min at 30°C, filtered through P81 paper, and washed with 50 mM sodium carbonate (pH 9.2). The amount of acetylated H3 peptide retained on the filter was measured by liquid scintillation.
Mass spectrometry.
Excised Coomassie-stained gel bands containing TAF7 were subjected to in-gel proteolytic digestion with trypsin as described previously (36). Following gel slice digestion, the digestion products were desalted using micro-C18 ZipTips (Millipore) per the manufacturer's instructions and dried by vacuum centrifugation. The resulting peptide sample was resuspended in 14 μl of 0.1% formic acid, and 5 μl was analyzed by liquid chromatography/electrospray ionization-mass spectrometry (LC/ESI-MS) with a two-dimensional (2D) nano-high-pressure liquid chromatography (HPLC) system (Eksigent) coupled to a LTQ-OrbiTrapXL (Thermo Scientific) mass spectrometer using an LC-MS ion source configuration as described previously (21). The protein database search engine MASCOT (Matrix Science) version 1.01 was used to search a protein database appended with the TAF7 sequence. Search parameters included variable phosphorylation modification on serine and threonine and oxidation modification on methionine. Phosphopeptides identified with high confidence were manually validated by checking the mass deviation of the phosphopeptide identification (less than 5 ppm) and checking the thoroughness and quality of the fragmentation spectra.
RESULTS
TAF7 inhibits transcription at a subset of cell cycle genes.
The histone acetyltransferase (HAT) activity of TAF1, the largest subunit of TFIID, is required for transcription of cyclin D1, E, and A genes as well as G1-to-S phase progression (7, 14). It has been reported that TAF7, another TFIID subunit, binds to TAF1 and inhibits the HAT activity of TAF1 (10). These findings have led us to investigate whether TAF7 functions as an inhibitor of cyclin gene transcription. We performed siRNA knockdown experiments of TAF7 in HeLa cells and examined the effect of decreasing TAF7 protein levels on the transcriptional activity of different TAF1-dependent and -independent genes by quantitative RT-PCR. After 72 h of siRNA treatment, TAF7 mRNA (Fig. 1A) and total protein levels (data not shown) were reduced by 80 to 90% compared to cells treated with the nontargeting control siRNA. More importantly, the amount of TAF7 found incorporated into endogenous TFIID complexes also was reduced by ∼50% compared to that in control-treated cells (Fig. 1B). We observed that loss of TAF7 resulted in a 40% increase in cyclin D1 and cyclin A transcript levels but unexpectedly had no significant effect on the expression level of cyclin E, another TAF1-dependent cell cycle gene (Fig. 1A). Knockdown of TAF7 also had no effect on c-fos and GAPDH expression levels (Fig. 1A). These data suggest that TAF7 serves as an inhibitor of gene transcription but only at a subset of RNA polymerase II promoters.
Fig 1.

TAF7 functions as a transcriptional repressor at a subset of promoters. (A) HeLa cells were treated with 50 nM control (white bars) or TAF7 (black bars) siRNA for 72 h. Total RNA was collected, and transcript levels for TAF7, cyclin D1, cyclin A, cyclin E, GAPDH, and c-fos were determined by qRT-PCR. Results are averaged from 3 independent experiments, each done in triplicate. *, P < 0.05; **, P < 0.005; ***, P < 0.001. (B) HeLa cells were treated with siRNA as described for panel A. TFIID complexes were immunoprecipitated using an anti-TBP antibody. Precipitated proteins were separated on SDS-polyacrylamide, and the indicated TFIID subunits were detected by immunoblotting. (C) HeLa cells were transfected with pGLUE empty vector (white bars) or TAP-TAF7 expression plasmid (black bars). Forty-eight hours posttransfection, total RNA was collected and analyzed by qRT-PCR as described for panel A. Results are averaged from 3 independent experiments, each done in triplicate. ***, P < 0.001. (D) HeLa cells were transfected with TAP-TAF7 expression plasmid. After 48 h, immunoprecipitations were carried out using an anti-TBP antibody. Precipitated proteins were analyzed by SDS-PAGE and immunoblotted for the indicated TFIID subunits.
To gather additional evidence that TAF7 functions as a transcriptional repressor, we overexpressed TAF7 in HeLa cells with the expectation that increasing TAF7 protein levels will lead to a reduction in cyclin D1 gene transcription. Immunoprecipitation followed by Western blotting revealed that the exogenously expressed tandem affinity purification (TAP)-tagged TAF7 was incorporated into endogenous TFIID complexes (Fig. 1D). We observed that most of the TAP-TAF7 overexpressed in the cells remained as free protein, and only a small fraction of the total TAP-TAF7 was incorporated into the endogenous TFIID (data not shown). Despite this low incorporation efficiency, overexpression of TAF7 decreased cyclin D1 and cyclin A transcript levels by 50 to 75% but had no significant effect on cyclin E, c-fos, and GAPDH transcript levels (Fig. 1C), consistent with our knockdown data. These complementary loss and gain of function studies strongly indicate that TAF7 is an inhibitor of gene transcription and that its repressor function displays promoter selectivity.
TAF7 is a cell cycle regulator.
Cyclin D1 is required for progression through G1 into S phase (32). Our data suggest that TAF7 is a negative regulator of cyclin D1 expression, which prompted us to ask if altering TAF7 protein levels would change the dynamics of cell cycle progression. TAF7, as a yellow fluorescent protein (YFP) fusion protein, was overexpressed in HeLa cells, and the DNA content and cell cycle profiles of YFP-positive and YFP-negative cells were determined using propidium iodide staining and flow cytometry. After 72 h, we observed a significant increase in the percentage of YFP-positive, TAF7-overexpressing cells accumulating in early S phase compared to YFP-negative cells (Fig. 2). This accumulation of S phase cells coincided with a decrease in the number of YFP-positive cells in the subsequent G2 phase of the cell cycle (Fig. 2C). One interpretation for these results is that TAF7 overexpression causes cells to arrest just past the G1/S phase boundary. Therefore, TAF7 can serve as a novel and unexpected regulator of early S phase progression.
Fig 2.
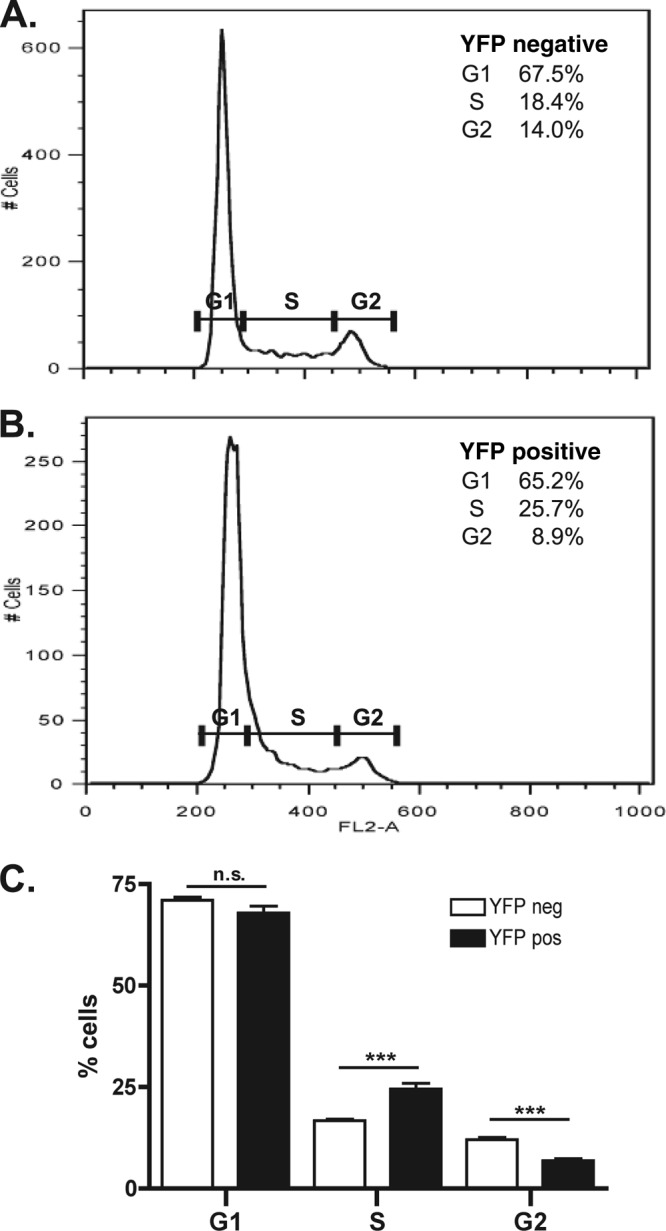
TAF7 is a regulator of early S phase progression. HeLa cells were transiently transfected with YFP-TAF7 expression plasmid. After 72 h, cells were fixed and stained with propidium iodide. YFP fluorescence and DNA content were determined by flow cytometry. The cell cycle profiles of YFP-negative (A) and YFP-positive (B) cells from one representative experiment are provided. Percentages of cells in each cell cycle phase, as defined by the indicated gates, are provided. (C) Cell cycle distribution of YFP-negative and YFP-positive cells from 11 independent transfections is shown. ***, P < 0.001; n.s., not significant.
TAF1 opposes TAF7 binding at the cyclin D1 and cyclin A promoters.
To elucidate the mechanism of TAF7's inhibitory function in gene transcription, the technique of chromatin immunoprecipitation (ChIP) was used to examine the localization of TAF1, TAF7, and TBP at the cyclin D1 and cyclin A promoters as cells progressed through the different stages of the cell cycle. HeLa cells were synchronized at G2/M phase using a thymidine/nocodazole block. At 4-hour intervals after removal of the drug, DNA content was analyzed by propidium iodide staining and flow cytometry. We observed that under these conditions, HeLa cells entered the G1 phase approximately 4 h after drug removal. Approximately 70% of the cells were in G1 at the 8-h time point and progressed into S phase at 12 h (Fig. 3A). ChIP experiments with anti-TAF1 and anti-TBP antibodies revealed peaks of TAF1 and TBP recruitment to the cyclin D1 promoter during G1, when cyclin D1 is highly expressed in proliferating cells (Fig. 3B, left panel). Peaks of TAF1 and TBP binding to the cyclin A promoter were observed at the G1/S phase boundary, consistent with the later time course of cyclin A induction during the cell cycle (Fig. 3B, right panel). ChIP analysis of TAF7 promoter occupancy was performed using an anti-TAF7 antibody (19TA) kindly provided by I. Davidson. The Davidson group showed that 19TA was able to immunoprecipitate both free and TFIID-bound TAF7 from cell lysates (19). We observed that at both the cyclin D1 and cyclin A promoters, high levels of TAF1 binding were associated with low levels of TAF7 (Fig. 3C). The decrease in TAF7 binding could not be explained by the release of TFIID, as TBP remained bound to the cyclin D1 and cyclin A promoters under conditions of low TAF7 promoter occupancy. The inverse correlation between TAF1 and TAF7 binding suggests that these two TFIID subunits have opposing functions in the regulation of cyclin D1 and cyclin A gene transcription.
Fig 3.

Inverse correlation between TAF1 and TAF7 binding at the cyclin D1 and cyclin A promoters. (A) HeLa cells were synchronized by thymidine/nocodazole block and collected at the indicated time points after drug removal. Percentage of G1 cells was determined by propidium iodide staining and flow cytometry. (B) Chromatin immunoprecipitation experiments were performed using anti-TAF1 or anti-TBP antibody, and samples were analyzed by qPCR using primers spanning the cyclin D1 and cyclin A promoters. The y axis shows the average percent input detected from one representative experiment carried out in triplicate. Time period when cells are predominantly in G1 is indicated by the shaded gray box. (C) ChIP experiments using anti-TAF7 antibody were carried out and analyzed as described for panel B.
TAF1 kinase activity opposes TAF7 binding at the cyclin D1 promoter.
Protein phosphorylation by TAF1 kinase has been shown to disrupt TAF7 binding to TAF1 in vitro (11). We asked if the near absence of TAF7 at the cyclin D1 promoter under conditions of high TAF1 occupancy could be mediated by TAF1 kinase activity. To address this question, TFIID was immunoprecipitated from HeLa nuclear extracts prepared from cells at different stages of the cell cycle. TAF1 kinase activity in the precipitated proteins was monitored using purified recombinant TAF7 as the exogenous substrate in an in vitro kinase assay. We found that phosphorylation of TAF7 was stimulated in early G1, approximately 4 h after release from the nocodazole block, and remained elevated for approximately 4 to 6 additional hours (Fig. 4A). Compared to the TAF7 occupancy profile shown in Fig. 3C, high levels of TAF1 kinase activity coincided with low levels of TAF7 binding to the cyclin D1 promoter and vice versa (Fig. 4B). These results are in agreement with our hypothesis that TAF1 phosphorylation of TAF7 regulates the association of TAF7 with TAF1 and thus with the TFIID complex.
Fig 4.

TAF7 phosphorylation by TAF1 kinase hinders their interaction. (A) HeLa cells were synchronized using a thymidine/nocodazole block and collected at the indicated time points after drug removal. For TAF1 kinase activity, HeLa nuclear extracts were prepared and immunoprecipitated with anti-TBP Sepharose beads. Bound proteins were added to in vitro kinase assays using recombinant His-TAF7 as the substrate. Phosphorylated TAF7 was detected by autoradiography (p-TAF7). Total TAF1 and His-TAF7 protein levels were monitored by Western blotting. (B) Phosphorylated TAF7 in kinase assays was quantified using ImageJ software. TAF1 kinase activity is expressed as intensity of TAF7 phosphorylation corrected for total TAF1 protein. Kinase activity was compared to TAF7 ChIP data presented in Fig. 3C. (C) Purified baculovirus-expressed TAF1 fragments, GST-CTK and GST-C.RAP, and bacterially expressed His-TAF7 were visualized by Coomassie blue staining. (D) Purified His-TAF7 preincubated with buffer (mock) or TAF1 CTK (pre-phos) was isolated using Ni-agarose beads (input) and incubated with C-terminal TAF1 fragment (GST-C.RAP). Proteins coprecipitating with GST-C.RAP (IP: Glut-Seph) were detected by immunoblotting with the indicated antibody. (E) TAF1 binding was quantified using ImageJ software. *, P < 0.05; n = 5. (F) Schematic diagram of full-length TAF1 and TAF1 fragments used in panels D and E are shown. Domains shown are as follows: NTK, N-terminal kinase; HAT, histone acetyltransferase domain; RAPiD, TAF7 interaction domain; and CTK, C-terminal kinase.
TAF1 phosphorylation of TAF7 disrupts their protein-protein interaction.
TAF1 possesses two independent serine/threonine kinase domains. Each of these domains is capable of autophosphorylation and transphosphorylation of substrates (6). It is unknown whether TAF1 autophosphorylation, transphosphorylation of TAF7, or both functions are necessary for the dissociation of TAF7 from TAF1 within the TFIID complex. Therefore, we examined the consequences of transphosphorylating TAF7 on TAF1 protein binding. Bacterially expressed and purified TAF7, with an N-terminal histidine tag (Fig. 4C, His-TAF7), was preincubated under kinase conditions in the absence or presence of purified GST-tagged TAF1 C-terminal kinase domain (Fig. 4C, GST-CTK). The GST-CTK fragment of TAF1 (Fig. 4F, CTK) lacks the interaction domain for TAF7 (RAPiD) but retains the ability to transphosphorylate TAF7. The mock phosphorylated and prephosphorylated His-TAF7 proteins were affinity purified away from the GST-CTK fragment using Ni-NTA-agarose beads. The isolated TAF7 proteins were subsequently incubated in the absence of any ATP with a purified C-terminal TAF1 fragment that includes the TAF7 interaction domain, CTK domain, and an N-terminal GST tag (Fig. 4C, GST-C.RAP; Fig. 4F, C.RAP). The amount of TAF7 that coimmunoprecipitated with the GST-C.RAP fragment was determined by immunoblotting. We observed that prephosphorylated TAF7 exhibited a 50% reduction in C.RAP TAF1 binding compared to the mock phosphorylated protein (Fig. 4D and E). These results demonstrate that transphosphorylation of TAF7 significantly lowers its binding affinity for the TAF1 protein.
Phosphorylation and release of TAF7 activate the TAF1 HAT domain.
We have found that transphosphorylation of TAF7 can disrupt the interaction of TAF7 with TAF1. Thus, will TAF1 phosphorylation of TAF7 be sufficient to stimulate release of TAF7 from the TFIID complex and lead to activation of TAF1 HAT activity? Intact TFIID complexes were immunoprecipitated from partially fractionated HeLa cell nuclear extracts enriched for TFIID using an anti-TBP polyclonal antiserum (38). Immunoprecipitated proteins, visualized by silver staining (Fig. 5A, SS), were incubated with radiolabeled ATP under kinase reaction conditions to allow for protein phosphorylation. Kinase reaction products were transferred to nitrocellulose following SDS-PAGE and analyzed by autoradiography and immunoblotting to identify the phosphorylated proteins (Fig. 5A, kinase and IB). As previously shown, TAF1 was autophosphorylated in the context of TFIID; we also detected transphosphorylation of TAF7. In parallel reactions, the TFIID immobilized on Sepharose beads was preincubated under kinase conditions in the absence or presence of 1 mM cold ATP to allow for protein phosphorylation, and the dissociation of TAF1, TBP, and TAF7 from TFIID was subsequently examined. The amount of each TFIID subunit either found in the unbound supernatant (S) or associated with TFIID complexes (B) was determined by immunoblotting and quantified using ImageJ software (Fig. 5B and C). We observed that incubation with ATP increased the amount of TAF7 found in the unbound supernatant by ∼50%, representing an increase in TAF7 subunits no longer associated with the TFIID complex. This increase in TAF7 in the supernatant was accompanied by a corresponding decrease in the amount of TAF7 bound to TFIID (Fig. 5C). In contrast, the level of TAF1 and TBP detected in the unbound supernatant remained unaffected by ATP. These findings further suggest that the association of TAF7 with the TFIID complex is regulated by protein phosphorylation.
Fig 5.

Phosphorylation disrupts TAF7 binding and stimulates TAF1 HAT activity. (A) TFIID was immunoprecipitated from fractionated HeLa nuclear extracts (enriched for TFIID) with an anti-TBP antibody and visualized by silver staining (SS). Proteins precipitated in the absence of antibody also are shown. In vitro kinase assays were performed with the precipitated proteins. Reaction products were separated on SDS-polyacrylamide, transferred to nitrocellulose, visualized by autoradiography (kinase), and subjected to immunoblotting (IB). The positions of molecular weight standards are shown. (B) TFIID was immunoprecipitated as described for panel A. In vitro kinase assays using cold ATP were carried out with TFIID immobilized on anti-TBP Sepharose beads. The supernatant (S) and bound (B) fractions were collected, and the amount of TAF1, TBP, and TAF7 present was determined by immunoblotting. (C) Signal intensities were quantified using ImageJ software, and the percent total for each protein in the bound and supernatant fractions was calculated. *, P < 0.05; n = 3. (D) Immunoprecipitated TFIID was incubated in the absence (TFIID) or presence (pre-phos TFIID) of cold ATP prior to assaying for HAT activity. [3H]acetyl-CoA incorporation into a human histone H3 peptide (aa 1 to 20) was measured by liquid scintillation. Results from one representative experiment are shown. **, P < 0.005; n = 3. (E) HeLa cells were treated with 50 nM control (white bars) or TAF7 (black bars) siRNA for 72 h. ChIPs were performed using anti-histone H3 K9K14Ac antibody and analyzed by qPCR at the indicated promoters. The y axis shows fold change in H3 K9K14Ac relative to control-treated cells. *, P < 0.05; ***, P < 0.001; n = 3.
The only component of TFIID shown to possess histone acetyltransferase activity is the TAF1 subunit, and histone H3 is a substrate for TAF1 HAT activity (24). We used these two pieces of information to investigate the role of TAF7 phosphorylation in regulating TAF1 HAT activity. TFIID, immunoprecipitated from phosphocellulose-fractionated HeLa nuclear extracts, was preincubated with ATP to promote TAF7 phosphorylation and release from the TFIID complex. The level of histone acetyltransferase activity was subsequently measured using a histone H3 N-terminal peptide (aa 1 to 20) as the substrate in an in vitro HAT assay (Fig. 5D). The expectation is that release of TAF7, an inhibitor of TAF1 HAT activity, would lead to an increase in acetylation of the H3 peptide. We found that preincubation of TFIID with ATP (prephosphorylated TFIID) increased the level of H3 acetylation compared to preincubation in the absence of ATP (TFIID). Our protein binding and acetyltransferase assays suggest that dissociation of TAF7 from the TFIID complex via a phosphorylation-dependent mechanism stimulates TAF1 HAT activity.
To gather evidence that TAF7 functions as a regulator of TAF1 HAT activity in vivo, we used TAF7 siRNA knockdown to decrease the amount of TAF7 within TFIID complexes and carried out ChIP experiments to examine the level of H3 acetylation at a number of different promoters. We reported previously that acetylation of histone H3-K9K14 by the TAF1 HAT domain is necessary for efficient cyclin D1 promoter function (7, 16). Treatment with TAF7 siRNA led to an increase in H3-K9K14 acetylation levels at both the cyclin D1 and cyclin A promoters (Fig. 5E). Changes in H3 acetylation levels were not observed at the cyclin E, c-fos, and GAPDH promoters (Fig. 5E), consistent with our earlier finding that the transcriptional activity of these promoters is not subject to TAF7 repression (Fig. 1C). These data suggest that TAF7 is a biologically significant negative regulator of TAF1 HAT activity in vivo.
TAF1 phosphorylates TAF7 on serine-264 to regulate protein binding of TFIID subunits.
To gain more insight into the molecular aspects of this regulatory mechanism, we set out to map the relevant sites of phosphorylation. Mass spectrometry analysis of peptides derived from TAF7 overexpressed in Sf9 and HEK293 cells identified two potential phosphosites on TAF7, serine-159 and serine-264 (Table 1). To determine if TAF1 is the kinase responsible for these posttranslational modifications, serine-to-alanine point mutants of TAF7 at residues 159 and 264 were created, expressed in bacteria, and used as the substrates for TAF1 in in vitro kinase assays. While the S159A mutant was phosphorylated at levels similar to those of WT TAF7, S264A exhibited a large reduction in total phosphorylation levels, suggesting that this amino acid is a substrate for TAF1 kinase activity (Fig. 6A). The level of phosphorylation for the S264 mutant, although reduced compared to that of WT TAF7, was still above background, suggesting that other serine or threonine residues in the TAF7 protein can serve as the substrates for TAF1 kinase activity. Next, we engineered a phosphomimetic serine-to-aspartic acid mutation at S264 (S264D) to test if phosphorylation of TAF7 S264 is sufficient to disrupt TAF1 binding. Comparable to what was observed for prephosphorylated TAF7 (Fig. 4E), the phosphomimetic S264D mutant showed a 50% reduction in TAF1 C.RAP binding compared to WT TAF7 and the S264A mutant, which cannot be phosphorylated on serine-264 (Fig. 6B and C). These data suggest that TAF1 phosphorylation of TAF7, more specifically at S264, can disrupt their protein-protein interaction.
Table 1.
TAF7 phosphopeptides identified by mass spectrometry
| Phosphopeptidea | Positionb | P value |
|---|---|---|
| YIEpSPDVEK | S159 | 0.920 |
| YIEpSPDVEKEVKR | S159 | 0.950 |
| LNEpSDEQHQENEGTNQLVMGIQK | S264 | 0.996 |
| QLQDKLNEpSDEQHQENEGTNQLVMGIQK | S264 | 0.991 |
A “p” precedes the phosphorylated amino acid.
The position of the phosphorylated amino acid in TAF7 protein.
Fig 6.
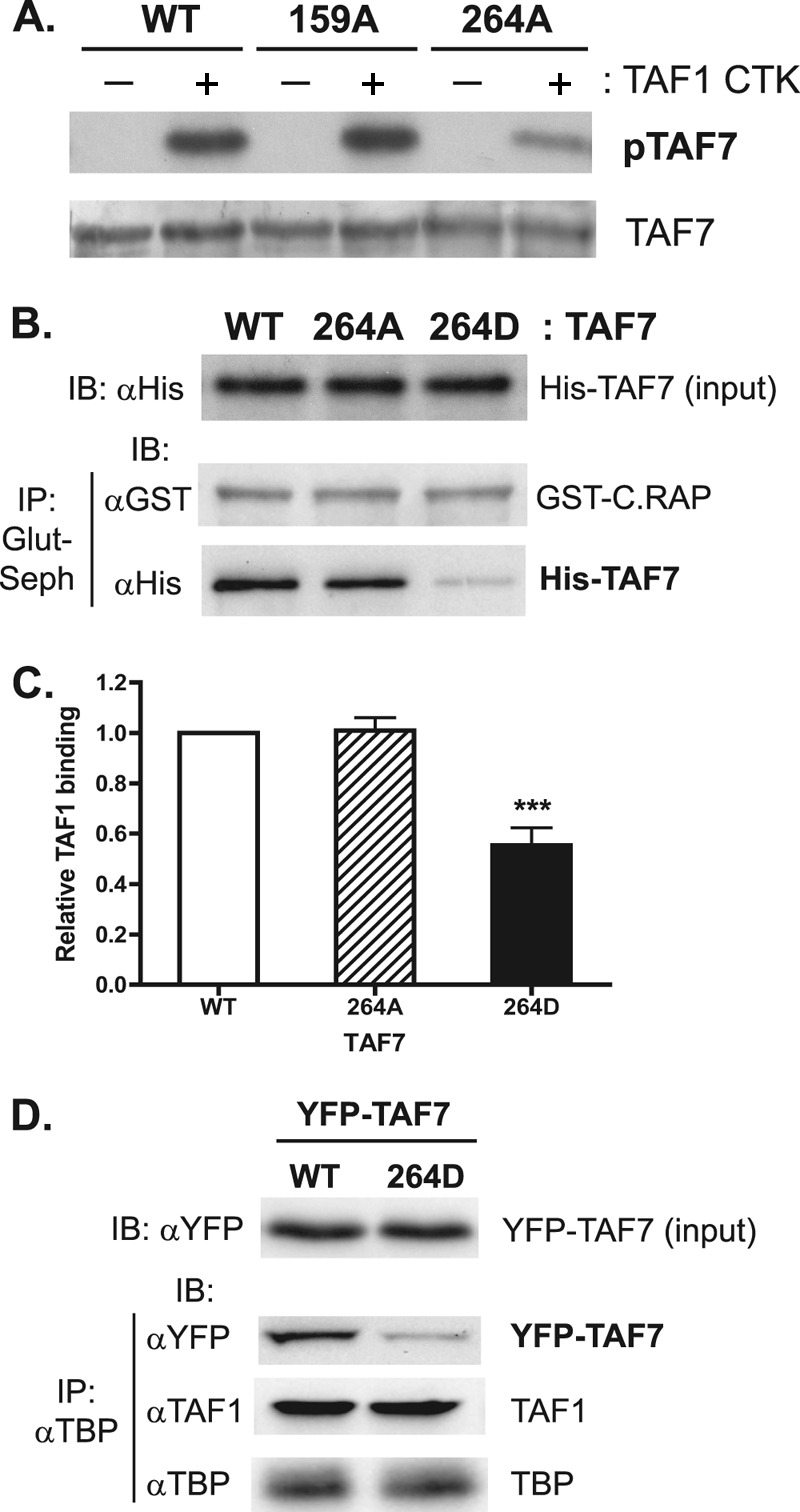
TAF1 phosphorylation of TAF7 at serine-264 disrupts their protein interaction. (A) His-tagged WT and TAF7 mutant proteins were expressed in bacteria, affinity purified, and used as the substrates for TAF1 CTK. Phosphorylated TAF7 was detected by autoradiography (pTAF7). Equal loading of TAF7 substrates was confirmed by silver staining (TAF7). (B) The indicated His-tagged WT and TAF7 mutant proteins (input) were incubated with GST-TAF1 C.RAP. Pulldowns with glutathione-Sepharose were performed, and precipitated proteins were subjected to SDS-PAGE and immunoblotting. (C) TAF1 binding was quantified as described for Fig. 4E. ***, P < 0.001; n = 4. (D) The YFP-tagged TAF7 WT or S264D mutant was transiently expressed in HeLa cells for 48 h. Nuclear extracts were prepared, and total levels of the YFP-tagged TAF7 WT and S264D mutant were determined by immunoblotting (input) using anti-YFP antibody. TFIID was immunoprecipitated using an anti-TBP antibody, and the indicated subunits were detected by immunoblotting.
To demonstrate that phosphorylation of S264 is biologically relevant, we examined the effect of this posttranslational modification on the efficiency of TAF7 incorporation into endogenous TFIID complexes in vivo. For these studies, YFP-tagged WT or the TAF7 S264D mutant was expressed in HeLa cells by transient transfection, and the amount of exogenously expressed TAF7 present in endogenously assembled TFIID complexes was determined. We found that when expressed at comparable levels, the phosphomimetic TAF7 S264D mutant was incorporated less efficiently into TFIID complexes than its WT counterpart (Fig. 6D). No change in the levels of TAF1 and TBP found in TFIID was observed with expression of the phosphomimetic mutant. These studies demonstrate that the phosphorylation state of TAF7 on S264 can dictate the subunit composition of TFIID in cells.
Regulation of gene expression and histone H3 acetylation by TAF7 serine-264 phosphorylation.
Next, we wanted to explore the consequences of overexpressing the TAF7 S264A and S264D mutants on cyclin D1 and cyclin A gene transcription. Following transfection of the empty vector, WT TAF7, S264A, or S264D expression plasmid into HeLa cells (Fig. 7A), cyclin D1, cyclin A, and c-fos transcript levels were measured by qRT-PCR. We found that the S264A mutant, which should be refractory to regulation by TAF1 phosphorylation, was even more effective at repressing cyclin D1 and cyclin A gene transcription than WT TAF7 (Fig. 7B and C, left panels). To our surprise, the S264D mutant actually had a stimulatory effect on cyclin D1 and cyclin A gene transcription and appeared to be functioning as a dominant negative mutant (Fig. 7B and C, left panels). One possible explanation for this unexpected increase in mRNA expression is that the S264D mutant competes with the endogenous WT TAF7 for assembly into TFIID complexes. Due to the reduced ability of S264D to remain associated with TFIID, the outcome is an increase in TFIID complexes lacking a TAF7 subunit, leading to cyclin D1 and cyclin A expression above normal levels. As expected, neither WT TAF7 nor the TAF7 mutants had any effect on the transcription levels of the c-fos gene (Fig. 7D, left panel). These data further support the hypothesis that the inhibitory function of TAF7 is regulated by serine-264 phosphorylation.
Fig 7.

TAF7 S264 phosphorylation stimulates cyclin D1 and cyclin A gene transcription and promoter H3 acetylation. (A) HeLa cells were transfected with TAP-tagged control (empty), TAP-TAF7 (WT), TAP-S264A, or TAP-S264D plasmid. After 48 h, whole-cell extracts were prepared and expression levels of TAF7 proteins were determined by immunoblotting using an anti-HA antibody. GAPDH protein levels were measured to ensure equal protein loading. (B to D) For RNA expression (left panels), total RNA was collected and transcript levels for cyclin D1 (B), cyclin A (C), and c-fos (D) were measured by qRT-PCR. The y axis shows mRNA levels relative to cells transfected with the empty control plasmid (n = 3). (A to D, right panels) In parallel transfections, H3 K9 acetylation levels at the indicated promoters were determined by ChIP using anti-histone H3 K9Ac antibody and qPCR. The y axis shows H3 acetylation levels relative to cells transfected with the empty control plasmid for each promoter (n = 3). *, P < 0.05; **, P < 0.005; ***, P < 0.001.
According to our model, the inability of TAF1 to phosphorylate the S264A mutant should produce TFIID complexes in which TAF7 cannot be dissociated by phosphorylation, thus inhibiting TAF1 HAT activity and reducing histone H3 acetylation at target promoters. We used ChIP to investigate the levels of histone H3 acetylation at the cyclin D1, cyclin A, and c-fos promoters following expression of the TAF7 WT and S264A and S264D mutants in HeLa cells (Fig. 7, right panels). In agreement with our gene expression results, promoter-selective inhibition of H3 acetylation was observed with the TAF7 WT at both cyclin loci; however, the TAF7 S264A mutant was the most effective at reducing H3 acetylation levels at these loci while having no effect at the c-fos promoter (Fig. 7B to D, right panels). Once again, the S264D mutant was an ineffective repressor of H3 acetylation in comparison to the TAF7 WT and S264A mutant. These ChIP results establish a connection between phosphorylation of TAF7, histone H3 acetylation, and gene regulation at the cyclin D1 and cyclin A promoters.
DISCUSSION
Our studies have identified the TFIID subunit TAF7 as a novel regulator of cyclin D1 and cyclin A gene transcription. We propose that activation of cyclin D1 and cyclin A gene transcription can occur via a phosphorylation-dependent mechanism in which TAF7 is phosphorylated by the TAF1 subunit of TFIID. This phosphorylation event results in release of TAF7 from the TFIID complex, stimulation of TAF1 HAT activity, acetylation of core promoter histones, and increased transcription. The phosphorylation and dissociation of TAF7 from TFIID complexes at the cyclin D1 and cyclin A promoters is most prevalent during the G1 phase of the cell cycle and potentially represents a novel downstream event of mitogenic signaling pathways that induce cyclin D1 and cyclin A gene transcription.
Acetylation of histones is well established as an important mechanism for activating gene transcription. Much effort has been placed on the characterization of histone acetyltransferases (HATs), enzymes that catalyze these posttranslational modifications. HATs can be regulated through a variety of molecular mechanisms. Autoacetylation within the activation loop of p300 and phosphorylation of CREB binding protein (CBP) stimulate their rates of histone acetylation (1, 13). CBP HAT activity also can be stimulated by its interaction with select transcriptional activators (2, 13). We have discovered that TAF1 HAT activity is regulated by an inhibitory protein interaction that is disrupted by protein phosphorylation. Intriguingly, the protein kinase responsible resides in TAF1, the same polypeptide that possesses the HAT domain subject to regulation. To our knowledge, TAF1 is the first example of a polypeptide that possesses two catalytic activities that are functionally connected. Whether this mode of regulation takes place within a single molecule in cis or between two TAF1 proteins in trans remains to be determined.
ts13 cells are conditional mutants that arrest in the late G1 phase of the cell cycle when shifted to the nonpermissive temperature of 39.5°C. Characterization of these mutant cells established that the G1/S phase cell cycle arrest is due to a missense mutation in the TAF1 HAT domain that abrogates catalytic activity (7, 14). Expression of WT TAF1 rescued the ts13 cell cycle arrest while expression of HAT-deficient mutants was ineffective (7). Unexpectedly, the introduction of TAF1 constructs containing disruptive kinase domain mutations also failed to complement the ts13 mutant phenotype (26). These data indicate that both TAF1 HAT and kinase activities are required for normal cell cycle progression. Our studies have extended these findings by placing both enzymatic activities of TAF1 in the same signaling pathway and adding to our molecular understanding of their function during G1 to S phase progression in mammalian cells.
We and others have reported previously that the HAT activity of TAF1 is required for efficient transcription from a subset of protein encoding genes (25, 31, 33, 39). TAF7 is a negative regulator of TAF1 HAT activity and inhibits transcription from only a subset of genes (10). Based on these results, we anticipated that transcription levels of genes driven by promoters dependent on TAF1 HAT activity also would be subject to inhibitory actions by TAF7. Unexpectedly, elevating TAF7 levels only inhibited transcription of the TAF1-dependent cyclin A and D1 genes and had no effect on cyclin E gene transcription, another gene that requires TAF1 HAT activity for efficient promoter activity. These data indicate that although the subset of genes dependent on TAF1 HAT activity overlaps genes regulated by TAF7, these two subpopulations are not identical. One possible explanation is that the inhibitory effects of TAF7 can be bypassed by the recruitment of another enzyme that catalyzes the histone modifications necessary for efficient gene transcription. Further studies will be necessary to identify additional TAF1-dependent genes resistant to the inhibitory effects of TAF7 and to determine the specific set of factors present at each of these promoters that accounts for their differential regulation.
Overexpression of TAF7 was sufficient to inhibit cyclin D1 and cyclin A mRNA expression levels. A question that comes to mind is “how does TAF7 overexpression mechanistically work in the cell to repress gene transcription?” One hypothesis is that excess levels of TAF7 protein would change the dynamics of TFIID assembly and create more complexes containing TAF7. This shift in TFIID composition would increase the number of DNA-bound TAF7-containing complexes at the cyclin D1 and cyclin A promoters, thereby inhibiting transcription of these genes.
When first isolated, it was generally thought that the subunit composition of a functional TFIID complex was invariant and contained a defined set of TAFs conserved from yeast to humans. Here we present data suggesting that TAF7 dissociates from the TFIID complex following phosphorylation by the TAF1 kinase, as cells transition through the G1 phase of the cell cycle (Fig. 4B and 5B). Immunoelectron microscopy performed with a TAF7 antibody mapped TAF7 at the periphery of the TFIID complex, near the TAF1 HAT domain (20). This peripheral localization suggests that TAF7 could easily enter and exit the TFIID complex. Our findings are consistent with recent studies that have demonstrated TFIID is variable and dynamic in both its subunit composition and overall structure. Several groups have identified TAF variants that incorporate into the TFIID complex at different stages of development or in different cell types (9, 15). The disruption and replacement of the canonical TFIID with a novel complex composed of the TBP-related factor 3 (TRF3) and TAF3 also have been reported in myoblasts undergoing differentiation into myotubes (4). In addition, TAF subunits originally identified in TFIID can be found in other transcription regulatory complexes (40). Therefore, regulating the composition of canonical and noncanonical TFIID complexes represents an important mechanism for controlling gene transcription.
Loss of the TAF2 subunit of TFIID relative to the other subunits has been repeatedly observed during the purification of TFIID from yeast and human cells, suggesting that the TAF2 subunit readily dissociates from the complex (3). Subsequent structural analysis of yeast TFIID by cryoelectron microscopy and electron tomography confirmed the existence of two subpopulations, TFIID complexes containing the TAF2 subunit and those lacking TAF2 (28). In the absence of TAF2, significant reorganization of different domains was observed, suggesting that TFIID demonstrates significant plasticity and is capable of varying its overall structure. However, the presence of TAF2 appeared to stabilize one of four abundant states identified for TFIID. The ts13 single missense mutation in TAF1, which disrupts the ability of TAF1 to acetylate histones, is thought to shift the TAF1 protein to an inactive state for HAT activity under nonpermissive conditions. These results have led us to hypothesize that TAF7 binds to TAF1 and locks the TFIID complex into a conformation in which the structure of the TAF1 HAT domain is no longer favorable for catalytic activity. To test this hypothesis, we have established a collaboration to determine the structure of a TAF1-TAF7 dimer using X-ray crystallography.
We have discovered a novel role for TAF7 as a negative regulator of transcription and cell cycle progression. Our studies have determined that activation of cyclin D1 and cyclin A gene transcription requires a previously uncharacterized phosphorylation-dependent switch in the composition of TFIID. Phosphorylation catalyzed by the TAF1 kinase leads to dissociation of the TAF7 subunit from TFIID and activation of TAF1 HAT activity. There is a growing body of evidence that TFIID is a highly dynamic molecule, and we now demonstrate that altering its subunit composition can have profound consequences on TFIID function and gene transcription.
ACKNOWLEDGMENTS
Many thanks are given to A. Weinmann and K. Bomsztyk for critical reading of the manuscript and providing insightful comments. We are indebted to R. Moon for the TAP-tagged expression plasmid, C. Hague for the YFP expression plasmid, and D. Singer for the FLAG-TAF7 expression construct. We also thank R. Tjian, X. Liu, and I. Davidson for generously providing antibodies for TBP, TAF1, and TAF7, respectively. We especially thank members of the Wang lab for invaluable technical support and discussions.
S.L.K was supported in part by Public Health Service, National Research Service Award, training grant 2T32 GM007270, from the National Institute of General Medical Sciences. This work was supported by research grant RSG-04-234-01-GMC from the American Cancer Society and by bridge funding from the University of Washington.
Footnotes
Published ahead of print 18 June 2012
REFERENCES
- 1. Ait-Si-Ali S, et al. 1998. Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature 396:184–186 [DOI] [PubMed] [Google Scholar]
- 2. Chen C, Deng Z, Kim A, Blobel G, Lieberman P. 2001. Stimulation of CREB binding protein nucleosomal histone acetyltransferase activity by a class of transcriptional activators. Mol. Cell Biol. 21:476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cler E, Papai G, Schultz P, Davidson I. 2009. Recent advances in understanding the structure and function of general transcription factor TFIID. Cell Mol. Life Sci. 66:2123–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deato M, Tjian R. 2007. Switching of the core transcription machinery during myogenesis. Genes Dev. 21:2137–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dignam JD, Martin PL, Shastry BS, Roeder RG. 1983. Eukaryotic gene transcription with purified components. Methods Enzymol. 101:582–598 [DOI] [PubMed] [Google Scholar]
- 6. Dikstein R, Ruppert S, Tjian R. 1996. TAF(II)250 is a bipartite protein kinase that phosphorylates the basal transcription factor RAP74. Cell 84:781–790 [DOI] [PubMed] [Google Scholar]
- 7. Dunphy E, Johnson T, Auerbach S, Wang E. 2000. Requirement for TAF(II)250 acetyltransferase activity in cell cycle progression. Mol. Cell Biol. 20:1134–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dynlacht BD, Hoey T, Tjian R. 1991. Isolation of coactivators associated with the TATA-binding protein that mediate transcriptional activation. Cell 66:563–576 [DOI] [PubMed] [Google Scholar]
- 9. Freiman R, et al. 2001. Requirement of tissue-selective TBP-associated factor TAF(II)105 in ovarian development. Science 293:2084–2087 [DOI] [PubMed] [Google Scholar]
- 10. Gegonne A, Weissman J, Singer D. 2001. TAFII55 binding to TAFII250 inhibits its acetyltransferase activity. Proc. Natl. Acad. Sci. U. S. A. 98:12432–12437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gegonne A, Weissman J, Zhou M, Brady J, Singer D. 2006. TAF7: a possible transcription initiation check-point regulator. Proc. Natl. Acad. Sci. U. S. A. 103:602–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grant P, et al. 1998. A subset of TAF(II)s are integral components of the SAGA complex required for nucleosome acetylation and transcriptional stimulation. Cell 94:45–53 [DOI] [PubMed] [Google Scholar]
- 13. Hamamori Y, et al. 1999. Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral oncoprotein E1A. Cell 96:405–413 [DOI] [PubMed] [Google Scholar]
- 14. Hayashida T, et al. 1994. The CCG1/TAF(II)250 gene is mutated in thermosensitive G1 mutants of the BHK-21 cell-line derived from golden-hamster. Gene 141:267–270 [DOI] [PubMed] [Google Scholar]
- 15. Hiller M, Lin T, Wood C, Fuller M. 2001. Developmental regulation of transcription by a tissue-specific TAF homolog. Genes Dev. 15:1021–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hilton T, Li Y, Dunphy E, Wang E. 2005. TAF1 histone acetyltransferase activity in Sp1 activation of the cyclin D1 promoter. Mol. Cell Biol. 25:4321–4332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huisinga K, Pugh B. 2004. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol. Cell 13:573–585 [DOI] [PubMed] [Google Scholar]
- 18. Imhof A, et al. 1997. Acetylation of general transcription factors by histone acetyltransferases. Curr. Biol. 7:689–692 [DOI] [PubMed] [Google Scholar]
- 19. Lavigne AC, et al. 1996. Multiple interactions between hTAFII55 and other TFIID subunits. Requirements for the formation of stable ternary complexes between hTAFII55 and the TATA-binding protein. J. Biol. Chem. 271:19774–19780 [DOI] [PubMed] [Google Scholar]
- 20. Leurent C, et al. 2004. Mapping key functional sites within yeast TFIID. EMBO J. 23:719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Licklider LJ, Thoreen CC, Peng J, Gygi SP. 2002. Automation of nanoscale microcapillary liquid chromatography-tandem mass spectrometry with a vented column. Anal. Chem. 74:3076–3083 [DOI] [PubMed] [Google Scholar]
- 22. Maile T, Kwoczynski S, Katzenberger R, Wassarman D, Sauer F. 2004. TAF1 activates transcription by phosphorylation of serine 33 in histone H2B. Science 304:1010–1014 [DOI] [PubMed] [Google Scholar]
- 23. Matangkasombut O, Buratowski R, Swilling N, Buratowski S. 2000. Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev. 14:951–962 [PMC free article] [PubMed] [Google Scholar]
- 24. Mizzen C, et al. 1996. The TAF(II)250 subunit of TFIID has histone acetyltransferase activity. Cell 87:1261–1270 [DOI] [PubMed] [Google Scholar]
- 25. O'Brien T, Tjian R. 2000. Different functional domains of TAF(II)250 modulate expression of distinct subsets of mammalian genes. Proc. Natl. Acad. Sci. U. S. A. 97:2456–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Brien T, Tjian R. 1998. Functional analysis of the human TAF(II)250 N-terminal kinase domain. Mol. Cell 1:905–911 [DOI] [PubMed] [Google Scholar]
- 27. Ogryzko V, et al. 1998. Histone-like TAFs within the PCAF histone acetylase complex. Cell 94:35–44 [DOI] [PubMed] [Google Scholar]
- 28. Papai G, et al. 2009. Mapping the initiator binding Taf2 subunit in the structure of hydrated yeast TFIID. Structure 17:363–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pham A, Sauer F. 2000. Ubiquitin-activating/conjugating activity of TAFII250, a mediator of activation of gene expression in Drosophila. Science 289:2357–2360 [DOI] [PubMed] [Google Scholar]
- 30. Pugh BF, Tjian R. 1991. Transcription from a TATA-less promoter requires a multisubunit TFIID complex. Genes Dev. 5:1935–1945 [DOI] [PubMed] [Google Scholar]
- 31. Ruppert S, Wang E, Tjian R. 1993. Cloning and expression of human TAFII250: a TBP-associated factor implicated in cell-cycle regulation. Nature 362:175–179 [DOI] [PubMed] [Google Scholar]
- 32. Sekiguchi T, et al. 1996. D-type cyclin expression is decreased and p21 and p27 CDK inhibitor expression is increased when tsBN462 CCG1/TAF(II)250 mutant cells arrest in G1 at the restrictive temperature. Genes Cells 1:687–705 [DOI] [PubMed] [Google Scholar]
- 33. Shen W, et al. 2003. Systematic analysis of essential yeast TAFs in genome-wide transcription and preinitiation complex assembly. EMBO J. 22:3395–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sherr C. 1996. Cancer cell cycles. Science 274:1672–1677 [DOI] [PubMed] [Google Scholar]
- 35. Sherr C. 1995. D-type cyclins. Trends Biochem. Sci. 20:187–190 [DOI] [PubMed] [Google Scholar]
- 36. Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68:850–858 [DOI] [PubMed] [Google Scholar]
- 37. Suzuki-Yagawa Y, Guermah M, Roeder RG. 1997. The ts13 mutation in the TAF(II)250 subunit (CCG1) of TFIID directly affects transcription of D-type cyclin genes in cells arrested in G1 at the nonpermissive temperature. Mol. Cell Biol. 17:3284–3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanese N, Pugh BF, Tjian R. 1991. Coactivators for a proline-rich activator purified from the multisubunit human TFIID complex. Genes Dev. 5:2212–2224 [DOI] [PubMed] [Google Scholar]
- 39. Wang EH, Tjian R. 1994. Promoter-selective transcriptional defect in cell cycle mutant ts13 rescued by hTAFII250. Science 263:811–814 [DOI] [PubMed] [Google Scholar]
- 40. Wieczorek E, Brand M, Jacq X, Tora L. 1998. Function of TAF(II)-containing complex without TBP in transcription by RNA polymerase II. Nature 393:187–191 [DOI] [PubMed] [Google Scholar]