

Abstract
The transactivation DNA-binding protein (TDP-43) pathology is associated with Fronto-Temporal Lobar Dementia (FTLD) with ubiquitinated inclusions and some cases of Alzheimer's disease (AD). Proteolytic fragments of β-amyloid precursor protein (βAPP) are detected in AD as well as the cerebrospinal fluid (CSF) from FTLD and Amyotrophic Lateral Sclerosis (ALS) patients, suggesting alteration in APP processing. Because of the overlap in TDP-43 pathology between FTLD and AD, we sought to determine whether there is a relationship between TDP-43 and APP metabolism. We generated gene transfer models using lentiviral delivery of human TDP-43 and Aβ1-42 into the rat primary motor cortex and examined their role 2 weeks post-injection. Expression of TDP-43 and/or Aβ1-42 increase pro-inflammatory markers, including Interleukin (IL)-6, tumor necrosis factor (TNF-α), glial neurofibrillary proteins (GFAP) and ionized calcium binding adaptor molecule 1 (IBA-1). Lentiviral Aβ1-42 up-regulates endogenous TDP-43 and promotes its phosphorylation, aggregation and cleavage into 35kDa fragments. Inversely, lentiviral TDP-43 expression increases the levels and activity of β-secretase (BACE), accelerating production of APP C-terminal fragments (C99) and Aβ1-40. Here we show that TDP-43 up-regulates APP metabolism and suggest a mechanistic link between TDP-43 and BACE.
Keywords: Amyloid precursor protein, Transactivation DNA-binding protein-43, β-secretase, neurodegeneration, Fronto-temporal lobar dementia, Alzheimer's disease
Introduction
Transactivation response (TAR) DNA-binding protein 43 (TDP-43) is the major constituent of the histopathological aggregates of Fronto-Temporal Lobar Degeneration with ubiquitin-positive inclusions (FTLD) and Amyotrophic Lateral Sclerosis (ALS) (Arai, et al., 2006, Neumann, et al., 2006). TDP-43 is a DNA/RNA binding protein of 414 amino acid residues in length (Ou, et al., 1995). It contains two highly conserved RNA recognition motifs (RRM1/RRM2) flanked on either side by N-terminal and glycine-rich C-terminal domains (Wang, et al., 2004). Under normal conditions, TDP-43 is localized predominantly to the nucleus. Under pathological conditions, the protein is mislocalized to the cytosol where it is ubiquitinated and/or phosphorylated and often cleaved into smaller fragments (Hasegawa, et al., 2008, Mackenzie, et al., 2007, Neumann, et al., 2007, Neumann, et al., 2007, Neumann, et al., 2006, Yoshiyama, et al., 2007, Zhang, et al., 2009). These attributes of TDP-43 pathology have become the hallmark characteristics in FTLD/ALS.
Alzheimer's disease (AD) is characterized by accumulation of β-amyloid (Aβ) in senile plaques, and Tau hyper-phosphorylation in neurofibrillary tangles (NFTs) (Hardy and Selkoe, 2002). Aβ is produced by cleavage of amyloid precursor protein (Li, et al.) by α- or β-secretases to yield C-terminal fragments α (CTFα) and β (CTFβ), respectively (Hardy and Selkoe, 2002). APP cleavage by α-secretase (Allinson, et al., 2003) precludes the formation of Aβ by cutting within the Aβ domain closer to the C-terminus of APP (C83), while cleavage by β-secretase (BACE) is closer to the N-terminus (C99) of APP (Li, et al., 1999). Subsequent cleavage of β-CTF by the γ-secretase complex (presenilin) generates Aβ peptides ranging in length from 39-43 amino acid residues (Jarrett, et al., 1993). Aβ1–40 and Aβ1–42 are present intracellularly and are prone to aggregation, but Aβ1-42 is thought to be the more toxic form of Aβ aggregates (Cook, et al., 1997, Gouras, et al., 2000, Greenfield, et al., 1999, Lee, et al., 1998, Skovronsky, et al., 1998, Xu, et al., 1997). While Aβ accumulation is necessary for AD diagnosis, TDP-43 pathology is also present in 20-36% of AD cases (Amador-Ortiz, et al., 2007, Arai, et al., 2009, Higashi, et al., 2007, Hu, et al., 2008, Uryu, et al., 2008), suggesting a possible link between TDP-43 and APP metabolism.
We previously reported that TDP-43 pathology is present in postmortem AD cortex and lentiviral Aβ1-42 incites the pathology of endogenous TDP-43 in animal models in vivo (Herman, et al., 2011). Because TDP-43 pathology is present in AD, and alteration of APP metabolism is reported in patients with FTLD/ALS (Schweikert, et al., 2009), we generated gene transfer animal models using lentiviral delivery of TDP-43 and Aβ1-42 into the primary rat motor cortex and examined crosstalk between these proteins. We specifically sought to determine the mechanistic effects of TDP-43 on APP processing, cell death and inflammation.
Materials and Methods
Stereotaxic injection
Lentiviral (Lv) constructs were used to generate the animal models. We used lentiviral delivery to target gene expression to specific brain regions and showed that targeting lentiviral Aβ1-42 to the endoplasmic reticulum results in intracellular protein accumulation (Rebeck, et al., 2010). We also generated gene transfer animal models using human wild type TDP-43 (Open Bio-systems) and Aβ1-42 into the motor cortex of two-month-old male Sprague-Dawley rats. Stereotaxic surgery was performed to inject the Lv constructs encoding either LacZ, Aβ1-42, or TDP-43 (Open Bio-systems) into the primary motor cortex of two-month-old male Sprague-Dawley rats weighing between 170-220g (Burns, et al., 2009). Animals were injected into one side of the brain with a Lv-LacZ vector at 2×108 m.o.i; or with 1×108 m.o.i Lv-Aβ1-42+1×108 m.o.i Lv-LacZ; or 1×108 m.o.i Lv-TDP-43+1×108 m.o.i Lv-LacZ; or 1×108 m.o.i Lv-Aβ1-42+1×108 m.o.i Lv-TDP-43;. Lv gene transfer rats were sacrificed two weeks post-injection. A total of 8 animals of each treatment (32 animals) were used for Western blot, ELISA and immunoprecipitation and 8 animals of each treatment (32 animals) were used for IHC. A total of 64 animals were used in these studies.
Western blot analysis
To extract the soluble fraction, the cortex was dissected out and homogenized in 1× STEN buffer (Herman, et al., 2011), then centrifuged at 5.000g and the supernatant was collected as a soluble fraction. To extract the insoluble or membrane fractions, the pellets were then re-suspended in 4M urea or 30% formic acid (FA) solution and analyzed by WB or ELISA. Human anti-Aβ1-42 was immunoprobed (1:1000) with mouse monoclonal (Zymed) antibody. Full length APP and CTF fragments were probed with C1/1.6 (1:1000) antibody, which detects C-terminal fragments C83 and C99 (Paul Mathews, Nathan Kline Institute). Anti-APP (6E10) antibody (1:600), which detects amino acids 3-8 was used to detect Aβ1-42 (Signet). Total TDP-43 was probed either with (1:1000) rabbit polyclonal antibody (ProteinTech) or (1:1000) mouse monoclonal (2E2-D3) TDP-43 (Abnova). Phosphorylated TDP-43 was probed (1:1000) with mouse monoclonal (1D3) phospho-TDP-43 (Millipore). BACE was probed (1:1000) with rabbit monoclonal anti-BACE1 antibody (Thermo Scientific). Tau changes were probed according to Herman et al. (Herman, et al., 2011). Western blots were quantified by densitometry using Quantity One 4.6.3 software (Bio Rad). Densitometry was obtained as arbitrary numbers measuring band intensity. N=8 animals were used to compare treatments and the data were analyzed as mean±Standard deviation, using ANOVA, with Neumann Keuls multiple comparison between treatment groups.
Immunohistology of brain sections
Immunohistochemistry was performed on 20μm-thick sections. TDP-43 was probed (1:200) with Rabbit polyclonal (ProteinTech) or (1:200) mouse monoclonal (Abnova) antibodies. BACE was immunostained (1:200) with rabbit polyclonal (Thermo Scientific) antibody. Staining against human Aβ1-42 (1:200), which detects amino acids 38-42, was performed using (1:200) rabbit polyclonal antibody (Zymed). Astrocytes were probed (1:200) with monoclonal anti-GFAP antibody (Millipore), and microglia were probed (1:200) with IBA-1 polyclonal antibody (Wako). Nissl staining was performed according to manufacturer's instructions (Sigma).
Stereological methods
were applied by a blinded investigator using unbiased stereology analysis (Stereologer, Systems Planning and Analysis, Chester, MD) to determine the total positive cell counts in 20 cortical fields on at least 10 brain sections (∼400 positive cells per animal) from each animal. These areas were selected across different regions on either side from the point of injection, and all values were averaged to account for the gradient of staining across 2.5 mm radius from the point of injection. An optical fractionator sampling method was used to estimate the total number of positive cells with multilevel sampling design. Cells were counted within the sampling frame determined optically by the fractionator and cells that fell within the counting frame were counted as the nuclei came into view while focusing through the z-axis. N=8 animals from each treatment condition were used for stereology.
Caspase-3 fluorometric activity assay
To measure caspase-3 activity in the animal models, we used EnzChek® caspase-3 assay kit #1 (Invitrogen) on cortical extracts and Z-DEVD-AMC substrate and the absorbance was read according to manufacturer's protocol.
Interleukin (IL)-6 and TNF-α enzyme-linked immunosorbent assay (ELISA)
Rat-specific IL-6 or TNF-α ELISA were performed using 50μl (1 μg/μl) of cortical rat brain lysates, detected with IL-6 or TNF-α primary antibody (3h) and 100μl anti-rabbit antibody (30min) at RT. Extracts were incubated with stabilized Chromogen for 30 minutes at RT and solution was stopped and read at 450 nm, according to manufacturer's (Invitrogen) protocol.
Aβ enzyme-linked immunosorbent assay (ELISA)
Human-specific Aβ1-40 and Aβ1-42 ELISAs (Invitrogen) were performed using 50μl (1μg/μl) of whole hemisphere brain lysates, detected with 50μl human Aβ1-40 or Aβ1-42 primary antibody (3h) and 100μl anti-rabbit antibody (30min.) at RT. Extracts were incubated with stabilized Chromogen for 30 minutes at RT and solution was stopped and read at 450nm, according to manufacturer's protocol.
BACE activity assay
Cortical tissues were homogenized as indicated above. All samples were immuno-precipitated with anti-BACE1 antibody (Thermo Scientific), and cortical lysates from BACE-/-mice were used to verify antibody specificity. Immuno-precipitation samples (50 μl) were then tested for BACE activity by the addition of a secretase-specific peptide conjugated to the reporter molecules EDANS and DABCYL (R&D Systems, Cat# FP002). In the uncleaved form the fluorescent emissions from EDANS are quenched by the physical proximity of the DABCYL moiety, which exhibits maximal absorption at the same wavelength (495nm). Cleavage of the peptide by the secretase physically separates the EDANS and DABCYL allowing for the release of a fluorescent signal. The level of secretase enzymatic activity in the cell lysate is proportional to the fluorometric reaction.
Results
Generation of gene transfer models using lentiviral delivery of TDP-43 and Aβ1-42
We generated gene transfer animal models using stereotaxic delivery of human wild type TDP-43 (Open Bio-systems) and Aβ1-42 into the motor cortex of two-month-old male Sprague-Dawley rats. Brains ipsilateral and contralateral to the injection site were analyzed 2-weeks post-injection. No human Aβ1-42 was detected in the motor cortex of animals injected with Lv-LacZ (Fig. 1A, N=8), but endogenous rat TDP-43 was observed throughout the cortex (Fig. 1B) predominantly within the nucleus (Fig. 1C), both ipsi- and contralateral to the injection site. The insert (Fig. 1C) is a high magnification image that shows intranuclear expression of TDP-43. Animals injected with Lv-Aβ1-42 had increased staining of both human Aβ1-42 and endogenous TDP-43 within the nucleus and the cytosol (data not shown), consistent with our previous results, which showed increased TDP-43 staining four weeks post-injection in Aβ1-42 expressing animals (Herman, et al., 2011). No Aβ1-42 staining was detected in the motor cortex of animals injected with Lv-TDP-43 (Fig. 1D), but increased a significant increase (61% by stereology) in TDP-43 staining (Fig. 1E) was observed both within the nucleus and the cytosol (Fig. 1F), compared to LacZ control. Insert (Fig. 1F) is a higher magnification image of cells that show cytoplasmic TDP-43 inclusions (arrow), compared to nuclear TDP-43 expression. Co-injection of Lv-Aβ1-42 and Lv-TDP-43 together resulted in a significant increase in Aβ1-42 (Fig.1G, 42% by stereology) and TDP-43 (Fig. 1H, 54% by stereology). Merged figures (Fig. 1I) show co-expression of Aβ1-42 and TDP-43 compared to LacZ control, and insert shows cells with co-localization of Aβ1-42 and TDP-43 in the cytosol (arrow) compared to cells that show cytosolic TDP-43 alone.
Figure 1. Histology shows that cortical expression of TDP-43 increases the levels of TDP-43 in motor cortex.
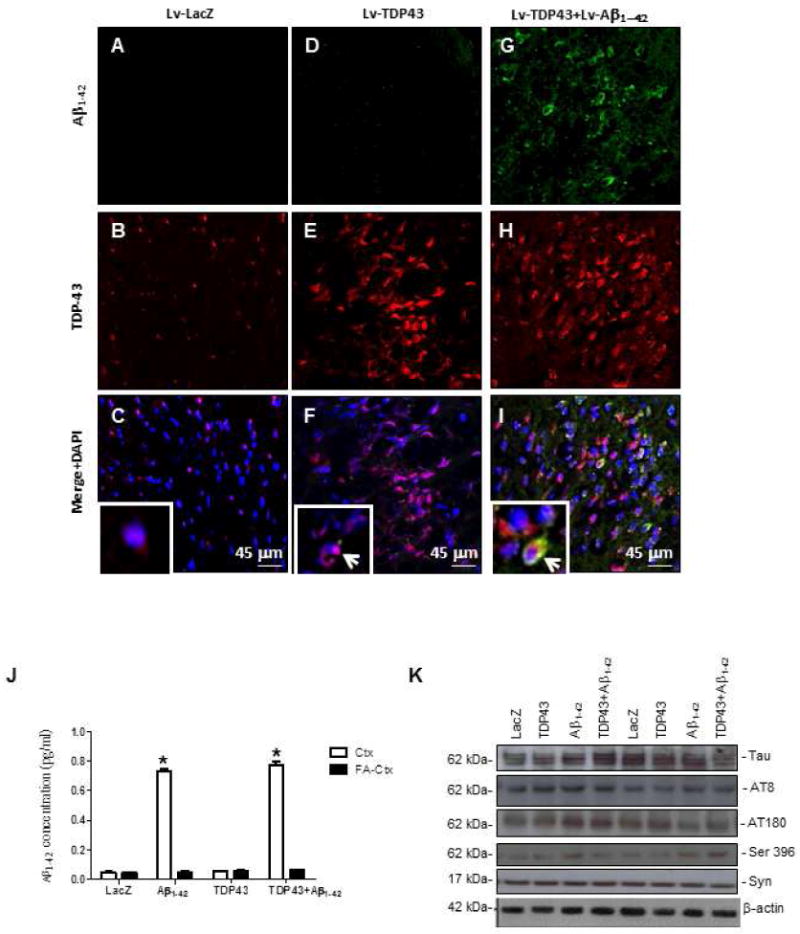
Staining with human-specific Aβ1-42 immunostaining of 20μm-thick sections of rat motor cortex injected with (A) Lv-LacZ, (D), Lv-TDP-43, or (G) Lv-TDP-43+Lv-Aβ1-42. Staining with TDP-43 of 20μm-thick cortical brain sections injected with (B) Lv-LacZ, (E) Lv-TDP-43, or (H) Lv-TDP-43+Lv-Aβ1-42. Co-localization of Aβ1-42 and TDP-43 in 4′,6-diamidino-2-phenylindole (DAPI)-positive cells in (C) Lv-LacZ, (F) Lv-TDP-43, or (I) Lv-TDP-43+Lv-Aβ1-42. Inserts show higher magnification images of DAPI stained sections to demonstrate cellular TDP-43 localization, arrows indicate cytosolic co-localization of TDP-43 and Aβ1-42 or cytosolic TDP-43 (I). (J) Human specific anti-Aβ1-42 ELISA showing levels in rat brains injected with Lv-LacZ, Lv-TDP-43, Lv-Aβ1-42, or Lv-TDP-43+Lv-Aβ1-42. (K) Western blot analysis of cortical brain lysates probed with total Tau, Tau epitopes (AT8, AT180, Ser 396), and α-Synuclein. N = 8 animals per treatment. Lentiviral lacZ: Lv-LacZ, Lentiviral Aβ1–42: Lv-Aβ1–42, Lentiviral TDP-43: Lv-TDP-43.
We also measured Aβ levels in brain extracts using human specific anti-Aβ1-42 ELISA. As expected, Aβ1-42 ELISA showed high levels of Aβ1-42 only in the cortex of animals injected with Lv-Aβ1-42. This Aβ1-42 was observed in the soluble, but not the formic acid-extracted samples (Fig. 1M). We also measured the levels of endogenous Tau and α-Synuclein but no differences in α-Synuclein levels or various Tau species were detected (Fig. 1N) after injection with Aβ1-42 or TDP-43 lentiviruses. Thus TDP-43 does not affect metabolism of endogenous Tau, α-Synuclein or exogenous Aβ1-42.
Lentiviral expression of Aβ1-42 and TDP-43 differentially activate microglia and increase levels of inflammatory markers
We sought to determine the effects of Aβ1-42 and TDP-43 on microglial marker activation and other inflammatory processes. Co-staining of TDP-43 and the microglial ionized calcium binding adaptor molecule 1 (IBA-1) revealed the presence of resting microglia with some evidence of endogenous neuronal and microglial TDP-43 in Lv-LacZ (Fig. 2A). Lv expression of TDP-43 either alone (Fig. 2B) or in the presence of Lv-Aβ1-42 (Fig. 2C) was associated with an increase in the level of TDP-43 in microglia, along with a change in microglial morphology. Co-expression of TDP-43 and Aβ1-42 showed microglial morphological changes suggestive of phagocytic cells (arrowhead). We stained each set of samples for astrocytic glial fibrillary acidic protein (GFAP) and microglial IBA-1 to determine if there is a change in glial number. Stereological counting of GFAP-stained astrocytes revealed a significant increase (P<0.05, N=8) in the number of astrocytes by 34% in Lv-TDP-43 (Fig. 2E) and 56% in Lv-Aβ1-42+Lv-TDP-43 (Fig. 2F) compared to Lv-LacZ (Fig. 2D) injected brains. Similarly, a significant increase in microglial number by 30% in Lv-TDP-43 (Fig. 2H) and 67% in Lv-Aβ1-42+Lv-TDP-43 (Fig. 2I) injected brains was observed compared to Lv-LacZ control (Fig. 2G). A significant increase (41%) in microglial density was detected in Lv-Aβ1-42+Lv-TDP-43 compared to Lv-TDP-43 injected brains alone. No differences in microglial (Fig. 2M) astrocyte (Fig. 2N) morphology or count were observed between LacZ injected and non-injected brains.
Figure 2. Histology shows lentiviral expression of intraneuronal Aβ1-42 and TDP-43 differentially activate microglia and increase levels of inflammatory markers.
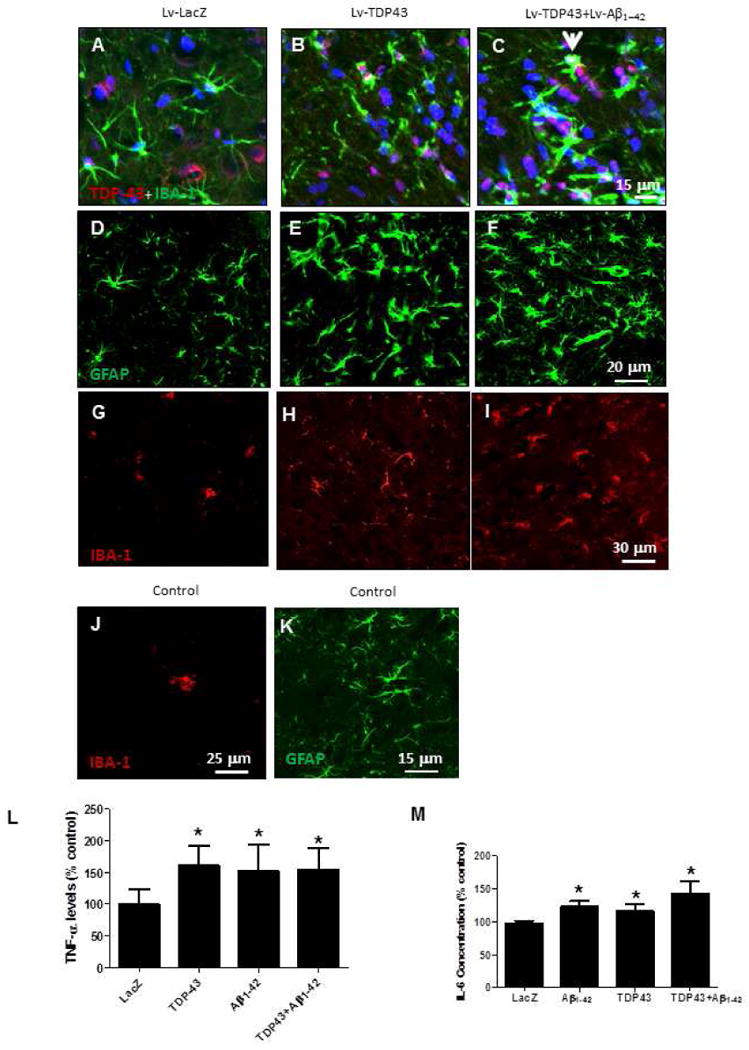
Staining of 20μm-thick primary motor cortex sections injected with (A) Lv-LacZ, (B) Lv-TDP-43, or (C) Lv-TDP-43+Lv-Aβ1-42 showing double staining of TDP-43 expression in DAPI-positive neurons and microglial IBA-1 marker. Arrowhead indicates phagocytosis of TDP-43-positive neuron by hyperactive microglia. Staining of 20μm-thick primary motor cortex sections injected with (D) Lv-LacZ, (E) Lv-TDP-43, or (F) Lv-TDP-43+Lv-Aβ1-42 showing GFAP-positive cells. Staining of 20μm-thick primary motor cortex sections injected with (G) Lv-LacZ, (H) Lv-TDP-43, or (I) Lv-TDP-43+Lv-Aβ1-42 showing microglial IBA-1 immunostaining. (J) Shows IBA-1 and (K) GFAP staining in control un-injected animals. ELISA detected levels of (L) TNF-α and (M) IL-6 in cortical extracts. Asterisk is significantly different to LacZ control, N = 8, P < 0.05. Lentiviral lacZ: Lv-LacZ, Lentiviral Aβ1–42: Lv-Aβ1–42, Lentiviral TDP-43: Lv-TDP-43.
We measured markers of neuro-inflammation using rat specific ELISA. A significant increase in TNF-α levels was detected in Lv-TDP-43 (64%, N=8), Lv-Aβ1-42 (53%) and Lv-Aβ1-42+Lv-TDP-43 (56%) compared to Lv-LacZ injected animals (Fig. 2O). A significant increase (N=8) in IL-6 concentration was detected in Lv-Aβ1-42 (25%), Lv-TDP-43 (19%) and Lv-Aβ1-42+Lv-TDP-43 (41%) compared to Lv-LacZ injected brains (Fig. 2P). Thus, Aβ1-42 and TDP-43 independently induce increases of glial markers and activation.
TDP-43 or Aβ1-42 expression increases caspase-3 activity and co-expression of TDP-43 and Aβ1-42 induces cell death
To determine whether TDP-43 or Aβ1-42 cause cell death two weeks post-injection, we counted Nissl-positive cells in the motor cortex and performed an independent fluorometric assay to measure caspase-3 activity. Stereological counting of Nissl-stained cortical brain sections showed no significant differences in cell count between Lv-LacZ (Fig. 3A&E) and either Lv-Aβ1-42 (Fig. 3B&E) or Lv-TDP-43 (Fig. 3C&E) injected animals (P<0.05, N=8). However, brains co-injected with both Lv-Aβ1-42 and Lv-TDP-43 showed a significant decrease (24%, P<0.05) in cell count compared to all other treatments (Fig. 3D&E). Significant increases were also observed in caspase-3 activation in Lv-Aβ1-42 (25%) Lv-TDP-43 (31%) and Lv-Aβ1-42+Lv-TDP-43 (44%) injected animals (Fig. 3F) compared to Lv-LacZ control.
Figure 3. TDP-43 or Aβ1-42 expression increases caspase-3 activity and co-expression of TDP-43 and Aβ1-42 induces cell death.
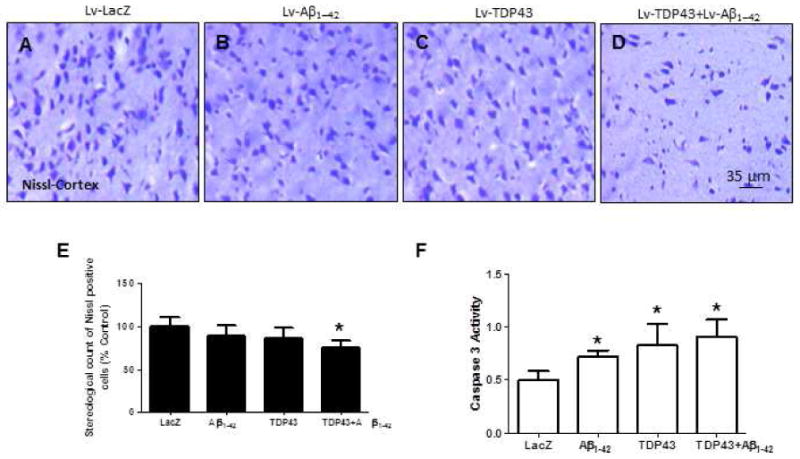
Nissl staining of 20μm-thick cortical brain sections injected with (A) Lv-LacZ, (B) Lv-Aβ1-42, (C) Lv-TDP-43, or (D) Lv-TDP-43+Lv-Aβ1-42. (E) Stereological counting of Nissl stained cells. (F) Caspase-3 activities. N = 8 animals per treatment. Lentiviral lacZ: Lv-LacZ, Lentiviral Aβ1–42: Lv-Aβ1–42, Lentiviral TDP-43: Lv-TDP-43.
TDP-43 pathology increases β-secretase levels and alters APP processing in gene transfer models
We measured changes in TDP-43 pathology two weeks post-injection either with Lv-TDP-43 or both to examine whether there is a mechanistic link between these proteins. Western blot analysis of cortical brain lysates was used to test levels of full length (43kDa) and cleaved (45kDa) TDP-43, which are recognized by an antibody to amino acid 260 of TDP-43. We found a significant increase (P<0.05, N=8) in the levels of full length 43kDa TDP-43 and 35kDa TDP-35 fragments (Fig. 4A&B) in brains injected with Lv-TDP-43 (41% and 53%, respectively), Lv-Aβ1-42 (38% and 51%, respectively), and Lv-TDP-43+Lv-Aβ1-42 (49% and 54%, respectively) compared to Lv-LacZ animals. Similar changes in TDP-43 were also detected (Fig. 4A, second panel) using a mouse antibody against the 261 amino acid N-terminal of TDP-43. We also detected a significant increase in phosphorylated TDP-43 (N=8) in Lv-Aβ1-42 (64%), Lv-TDP-43 (28%) and Lv-TDP-43+Lv-Aβ1-42 (69%) injected cortices (4th panel, Fig. 4A&B) using an antibody against serine 409-410, compared to Lv-LacZ injected animals. We probed for changes in TDP-43 in other brain areas, including the hippocampus and we did not observe any noticeable changes with TDP-43 (data not shown).
Figure 4. Western blot and ELISA reveal that TDP-43 pathology increases β-secretase levels and alters APP processing in gene transfer models.
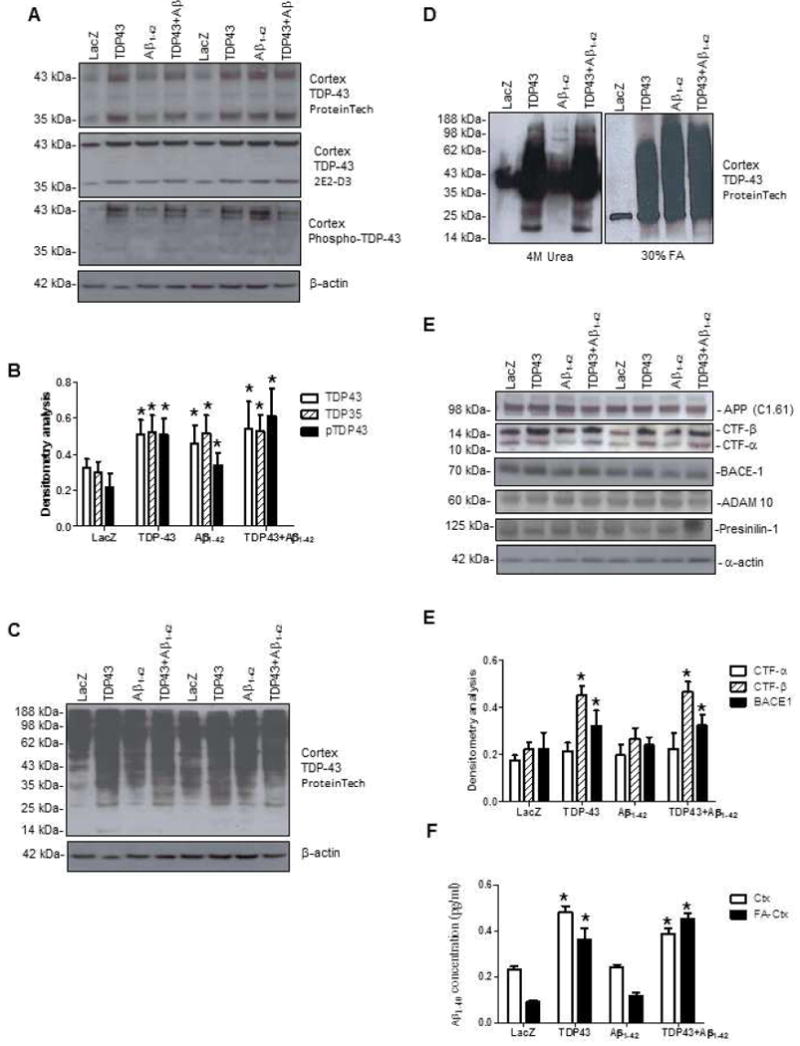
(A) Western blots of rat cortical brain lysates injected with Lv-LacZ, Lv-TDP-43, Lv-Aβ1-42, or Lv-TDP-43+Lv-Aβ1-42 probed with (Protein Tech and 2E2-D3) TDP-43 (1st, 2nd panels), phospho TDP-43 (3rd panel) and (B) Densitometry for Western blots. (C) Overexposed blot showing aggregation of TDP-43. D). Western blot analysis of 4M urea or 30% formic acid cortical extracts. (E) Immunoblots of rat cortical brain lysates probed for amyloid precursor protein (Top panel) and its C-terminal fragments (2nd panel), β-secretase (BACE-1), α-secretase (ADAM-10), γ-secretase complex (Presenilin-1) and F). densitometry analysis. (G) Detergent and formic acid extracted Aβ1-40 levels in rat brains injected with Lv-LacZ, Lv-TDP-43, Lv-Aβ1-42, or Lv-TDP-43+Lv-Aβ1-42. Asterisk is significantly different to LacZ control, ANOVA, with Neumann Keuls multiple comparison N = 8, P < 0.05. Lentiviral lacZ: Lv-LacZ, Lentiviral Aβ1–42: Lv-Aβ1–42, Lentiviral TDP-43: Lv-TDP-43.
To observe whether TDP-43 up-regulation in Aβ1-42 expressing animals, or expression of Lv-TDP-43 results in protein aggregation, we over-exposed a blot probed with TDP-43 antibody and observed more aggregation in cortices injected with Lv-TDP-43, compared to Lv-Aβ1-42 and Lv-LacZ injected animals (Fig. 4C). Thus, Aβ1-42 increased the levels of various TDP-43 species in vivo. To further ascertain whether TDP-43 aggregation results in alteration of protein solubility we homogenized cortical pellets in 4 M urea (Fig. 4D, left panel) or 30% formic acid (right panel). Protein smears were detected in the TDP-43 expressing animals in urea samples, but formic acid extracts showed protein aggregation in cortices expressing both TDP-43 and/or Aβ1-42, suggesting changes in protein solubility.
To demonstrate whether TDP-43 affected APP processing and Aβ generation, we probed for full length and other proteolytic fragments of endogenous APP. No changes in full length APP were observed in the cortex (or hippocampus, data not shown) injected with Lv-TDP-43, Lv-Aβ1-42, or Lv-TDP-43+Lv-Aβ1-42 (Fig. 4E, top panel). A significant increase in CTF-β (C99), but not CTF-α (C83), was observed (Fig. 4E&F, 2nd panel) in Lv-TDP-43 (64%) and Lv-TDP-43+Lv-Aβ1-42 (61%) compared to Lv-Aβ1-42 and Lv-LacZ injected brains (N=8). We also observed a significant increase in the levels of β-secretase (BACE 1) in Lv-TDP-43 (28%) and Lv-TDP-43+Lv-Aβ1-42 (31%) compared to Lv-LacZ. No significant differences were detected in the levels of α- or γ-secretase related proteins ADAM10 and presenilin-1 (Fig. 4E). Further analysis using rat-specific Aβ1-40 ELISA on cortical extracts revealed a significant increase in the levels of soluble Aβ1-40 in animals injected with Lv-TDP-43 (42%) and Lv-TDP-43+Lv-Aβ1-42 (35%) compared to Lv-Aβ1-42 and Lv-LacZ injected animals (Fig. 4G). We also detected a significant increase in FA-extracted Aβ1-40 in Lv-TPD-43 (64%) and Lv-TDP-43+Lv-Aβ1-42 (81%) compared to Lv-Aβ1-42 and Lv-LacZ injected animals (Fig. 4G).
TDP-43 expression leads to up-regulation of BACE levels and activity in gene transfer animal models
We pursued an independent approach to examine alterations in BACE levels with TDP-43 over-expression, using immunohistochemistry. Brain slices from animals injected with Lv-LacZ revealed under 100 × magnifications some TDP-43 (Fig. 5A) with no distinguishable BACE staining (Fig. 5B&C) in the cortex. However, over-expression of Lv-TDP-43 (Fig. 5D) resulted in up-regulation of BACE levels (Fig. 5E). No conclusive localization was detected between BACE and TDP-43 (Fig. 5F). To determine whether the increase in BACE levels were associated with changes in enzyme function, we normalized BACE levels and performed immuno-precipitation using 100 μg cortical lysates compared to BACE-/- mice, and then performed BACE activity assay (Fig. 5G). Significant increases in BACE activity were detected with over-expression of TDP-43 alone (39%. P<0.05, N=8) or together with Aβ1-42 (37%), compared to LacZ or Aβ1-42 injected cortex (Fig. 5G). No signals were detected in BACE-/- mice cortical lysates, suggesting assay specificity to BACE activity.
Figure 5. TDP-43 increases BACE activity and levels in gene transfer animal models.
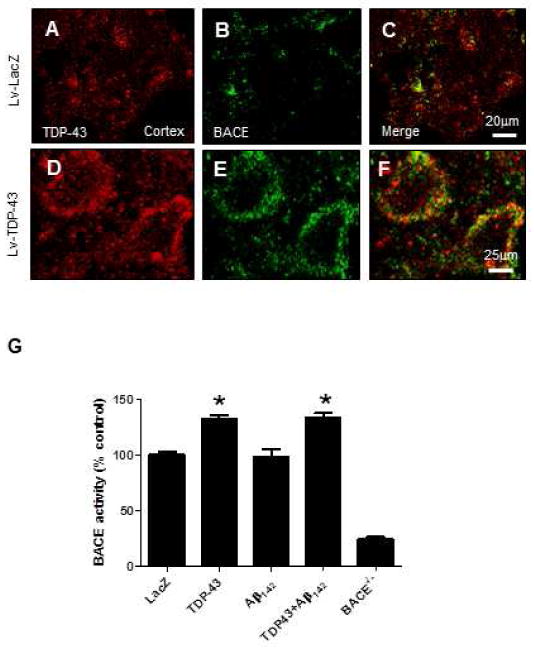
A) Immunohistochemistry of 20 μm thick motor cortex brain sections showing immunoreactivity to A) TDP-43, B) BACE and C) merged figures of BACE and TDP-43 in lentiviral Lac-Z injected animals. Immunoreactivity to D) TDP-43, E) BACE and F) merged BACE and TDP-43 figures in lentiviral TDP-43 injected animals. G) BACE activity assay in cortex injected with Lv-LacZ, Lv-TDP-43, Lv-Aβ1-42, or Lv-TDP-43+Lv-Aβ1-42. Asterisk is significantly different to LacZ control, ANOVA, with Neumann Keuls multiple comparison N = 8, P < 0.05. Lentiviral lacZ: Lv-LacZ, Lentiviral Aβ1–42: Lv-Aβ1–42, Lentiviral TDP-43: Lv-TDP-43.
Discussion
TDP-43 mutations are detected in FTLD, which is the most common type of dementia among young adults (Cairns, et al., 2007, Hodges, et al., 2004, Johnson, et al., 2005, Lipton, et al., 2004, Shi, et al., 2005). TDP-43 pathology also features prominently in ALS (Dickson, et al., 2007, Geser, et al., 2008, McCluskey, et al., 2009, Mitchell and Borasio, 2007). FTLD mainly affects neurons of the frontal and temporal lobes (Cairns, et al., 2007), similar to AD, which also exhibits TDP-43 pathology (Amador-Ortiz, et al., 2007). Analysis of cerebrospinal fluid from patients diagnosed with FTLD shows APP fragments, suggesting alteration of APP processing (Schweikert, et al., 2009). The present studies show for the first time that human TDP-43 affects APP processing via increased activity and levels of BACE and cleavage of full length APP, yielding CTF-β and Aβ1-40. These results are consistent with other findings, which show that BACE up-regulation occurs in the brain under stress conditions (Rossner, et al., 2006, Tabaton, et al., 2010). Overexpression and accumulation of TDP-43 in the cytosol may increase the bioavailability of this predominately nuclear protein (Hasegawa, et al., 2008, Mackenzie, et al., 2007, Neumann, et al., 2007, Neumann, et al., 2007, Neumann, et al., 2006, Yoshiyama, et al., 2007, Zhang, et al., 2009), leading to gain of cytosolic TDP-43 function and possible interaction with BACE to modulate processing and cleavage of APP. TDP-43 may alter BACE levels via increased stabilization to the cytosolic membrane, thus increasing APP cleavage. Inversely, future studies are necessary to determine the interaction of BACE with TDP-43 and whether BACE can cleave cytosolic TDP-43.
Expression of Lv-Aβ1-42 was associated with up-regulation of endogenous TDP-43, including cleavage, cytosolic accumulation, phosphorylation and aggregation, consistent with our previously reported data (Herman, et al., 2011). Expression of Lv-TDP-43 either alone or in the presence of Lv-Aβ1-42, resulted in similar changes in TDP-43 in the rat cortex. Taken together, these data suggest that Aβ1-42 can up-regulate endogenous TDP-43 protein levels and that TDP-43 alters APP processing, perhaps leading to increased levels of amyloidogenic proteins. These data are consistent with previous findings that cleavage of full length TDP-43 increases its propensity to aggregate (Rutherford, et al., 2008, Zhang, et al., 2009, Zhang, et al., 2007). TDP-43 protein aggregation was also independent of Tau and α-Synuclein, which did not change in these animal models. Our laboratory and others have shown in 3×Tg-AD mice, which exhibit characteristic AD pathology including amyloid plaques and NFTs, that TDP-43 is increased in the presence of Aβ, independent of Tau pathology (Caccamo, et al., 2010, Herman, et al., 2011). Interestingly, Aβ1-42 does not trigger alteration in Tau metabolism two weeks post-injection, but Tau is hyper-phosphorylated in the same model four weeks post-injection (Rebeck, et al., 2010).
No significant cell loss is observed in Lv-TDP-43 and Lv-Aβ1-42 injected animals, but caspase-3 activity is increased two weeks post-injection and is associated with increased levels of pro-inflammatory markers, suggesting that apoptotic cells may trigger an inflammatory response prior to significant cell death. The number of astrocytes and microglia is elevated along with an increase in the levels of TNF-α and IL-6 in TDP-43 and Aβ1-42 expressing animals, suggesting that these proteins may induce inflammation prior to significant cell loss. However, co-injection of Lv-TDP-43 and Lv-Aβ1-42 results in significant cell loss and further increase in pro-inflammatory markers, indicating that cell loss exacerbates the inflammatory reaction. These data are consistent with the presence of inflammation in the brains of transgenic mice and human patients with AD and FTLD (Akiyama, et al., 2000). Further analysis of the immunologic profile associated with TDP-43 changes remains to be elucidated to determine the role of this intracellular protein in microglial activation.
We did not detect any behavioral changes using strength and basal locomotion tests in these animals 2 weeks post-injection of Aβ1-42 or TDP-43. However, it would be very interesting to test these animals at later time points to determine any behavior abnormalities with TDP-43 over-expression. Furthermore, the data show that changes in TDP-43, including aggregation, phosphorylation and cleavage resemble the pathological changes, which are observed in FTLD with TDP-43 mutations, suggesting that over-expression of wild type TDP-43 may have toxic effects.
In conclusion, TDP-43 activates BACE and increases its expression levels, suggesting a mechanistic link between these proteins, leading to alteration of APP metabolism. Aβ1-42 increases full length, cleaved and phosphorylated TDP-43 levels, while TDP-43 increases APP processing to produce Aβ. Together these findings demonstrate interactions between pathological proteins and suggest future areas of investigation, including the relationship between BACE and TDP-43 cleavage and the role of TDP-43 accumulation in microglial activation.
Research Highlights.
We describe novel mechanisms of interaction between TDP-43 and APP
Lentiviral gene delivery of TDP-43 alters APP processing
TDP-43 increases BACE levels and activity
TDP-43 is associated with cleavage of APP into CTFβ and Aβ1-40
TDP-43 is associated with increased levels of neuro-inflammatory markers
Acknowledgments
This work was supported by NIH grant AG30378 to Charbel E-H Moussa.
Abbreviations
- TDP-43
Tar DNA binding protein
- Aβ
β-amyloid
- FTLD
Fronto-Temporal Lobar Dementia
- ALS
Amyotrophic Lateral Sclerosis
- Lv
Lentivirus
- GFAP
glial neurofibrillary proteins
- IBA-1
ionized calcium binding adaptor molecule 1
- TNF-α
Tumor Necrosis Factor
- IL-6
Interleukin-6
- APP
amyloid precursor protein
- m.o.i
multiplicity of infection
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allinson TM, Parkin ET, Turner AJ, Hooper NM. ADAMs family members as amyloid precursor protein alpha-secretases. J Neurosci Res. 2003;74:342–352. doi: 10.1002/jnr.10737. [DOI] [PubMed] [Google Scholar]
- Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, Iritani S, Onaya M, Akiyama H. Phosphorylated TDP-43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117:125–136. doi: 10.1007/s00401-008-0480-1. [DOI] [PubMed] [Google Scholar]
- Burns MP, Zhang L, Rebeck GW, Querfurth HW, Moussa CE. Parkin promotes intracellular Abeta1-42 clearance. Hum Mol Genet. 2009;18:3206–3216. doi: 10.1093/hmg/ddp258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Magri A, Oddo S. Age-dependent changes in TDP-43 levels in a mouse model of Alzheimer disease are linked to Abeta oligomers accumulation. Mol Neurodegener. 2010;5:51. doi: 10.1186/1750-1326-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL, 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL, 3rd, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM, Mackenzie IR. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171:227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW. Alzheimer's A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Josephs KA, Amador-Ortiz C. TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol. 2007;114:71–79. doi: 10.1007/s00401-007-0234-5. [DOI] [PubMed] [Google Scholar]
- Geser F, Brandmeir NJ, Kwong LK, Martinez-Lage M, Elman L, McCluskey L, Xie SX, Lee VM, Trojanowski JQ. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol. 2008;65:636–641. doi: 10.1001/archneur.65.5.636. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci U S A. 1999;96:742–747. doi: 10.1073/pnas.96.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70. doi: 10.1002/ana.21425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman AM, Khandelwal PJ, Stanczyk BB, Rebeck GW, Moussa CE. beta-Amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res. 2011;1386:191–199. doi: 10.1016/j.brainres.2011.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, Togo T, Katsuse O, Uchikado H, Furukawa Y, Kosaka K, Arai H. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–294. doi: 10.1016/j.brainres.2007.09.048. [DOI] [PubMed] [Google Scholar]
- Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, Kril JJ, Halliday GM. Clinicopathological correlates in frontotemporal dementia. Ann Neurol. 2004;56:399–406. doi: 10.1002/ana.20203. [DOI] [PubMed] [Google Scholar]
- Hu WT, Josephs KA, Knopman DS, Boeve BF, Dickson DW, Petersen RC, Parisi JE. Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol. 2008;116:215–220. doi: 10.1007/s00401-008-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- Johnson JK, Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, Chute DJ, Roberson ED, Pace-Savitsky C, Neumann M, Chow TW, Rosen HJ, Forstl H, Kurz A, Miller BL. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. 2005;62:925–930. doi: 10.1001/archneur.62.6.925. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Jr, Kosik KS. A detergent-insoluble membrane compartment contains A beta in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- Li QX, Maynard C, Cappai R, McLean CA, Cherny RA, Lynch T, Culvenor JG, Trevaskis J, Tanner JE, Bailey KA, Czech C, Bush AI, Beyreuther K, Masters CL. Intracellular accumulation of detergent-soluble amyloidogenic A beta fragment of Alzheimer's disease precursor protein in the hippocampus of aged transgenic mice. J Neurochem. 1999;72:2479–2487. doi: 10.1046/j.1471-4159.1999.0722479.x. [DOI] [PubMed] [Google Scholar]
- Lipton AM, White CL, 3rd, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol. 2004;108:379–385. doi: 10.1007/s00401-004-0900-9. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM, Trojanowski JQ. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- McCluskey LF, Elman LB, Martinez-Lage M, Van Deerlin V, Yuan W, Clay D, Siderowf A, Trojanowski JQ. Amyotrophic lateral sclerosis-plus syndrome with TAR DNA-binding protein-43 pathology. Arch Neurol. 2009;66:121–124. doi: 10.1001/archneur.66.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JD, Borasio GD. Amyotrophic lateral sclerosis. Lancet. 2007;369:2031–2041. doi: 10.1016/S0140-6736(07)60944-1. [DOI] [PubMed] [Google Scholar]
- Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol. 2007;64:1388–1394. doi: 10.1001/archneur.64.10.1388. [DOI] [PubMed] [Google Scholar]
- Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66:152–157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeck GW, Hoe HS, Moussa CE. Beta-amyloid1-42 gene transfer model exhibits intraneuronal amyloid, gliosis, tau phosphorylation, and neuronal loss. J Biol Chem. 2010;285:7440–7446. doi: 10.1074/jbc.M109.083915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossner S, Sastre M, Bourne K, Lichtenthaler SF. Transcriptional and translational regulation of BACE1 expression--implications for Alzheimer's disease. Prog Neurobiol. 2006;79:95–111. doi: 10.1016/j.pneurobio.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, Stewart H, Kelley BJ, Kuntz K, Crook RJ, Sreedharan J, Vance C, Sorenson E, Lippa C, Bigio EH, Geschwind DH, Knopman DS, Mitsumoto H, Petersen RC, Cashman NR, Hutton M, Shaw CE, Boylan KB, Boeve B, Graff-Radford NR, Wszolek ZK, Caselli RJ, Dickson DW, Mackenzie IR, Petrucelli L, Rademakers R. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweikert B, Hahmann H, Steinacker JM, Imhof A, Muche R, Koenig W, Liu Y, Leidl R. Intervention study shows outpatient cardiac rehabilitation to be economically at least as attractive as inpatient rehabilitation. Clin Res Cardiol. 2009;98:787–795. doi: 10.1007/s00392-009-0081-6. [DOI] [PubMed] [Google Scholar]
- Shi J, Shaw CL, Du Plessis D, Richardson AM, Bailey KL, Julien C, Stopford C, Thompson J, Varma A, Craufurd D, Tian J, Pickering-Brown S, Neary D, Snowden JS, Mann DM. Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol. 2005;110:501–512. doi: 10.1007/s00401-005-1079-4. [DOI] [PubMed] [Google Scholar]
- Skovronsky DM, Doms RW, Lee VM. Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. J Cell Biol. 1998;141:1031–1039. doi: 10.1083/jcb.141.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabaton M, Zhu X, Perry G, Smith MA, Giliberto L. Signaling effect of amyloid-beta(42) on the processing of AbetaPP. Exp Neurol. 2010;221:18–25. doi: 10.1016/j.expneurol.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, Miller BL, Kretzschmar HA, Lee VM, Trojanowski JQ, Neumann M. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol. 2008;67:555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83:130–139. doi: 10.1016/s0888-7543(03)00214-3. [DOI] [PubMed] [Google Scholar]
- Xu H, Sweeney D, Wang R, Thinakaran G, Lo AC, Sisodia SS, Greengard P, Gandy S. Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc Natl Acad Sci U S A. 1997;94:3748–3752. doi: 10.1073/pnas.94.8.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, Gass J, Rangachari V, Buratti E, Baralle F, Golde TE, Dickson DW, Petrucelli L. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2009;106:7607–7612. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, Pickering-Brown S, Dickson D, Petrucelli L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–10534. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]