

Abstract
Lipid nanoparticles (LNP) containing ionizable cationic lipids are the leading systems for enabling therapeutic applications of siRNA; however, the structure of these systems has not been defined. Here we examine the structure of LNP siRNA systems containing DLinKC2-DMA(an ionizable cationic lipid), phospholipid, cholesterol and a polyethylene glycol (PEG) lipid formed using a rapid microfluidic mixing process. Techniques employed include cryo-transmission electron microscopy, 31P NMR, membrane fusion assays, density measurements, and molecular modeling. The experimental results indicate that these LNP siRNA systems have an interior lipid core containing siRNA duplexes complexed to cationic lipid and that the interior core also contains phospholipid and cholesterol. Consistent with experimental observations, molecular modeling calculations indicate that the interior of LNP siRNA systems exhibits a periodic structure of aqueous compartments, where some compartments contain siRNA. It is concluded that LNP siRNA systems formulated by rapid mixing of an ethanol solution of lipid with an aqueous medium containing siRNA exhibit a nanostructured core. The results give insight into the mechanism whereby LNP siRNA systems are formed, providing an understanding of the high encapsulation efficiencies that can be achieved and information on methods of constructing more sophisticated LNP systems.
1. Introduction
Lipid nanoparticles (LNP) are the leading delivery systems for enabling the therapeutic potential of siRNA for systemic applications.1,2 LNP siRNA systems containing optimized ionizable cationic lipids can exhibit remarkable in vivo potencies at doses as low as 0.02 mg siRNA/kg body weight for silencing liver (hepatocyte) target genes in rodents following intravenous (i.v.) injection.2 These systems are relatively nontoxic, leading to therapeutic indices in mice approaching 1000, indicating potential clinical utility.
Despite this progress, the structure of these LNP siRNA systems is unclear. Some models of LNP siRNA suggest a bilayer vesicle structure of the LNP with siRNA on the inside in an aqueous interior,3 however other observations suggest that such models may be incorrect. For example, recent cryo-transmission electron microscopy (cryo-TEM) studies of LNP siRNA systems formed by mixing an ethanol stream containing lipid with an aqueous stream containing siRNA using a T-tube mixing system results in LNP that have electron-dense cores4 rather than the less dense aqueous cores observed for vesicular systems.5 In addition, formulation of LNP siRNA systems using the T-tube mixer6 can result in siRNA encapsulation efficiencies above 70%, an observation that is difficult to reconcile with bilayer vesicular structure. This is because encapsulation depends on the presence of cationic lipid and it would therefore be expected that the maximum encapsulation efficiency should be approximately 50% for bilayer systems, assuming that the cationic lipid is equally distributed on both sides of the bilayer.
In this work, we characterize the structure of LNP siRNA systems produced using a rapid microfluidic mixing technology7 by employing a variety of biophysical assays as well as in silico simulations. Cryo-TEM studies show that these LNP siRNA systems exhibit an electron-dense core (in contrast to bilayer vesicle systems), and that “limit size” systems can be generated at high PEG-lipid contents that are consistent with the ability of siRNA to stimulate formation of inverted micelles in association with cationic lipid. Fluorescent energy resonance transfer (FRET), 31P NMR, and RNase digestion studies show that encapsulated siRNA is associated with internalized cationic lipid, is effectively immobilized on the NMR time scale and is fully protected from external RNase. Density gradient studies show that the density of LNP siRNA systems can vary from being significantly less dense than bilayer vesicle systems to exhibiting increased densities at higher siRNA contents. Taken together, these experimental results suggest that siRNA resides in inverted micelles within the overall LNP siRNA structure. These results are consistent with molecular modeling studies that indicate these LNP siRNA systems have a nanostructured core consisting of a periodic arrangement of aqueous compartments, some of which contain siRNA duplexes.
2. Experimental Section
2.1. Materials
The lipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD-PE), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rh-PE) were obtained from Avanti Polar Lipids (Alabaster, AL). 4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES), cholesterol, Sephadex G-50, tetrasodium EDTA, Trizma Base, and xylene cyanol were obtained from Sigma-Aldrich (St. Louis, MO). Triton X-100 and 2-(N-morpholino)ethanesulfonic acid (MES) were obtained from BDH (Westchester, PA). Ammonium acetate, boric acid, sodium acetate, and sodium chloride were obtained from Fisher Scientific (Fair Lawn, NJ). Bovine pancreatic RNase A was purchased from Applied Biosystems/Ambion (Austin, TX). Deionized formamide was obtained from GIBCO BRL (Grand Island, NY). Bromophenol blue was obtained from BioRad (Hercules, CA). The phosphodiester siRNA used in this study was obtained from Invitrogen (Carlsbad, CA), and phosphorothioate siRNA were 25-mer blunt-end duplexes (sense -5′ -GCCUUAACUUUGGUGAUCAAGGAUA-3′) and were obtained from Dharmacon (Lafayette, CO). The ionizable cationic lipid 2,2-dilinoleyl-4-(2-dimethylaminoethyl)-[1,3]-dioxolane (DLinKC2-DMA) and N-[(methoxy polyethylene glycol 2000 carbamyl]-1,2-dimyristyloxlpropyl-3-amine (PEG-c-DMA) were obtained from AlCana Technologies Inc., Vancouver, BC. Cholesterol E total cholesterol assay kit was obtained from Wako Diagnostics (Richmond, VA). Quant-it RiboGreen RNA assay kit was obtained from Molecular Probes (Eugene, OR).
2.2. Preparation of POPC/Cholesterol Bilayer Vesicles
Bilayer POPC/cholesterol (1:1; mol/mol) vesicles were prepared by hydration of a dried lipid film with PBS, and the dispersion was then freeze–thawed five times using liquid nitrogen. Unilamellar vesicles were then prepared by extruding the frozen and thawed lipid suspension 10 times through two stacked 80 nm pore size polycarbonate filters.
2.3. Preparation of LNP siRNA Systems
LNP were prepared by mixing appropriate volumes of lipid stock solutions in ethanol buffer with an aqueous phase containing siRNA duplexes employing a microfluidic micromixer as described elsewhere.7 For the encapsulation of siRNA, the desired amount of siRNA was dissolved in 25 mM sodium acetate, pH 4.0. Equal volumes of the lipid in ethanol and the siRNA in buffer were combined in the microfluidic micromixer using a dual-syringe pump (model S200, KD Scientific, Holliston, MA) to drive the solutions through the micromixer at a combined flow rate of 2 mL/min (1 mL/minute for each syringe). A herringbone micromixer8 was employed. The mixed material was diluted into an equal volume of 25 mM sodium acetate buffer, pH 4.0, upon leaving the micromixer outlet, thus reducing the ethanol content to 25%. The lipid mixture was then dialyzed for 4 h against 1000 volumes of 50 mM MES/50 mM sodium citrate buffer (pH 6.7) followed by an overnight dialysis against 1000 volumes of 1× phosphate buffered saline, pH 7.4 (GIBCO, Carlsbad, CA) using Spectro/Por dialysis membranes (molecular weight cutoff 12 000–14 000 Da, Spectrum Laboratories, Rancho Dominguez, CA). The mean diameter of the LNP after dialysis was 41.3 ± 14.9 nm as determined by dynamic light scattering (number mode; NICOMP 370 submicrometer particle sizer, Santa Barbara, CA). Lipid concentrations were determined by measuring total cholesterol using the Cholesterol E enzymatic assay from Wako Chemicals USA (Richmond, VA). Encapsulation efficiency was calculated by determining unencapsulated siRNA content by measuring the fluorescence upon the addition of RiboGreen (Molecular Probes, Eugene, OR) to the siRNA-LNP (Fi) and comparing this value to the total siRNA content that is obtained upon lysis of the LNP by 1% Triton X-100 (Ft): % encapsulation = (Ft – Fi)/Ft × 100.
Limit size particles were prepared by mixing 20 mM DLinKC2-DMA/PEG-c-DMA (90/10, mol %) dissolved in ethanol with five volumes of 25 mM sodium acetate, pH 4.0 containing siRNA. Mixing of the two streams are accomplished using the above-mentioned herring micromixer device with a flow-rate of 0.5 mL/min for the lipid/ethanol stream and 2.5 mL/min for the aqueous stream. Ethanol was removed by first diluting the lipid mixture with sodium acetate buffer, pH 4.0 and then removing the ethanol-containing buffer with an Amicon Ultra, 10 000 MWCO, regenerated cellulose concentrator (Millipore, Billerica, MA). The process was repeated five times to ensure all residual ethanol was removed.
2.4. Cryo-TEM
Cyro-TEM samples were prepared by applying 3 μL LNP at 10–20 mg/mL total lipid to a standard electron microscopy grid with a perforated carbon film. Excess liquid was removed from the grid by blotting and then the grid was plunge-frozen in liquid ethane to rapidly freeze the sample using a Vitrobot system (FEI, Hillsboro, Oregon). Images were taken under cryogenic conditions (∼88 K) at a magnification of 50 000× with an AMT HR CCD side mount camera. Samples were loaded with a Gatan 70 degree cryo-transfer holder in an FEI G20 Lab6 200 kV TEM (FEI, Hillsboro, OR) under low dose conditions with an under-focus of 4–6 μm to enhance image contrast. Experiments were performed at the University of British Columbia Bioimaging Centre (Vancouver, BC). Particle diameters were measured from the micrographs with the aid of ImageJ (National Institute of Health, Bethesda, MD). Average diameters and standard deviations were calculated from more than 150 particles.
2.5. RNase Protection Assay
Factor VII siRNA was encapsulated with LNP formulations containing 40% DLinKC2-DMA, 11.5% DSPC, 47.5% cholesterol, and 1% PEG-c-DMA (mol %). An amount of 1.0 μg of siRNA (entrapped in LNP) was incubated with 0.05 μg of bovine pancreatic RNase A (Ambion, Austin, TX) in 50 μL of 20 mM HEPES (pH 7.0) at 37 °C for 1 h. At the end of the incubation, a 10 μL aliquot of the reaction mix was added to 30 μL of FA dye (deionized formamide, TBE, PBS, xylene cyanol, bromophenol blue, triton X-100) to halt the RNase reaction. Gel electrophoresis was performed using 20% native polyacrylamide gel, and nucleic acids were visualized by staining with SYBR-Safe (Invitrogen, Carlsbad, CA).
2.6. 31P NMR Studies
Proton-decoupled 31P NMR spectra were obtained using a Bruker AVII 400 spectrometer operating at 162 MHz. Free induction decays (FID) corresponding to ∼104 scans were obtained with a 15 μs, 55 degree pulse with a 1 s interpulse delay and a spectral width of 64 kHz. An exponential multiplication corresponding to 50 Hz line broadening was applied to the FID prior to Fourier transformation. The sample temperature was regulated using a Bruker BVT 3200 temperature unit. Measurements were performed at 25 °C. Experiments were performed at the Centre for the Drug Research and Development, Vancouver, BC.
2.7. FRET Membrane Fusion Studies
Fusion between LNP siRNA nanoparticles and anionic DOPS bilayer vesicles was assayed by employing a fluorescence resonance energy transfer lipid mixing assay.9,10 Labeled DOPS vesicles containing NBD-PE and Rh-PE (1 mol % each) were prepared by hydration of the lipid in a thin film with 20 mM HEPES buffer at pH 7.0 followed by 10 extrusions through two stacked 100 nm pore size polycarbonate filters (Nuclepore) using the Extruder (Northern Lipids, Vancouver, BC). LNP composed of 40% DLinKC2-DMA, 11.5% DSPC, 47.5% cholesterol, and 1% PEG-c-DMA were prepared with an siRNA-to-total lipid ratio (D/L ratio, wt/wt) of 0, 0.06, and 0.24. A D/L ratio of 0.24 represents a charge ratio of negative (siRNA–phosphate) to positive (cationic lipid–amine) of one. Lipid mixing experiments were conducted as previously described.10 Unlabeled LNP were added to a stirred cuvette containing NBD-PE/Rh-PE labeled DOPS vesicles at a 2:1 lipid molar ratio (200 μM LNP/100 μM DOPS vesicles) in 2 mL of 10 mM ammonium acetate, 10 mM MES, 10 mM HEPES, and 130 mM NaCl equilibrated to pH 5.5. Fluorescence of NBD-PE was monitored using 465 nm excitation and 535 nm emission using a Perkin-Elmer LS-55 fluorimeter with a 1 × 1 cm cuvette under continuous low speed stirring. Lipid mixing was monitored for approximately 10 min, after which 20 μL of 10% Triton X-100 was added to disrupt all lipid vesicles, representing infinite probe dilution (0.1% Triton X-100 vol/vol final). Lipid mixing was expressed as a percentage of infinite probe dilution determined using the equation: % lipid mixing = (F – F0)/(Fmax – F0) × 100, where F is the fluorescence intensity measured by the assay, F0 is the initial fluorescence intensity of NBD-PE/Rh-PE/DOPS vesicles, and Fmax is the maximum fluorescence intensity at infinite probe dilution after the addition of Triton X-100 (0.1% v/v final).
2.8. Sucrose Density Gradient Centrifugation
Solutions of 1%, 2.5%, 5%, 10% and 15% sucrose (wt/vol) were prepared in distilled water and used to make a 10 mL step gradient. Gradients were prepared successively overlaying 2 mL of less concentrated sucrose on top of the more concentrated ones. LNP and POPC/cholesterol vesicles were prepared as described above. Five hundred μL of sample was applied to the gradient and was centrifuged at 39000 rpm in a Beckman SW41 swing bucket rotor using a Beckman Coulter Optima LE-80K ultracentrifuge (Brea, CA) for 18 h. These conditions results in an average centrifugal force of approximately 190 000g in the middle of the tube. After centrifugation, 500 uL fractions were successively removed from the top. Lipid content from each fraction was determined using the Cholesterol E enzymatic assay from Wako Chemicals USA (Richmond, VA) described above.
2.9. Computer Simulation of LNP-siRNA Systems
The LNP containing nucleic acids was formed by first simulating the self-assembly of a smaller building block, then creating a large particle by spatial translations of the building block, coating the large particle with a polymer-grafted lipid, and resolvating the system. The building block was self-assembled starting from random configurations in a small system (Figure 7A). The small system contained all components excluding the polymer lipid and was prepared at low, intermediate, and high hydration levels (12, 20, and 40 water molecules per lipid, respectively). Self-assembly resulted in a periodic nanostructure with enclosed water compartments (Figure 7B). We also simulated self-assembly of a medium-sized system with all components including the polymer-grafted lipid as a test case. It resulted in the structure similar to the small system at low hydration, and the low-hydration building block was selected for simulations of the large system. The solvent was removed, and the building block was multiplied 3 × 3 × 3 times to obtain a large nanoparticle, retaining a small spacing (<1 nm) between the translated unit cells. This nanoparticle was coated with a polymer-grafted lipid. The coating layer contained the polymer lipid in random conformations distributed within a thin slab (∼3 nm), and was placed in close proximity (∼1 nm) at all facets of the nanoparticle. This system was then resolvated (water placed inside and outside of LNP) in a large simulation box.
Figure 7.
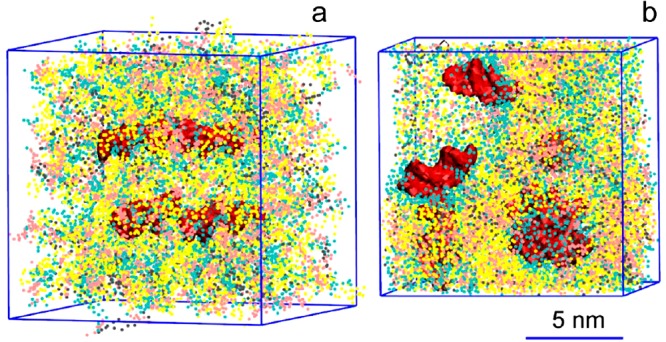
Self-assembly from a random configuration (a) into a building block (b) for a lipid nanoparticle (LNP). A mixture of DLinKC2-DMA, DSPC and cholesterol (576 DLinKC2-DMAlipids, 144 DSPC lipids and 576 cholesterol molecules; 44/11/44; mol/mol) is placed in a small simulation box at a low hydration level; see text. DLinKC2-DMAis shown in yellow, cholesterol in pink, DSPC in gray, lipid polar moiety in cyan, and nucleic acids (12 bp duplex DNA) in red; water not shown for clarity.
The small systems contained 8 duplex 12 bp DNA complexes, 576 DLinKC2-DMAlipids, 144 DSPC lipids, and 576 cholesterol molecules. The medium-sized system contained 64 DNAs, 4608 DLinKC2-DMAlipids, 1152 DSPC, 4608 cholesterols, and 1152 PEG-lipids. The large system contained 216 DNAs, 15552 DLinKC2-DMAlipids, 3888 DSPC, 15 552 cholesterols, and 3888 PEG-lipids. The total ratio in the medium and large systems was DLinKC2-DMA/DSPC/cholesterol/PEG-lipid 4:1:4:1 (molar), and DNA to lipid ratio ∼0.05 wt/wt, similar to experimental conditions.2 The small system at low, intermediate, and high hydration levels contained 4030, 6530, and 12 560 water particles, respectively; the medium and large systems contained ∼400 000 and ∼900 000 water particles, respectively; Cl– ions were added to all systems to neutralize the charge. The large system included ∼1 400 000 particles in total, had a box size of 54 × 54 × 54 nm3, and was simulated for 10 μs (actual simulation time is indicated). Two independent runs of 5 μs each were performed for the medium-sized system, and six copies of the small system (two for each level of hydration) were simulated for 1 μs.
The MARTINI coarse-grained (CG) force field was employed.11 In this force field, typically one MARTINI particle represents 4 heavy atoms. siRNA duplexes were represented by 12 base pair double stranded DNAs; the RNA molecules would have minor differences in the Lennard–Jones interactions, and lower chain flexibility which would be important for longer strands. Models for DNA12 and DSPC lipid were downloaded from the MARTINI Web site (http://md.chem.rug.nl/cgmartini/index.php/downloads). The headgroup of DLinKC2-DMAlipid comprised charged (Qd type) and apolar (C4) particles, and the linker was constituted by a ring of SC4 and 2 SP1 particles. Available models for sphingomyelin and PEG polymer13 were combined to produce the PEG-grafted ceramide lipid (containing a chain of 7 beads). While longer polymer chains are used in experimental studies to control the nanoparticle size, here a short chain length was incorporated to avoid unnecessary and computationally expensive increase of the simulation box volume (to accommodate longer chains). Short polymers combined with a moderate water volume surrounding the nanoparticle were sufficient to keep it disconnected from its periodic image in our simulations. However, a somewhat larger molar concentration of the short PEG-lipid (as compared to longer polymers in the experimental setups) was required to cover the whole nanoparticle surface.
Simulations were performed with the Gromacs v.4 software package.14 For nonbonded interactions, the standard cutoffs for the MARTINI force field were used: the Lennard–Jones potential was shifted to zero between 0.9 and 1.2 nm, the Coulomb potential was shifted to zero between 0 and 1.2 nm, with a relative dielectric constant of 15. The time step was 10 fs with the neighbor list updates every 10 steps. The system was coupled to an isotropic pressure of 1 bar using the Berendsen barostat15 with a time constant of 4 ps. Lipids, water, and nucleic acids were coupled separately to a temperature of 310 K using the velocity rescaling thermostat16 with a time constant of 1 ps.
3. Results and Discussion
3.1. Microfluidic Mixing Allows Highly Efficient Encapsulation of siRNA in LNP-siRNA Systems over a Wide Range of siRNA/Lipid Charge Ratios
As noted above, high siRNA encapsulation levels for siRNA in LNP systems are inconsistent with bilayer structure, where a maximum encapsulation efficiency of 50% would be expected. Here we characterized the encapsulation efficiencies of LNP siRNA systems over a range of siRNA/cationic lipid charge ratios using the microfluidic mixing process as described in the Experimental Section and employing the lipid mixture DLinKC2-DMA/DSPC/Chol/PEG-lipid (40/11.5/47.5/1; mol/mol). The resulting LNP siRNA systems exhibited diameters of approximately 50 nm (number mode) as measured by dynamic light scattering. Essentially complete (∼ 95% as indicated using the Ribo-Green assay) encapsulation was achieved over a wide range of siRNA/cationic charge ratios, including charge ratios as high as 1.25 (see Figure 1, Supporting Information). These observations suggest that the microfluidic mixing technique for formulating siRNA allows somewhat more efficient encapsulation than the T-tube mixing technique where trapping efficiencies of ∼70% have been noted.6
3.2. LNP Systems Exhibit an Electron Dense Core Structure As Indicated by Cryo-TEM
In the next set of experiments, the cryo-TEM characteristics of the LNP siRNA systems produced by microfluidic mixing were investigated for the DLinKC2-DMA/DSPC/Chol/PEG-lipid (40/11.5/47.5/1; mol/mol) lipid composition. As shown in Figure 1A and B, LNP siRNA systems prepared at siRNA/total lipid ratios of 0.06 and 0.24, corresponding to siRNA-to-cationic charge ratios of 0.25 and 1, exhibited an electron dense core similar to that observed for LNP siRNA systems formulated using the T-tube apparatus.4 The electron dense LNP siRNA structure contrasts with the less dense interior of a vesicle system with an aqueous interior generated from POPC/cholesterol (1:1; mol/mol) (Figure 1C) and is visually similar to the electron dense interior exhibited by cryo-TEM of a colloidal fat emulsion as reported by Kuntsche et al.17
Figure 1.

LNP containing DLinKC2-DMA exhibit electron dense cores both in the presence and absence of encapsulated siRNA as indicated by cryo-TEM. LNP were prepared by microfluidic mixing employing a herringbone mixer as indicated in the Experimental Section. POPC/cholesterol (50/50; mol/mol) bilayer vesicles were prepared by extrusion through polycarbonate filters with 80 nm pore size. (A) Cryo-TEM micrograph obtained from LNP siRNA with lipid composition DLinKC2-DMA/DSPC/Chol/PEG-lipid (40/11.5/47.5/1; mol/mol) and siRNA at a siRNA/lipid ratio of 0.06, wt/wt, corresponding to an siRNA/cationic lipid charge ratio of 0.25. (B) LNP with the same lipid composition as for (A) but prepared at an siRNA/lipid ratio of 0.24 wt/wt, corresponding to an siRNA/cationic lipid charge ratio of 1. (C) Cryo-TEM micrograph of POPC/cholesterol (1:1) vesicles. (D) LNP with the same lipid composition as for (A) and (B) but prepared in the absence of siRNA.
The LNP siRNA formulation employed in Figure 1A contains siRNA at a 0.06 siRNA/lipid (w/w) ratio which corresponds to an siRNA-to-cationic lipid charge ratio of 0.25. As a result, when the LNP siRNA is formulated at pH 4.0, approximately 75% of the cationic lipid is not complexed to siRNA in the LNP. It is therefore of interest that the LNP siRNA particles observed in Figure 1A exhibit an electron dense interior with no evidence of an internal aqueous core. This suggests that the cationic lipid may contribute to the electron dense interior even when not complexed to siRNA. In order to determine whether this is the case, LNP systems with the same lipid composition but no siRNA were formulated employing the microfluidic process and characterized by cryo-TEM. As shown in Figure 1D, an electron dense core was observed in the absence of siRNA, indicating that cationic lipids such as DLinKC2-DMA, possibly in combination with DSPC and cholesterol, contribute to electron dense structures in the LNP interior.
3.3. LNP Containing Cationic Lipid Exhibit Limit Sizes Consistent with the Formation of Inverted Micellar Structures in the LNP Interior Both in the Presence and Absence of siRNA
The structures that could give rise to the electron dense cores detected by cryo-TEM are of interest. In the absence of siRNA it may be proposed that the cationic lipid, in association with a counterion, adopts an inverted structures such as inverted micelles, consistent with the propensity of these lipids for highly curved “inverted” structures such as the hexagonal HII phase in mixtures with anionic lipids.18,19 These structures are inverted in the sense that the polar headgroups are oriented toward interior aqueous cores with diameters as small as 3 nm.20 The actual equilibrium radius of an inverted micelle could be larger as dictated by the intrinsic or spontaneous radius of curvature of the constituent lipids.21 In turn, assuming a bilayer thickness of 4 nm this would suggest that LNP systems composed of pure cationic lipid should exhibit limit sizes with diameters in the range of 11 nm or larger, which is essentially a bilayer surrounding an aqueous interior with diameter as small as 3 nm. Alternatively, in the presence of siRNA, it is logical to suppose that the limit size particle consists of a distorted inverted micelle of cationic lipid surrounding the siRNA oligonucleotide. In turn, this would suggest a limit size system as small as 14 nm diameter, assuming that the siRNA contained in this inverted micelle is surrounded by an inner monolayer of cationic lipid within an outer monolayer of surface lipid and that the dimensions of the siRNA are 2.6 nm in diameter and 5.8 nm in length.22
Here we explored whether limit size particles compatible with such structures could be generated using PEG-lipid as the surface lipid. In this regard, a vesicle containing an internal aqueous core of 3 nm diameter has an outside-to-inside surface area ratio of 6.8 (assuming a bilayer thickness of 4 nm), indicating that the outer monolayer requires the presence of lipids that provide an interfacial area approximately 7 times larger than the inner monolayer area. Assuming that the interfacial area for a lipid such as DLinKC2-DMA is similar to that of dioleoylphosphatidylcholine (0.7 nm2),23 it is straightforward to show that approximately 10 mol % PEG2000-lipid (surface area per molecule 36 nm2)24 would be required to coat inverted micelles composed of DLinKC2DMA with an aqueous core 3 nm in diameter.
We therefore examined the limit size LNP that could be achieved for a DLinKC2-DMA/PEG-c-DMA system (90:10, mol:mol) produced by the microfluidic mixing process. The size determined from cryo-TEM micrographs (Figure 2A) was 14.7 ± 6.9 nm, consistent with an ability of the cationic lipid to form inverted micellar structures with interior aqueous diameters in the range of 8 nm. On the other hand, the limit size of LNP siRNA systems formulated at an siRNA-to-cationic lipid charge ratio of one resulted in limit size systems of 22.7 ± 6.1 nm in diameter (Figure 2B), consistent with the presence of inverted micelles with interior diameters of approximately 15 nm consisting of cationic lipid complexed to internalized siRNA.
Figure 2.
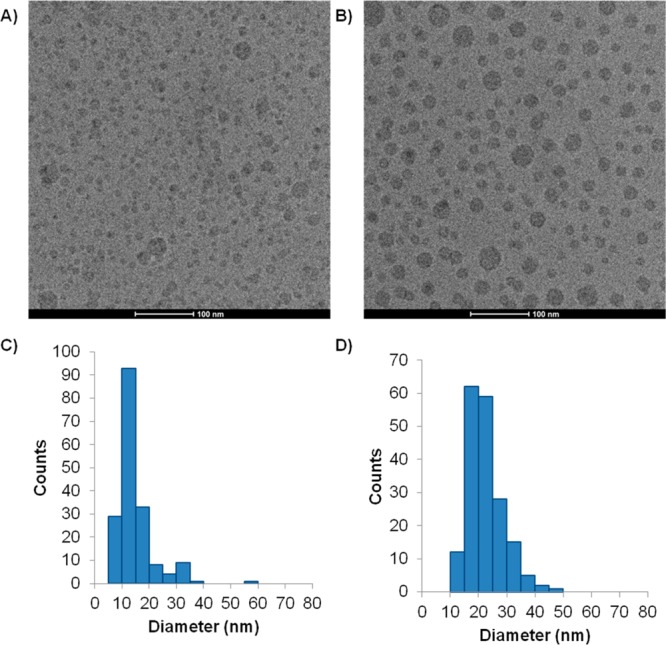
LNP exhibit limit sizes consistent with inverted micelle structure in presence and absence of siRNA. Limit size LNP were prepared by microfluidic mixing as indicated in the Experimental Section. (A) Cryo-TEM micrograph obtained from LNP with lipid composition DLinKC2DMA/PEG-lipid (90/10; mol/mol) in the absence of siRNA. (B) LNP with the same composition as for (A) but prepared with siRNA at an siRNA-to-cationic lipid charge ratio of 1. (C) Size distribution of LNPs in Figure 2A and (D) size distribution of LNPs in (B). Particle diameters are determined with the aid of Image J (NIH, Bethesda, MD), and average diameters are calculated from over 150 particles.
3.4. Encapsulated siRNA Is Immobilized in the LNP siRNA System
If the siRNA is complexed to cationic lipid and localized in an inverted micelle inside the LNP, it would be expected to be less mobile than if freely tumbling in the aqueous interior of a bilayer vesicle system. The mobility of the siRNA can be probed using 31P NMR techniques. In particular, it would be expected that limited motional averaging would be possible for complexed siRNA, leading to very broad “solid state” 31P NMR resonances due to the large chemical shift anisotropy of the phosphate phosphorus.25 If, on the other hand, the siRNA is able to freely tumble in an aqueous environment, rapid motional averaging would be expected to lead to narrow, readily detectable 31P NMR spectra. In order to avoid conflicting 31P NMR signals arising from phosphorus in phospholipids and siRNA, phosphorothoiate siRNA, which gives a 31P NMR signal that is shifted downfield from the normal phosphate resonance, was used to ascertain the motional environment of the siRNA.
As shown in Figure 3A, the 31P NMR signal from free phosphorothioate siRNA is a doublet peak, shifted approximately 56 ppm downfield from the phosphate resonance.26,27 The doublet structure may be attributed to the presence of Rp and Sp isomers of the siRNA strands.28 For LNP siRNA systems with the lipid composition DLinKC2-DMA/DSPC/Chol/PEG-c-DMA (40/11.5/47.5/1 mol %) and containing siRNA (0.06 siRNA/lipid; wt/wt) no 31P NMR signal was observable for the encapsulated siRNA (Figure 3B), consistent with immobilization within the LNP core. Detergent solubilization of the LNP particle using sodium dodecyl sulfate (1% vol/vol) resulted in recovery of the siRNA signal (Figure 3C).
Figure 3.

Encapsulated siRNA is immobilized on the NMR time scale. (A) 31P signal from free (phosphothioate) siRNA. Note that phosphothioate siRNA, which gives rise to a 31P NMR resonance ∼56 ppm downfield of the phosphodiester peak, was used to avoid overlap with the 31P NMR signal arising from the DSPC phosphorus. (B) 31P NMR spectrum of phosphothioate siRNA encapsulated at a siRNA/lipid ratio of 0.06 (w/w) in LNP containing DLinKC2-DMA/DSPC/Chol/PEG-lipid (40/11.5/47.5/1; mol/mol). (C) 31P NMR signal arising from the same sample as (B) after the addition of 1% SDS to solubilize the particle. The spectra depicted were obtained from 15 000 transients as described in the Experimental Section.
3.5. Encapsulated siRNA Is Fully Protected from Degradation by External RNase A
siRNA sequestered in the LNP core should be fully protected from degradation by externally added RNase. LNP siRNA systems with the lipid composition DLinKC2-DMA/DSPC/Chol/PEG-c-DMA (40/11.5/47.5/1, mol %) were incubated with bovine pancreatic RNase A to determine the protection of encapsulated siRNA. As shown in Figure 4, gel electrophoresis indicates that free siRNA is degraded, while the siRNA encapsulated in the LNP particles formulated by the microfluidic method is completely protected. Addition of the detergent Triton X-100 dissolves the LNP, releases the siRNA, and results in siRNA degradation in the presence of RNase.
Figure 4.

siRNA encapsulated in LNP is fully protected from external RNase. siRNA was either employed in the free form or encapsulated in LNP containing DLinKC2-DMA/DSPC/Chol/PEG-lipid (40/11.5/47.5/1; mol/mol) at an siRNA/lipid ratio of 0.06 (w/w). Encapsulation was performed using the microfluidic mixer as indicated in the Experimental Section. The integrity of the siRNA was challenged with 1 μg/mL bovine pancreatic RNase A. 5% Triton X-100 was added to solubilize the LNP. Gel electrophoresis was performed on 20% native polyacrylamide gel and siRNA visualized by staining with SYBR-Safe.
3.6. Encapsulated siRNA Is Complexed with Internalized Cationic Lipid
As indicated above, the electron dense core of the LNP siRNA systems may be suggested to consist of encapsulated siRNA complexed to cationic lipid and the remaining lipid (cationic lipid, phospholipid, cholesterol and PEG-lipid) is either present in the core in inverted micellar or related nanostructures, or is resident on the LNP exterior. It would then be expected that at high siRNA contents corresponding to siRNA-to-cationic lipid charge ratios of 1, where all the cationic lipid is complexed with internalized siRNA, little or no cationic lipid would be present on the external monolayer. In order to test whether this is the case , a fluorescence resonance energy transfer (FRET) assay for exterior cationic lipid was developed. This assay, which is essentially a membrane fusion assay, utilized negatively charged bilayer lipid vesicles composed of dioleoylphosphatidylserine (DOPS) that contained the FRET pair, NBD-PE and Rh-PE at 1 mol % each. LNP siRNA systems consisting of DLinKC2-DMA/DSPC/Chol/PEG-lipid (40/11.5/47.5/1 mol %) were added to the DOPS vesicles and incubated at pH 5.5. The pKa of DLinKC2-DMA is 6.72 and thus more than 90% of the DLinKC2-DMA on the outside of the LNP will be charged at pH 5.5, potentially promoting fusion with negatively charged DOPS LNP. As indicated elsewhere9,10 fusion is observed as an increase in the NBD-PE fluorescence at 535 nm as the NBD-PE and Rh-PE probes become diluted following lipid mixing.
As shown in Figure 5, when the LNP systems contained no siRNA, substantial fusion was observed, consistent with a considerable proportion of the DLinKC2-DMAresiding on the outer monolayer of the LNP system. When the LNP systems contained siRNA at a siRNA-to-total lipid ratio of 0.06 (w/w), which corresponds to an siRNA-to-cationic charge ratio of 0.25, however, the extent of fusion was reduced (Figure 5), and for LNP siRNA systems prepared with an siRNA-to-cationic lipid charge ratio of one little or no fusion was observed, indicating that little or no DLinKC2-DMAwas present on the LNP siRNA exterior. This supports the hypothesis that in LNP with high siRNA content all the cationic lipid is complexed with siRNA and sequestered in the LNP interior.
Figure 5.

Cationic lipid is associated with internalized siRNA in LNP siRNA systems. The amount of external cationic lipid in LNP siRNA systems was assayed as a function of the siRNA phosphate-to-cationic lipid charge ratio using the FRET lipid mixing assay described in the Experimental Section. Three LNP systems DLinKC2-DMA/DSPC/Chol/PEG-lipid (40/11.5/47.5/1; mol/mol) were prepared at charge ratios of 0 (solid line), 0.25 (dotted line), and 1 (dash line). The lipid mixing assay was performed at pH 5.5 to ensure that essentially all external DLinKC2-DMA was positively charged. The reaction was initiated by injecting the LNP (at t = 30 s) into a stirred cuvette containing the anionic DOPS/NBD-PE/Rh-PE (98:1:1 molar ratio) vesicles.
A remaining question concerns why the maximal dequenching for the unloaded sample plateaus at approximately 25%. It is logical to suppose that the DOPS vesicles fuse with positively charged LNP to the point that all available cationic lipid is complexed with DOPS, after which there is no electrostatic attraction driving LNP-DOPS vesicle fusion and therefore no further dilution of the FRET pairs. The above experiments were performed using 200 μmol LNP (80 μmol cationic lipid) and 100 μmol DOPS vesicles, indicating that when all possible fusion has occurred, approximately 20% of the DOPS vesicles would remain intact and the FRET pairs in the remaining 80% of the DOPS vesicles would experience a dilution of approximately 3.5-fold. This may be insufficient to produce maximum dequenching. In order to test this hypothesis, the concentration of FRET pairs in the DOPS vesicles was lowered to maximize dequenching effects. As shown in Supporting Information Figure 2, the maximal dequenching then increased to approximately 80% on fusion with unloaded LNP, consistent with hypothesis.
3.7. LNP siRNA Systems Have a Different Density than Aqueous Core Bilayer Vesicles
If the LNP siRNA systems exhibit a lipid core, they would be expected to exhibit a different density as compared to vesicles with an aqueous core. In the absence of siRNA where the interior consists of inverted micelles of cationic lipid the density should be less than vesicular systems, and the density should increase as more siRNA is encapsulated. As shown in Figure 6, when empty LNPs without siRNA and LNPs containing siRNA at 0.06 siRNA-to-total lipid ratio are centrifuged on a 1–15% sucrose step gradient as described under in the Experimental Section, the LNPs remained on the top of the gradient (Figure 6). In a parallel experiment, POPC/cholesterol (1:1; mol/mol) bilayer vesicles were centrifuged on an identical gradient and the vesicles distributed as a broad peak centered at around fraction 7 of the gradient. Increasing siRNA content in the LNP results in an increase in density, as LNPs containing siRNA at a 0.24 siRNA-to-total lipid ratio (corresponding to a 1:1 siRNA/cationic lipid charge ratio) were much denser, exhibiting a peak centered at fraction 15 of the column. In an additional experiment (data not shown) empty LNPs and LNP containing siRNA at 0.06 siRNA-to-lipid ratio were introduced first, at the bottom of the centrifuge tube, and a 1–10% sucrose step gradient layered on top. After centrifugation, the LNP were found to have redistributed to the top of the gradient. These results provide strong evidence that LNP siRNA systems contain a lipid core with a density dependent on the amount of siRNA encapsulated.
Figure 6.

The density of LNP siRNA systems is consistent with a hydrophobic lipid core as indicated by density gradient ultracentrifugation. A 1–15% sucrose step gradient was used as described in the Experimental Section. Fractions (500 μL) were successively removed from the top of gradient following centrifugation at 190 000g for 18 h and were assayed for cholesterol in POPC/cholesterol bilayer vesicles (open circles), empty LNP system (filled squares), LNP siRNA systems at a siRNA/cationic lipid charge ratios of 0.25 and 1 (filled triangles and filled diamonds, respectively).
3.8. Simulation Results Indicate that LNP siRNA Systems Exhibit a Nanostructured Core
The first step in computer modeling was to simulate the self-assembly of a putative unit cell for a lipid nanoparticle. To this end, a mixture of DLinKC2-DMA, distearoylphosphatidylcholine (DSPC), cholesterol, and nucleic acids was placed in a small box in a random configuration (Figure 7a). The self-assembly was performed in several independent simulations at different hydration levels to explore possible structures formed in the mixture. We also simulated self-assembly of a medium-sized system containing a polymer-grafted lipid in addition to other components. Comparing resulting structures, we found that a small system at low hydration (Figure 7b) was similar to the core of the self-assembled medium sized system and selected it as a building block for a larger LNP. The large LNP was constructed by multiplying the building block, coating the resulting structure with a PEG-lipid layer, and resolvating the system; see the Experimental Section. Small spacing between the unit cells and between the polymer layer and the nanoparticle was allowed to adjust the water contents upon equilibration of the LNP.
During the equilibration (∼1 μs), the water compartments closed and the PEG-grafted lipids adsorbed on its surface with polymer chains oriented toward the solution. On the simulation time, the LNP gradually transformed into a smooth rounded capsule with an averaged diameter of ca. 44 nm (see Figure 8a). The outer layer of the LNP is constituted by a roughly homogeneous coating of PEG-lipids. Inside the LNP, irregular water filled compartments of diameters ranging from ca. 3 to 9 nm are separated by bilayer membranes with DNAs bound to the membrane surface (see Figure 8b–d). The structure of the LNP core resembles an inverted hexagonal phase (HII) distorted at a high hydration level.29 The volume of water trapped inside the LNP constitutes ca. 6 waters/lipid which corresponds to 0.10 μL/μmol lipid, in good agreement with experimental data (data not shown). Given the significant fraction of lipids on the LNP surface (∼1/4), we expect this number to be slightly higher for a larger size LNP.
Figure 8.
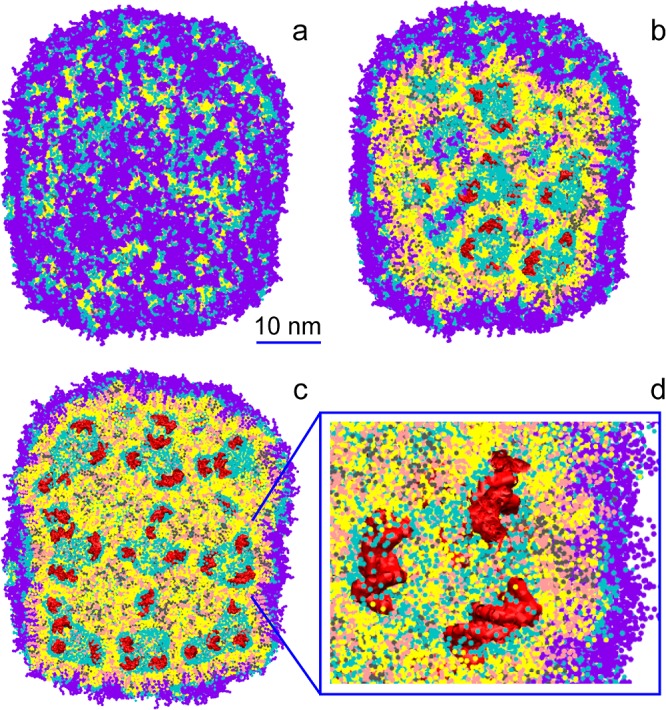
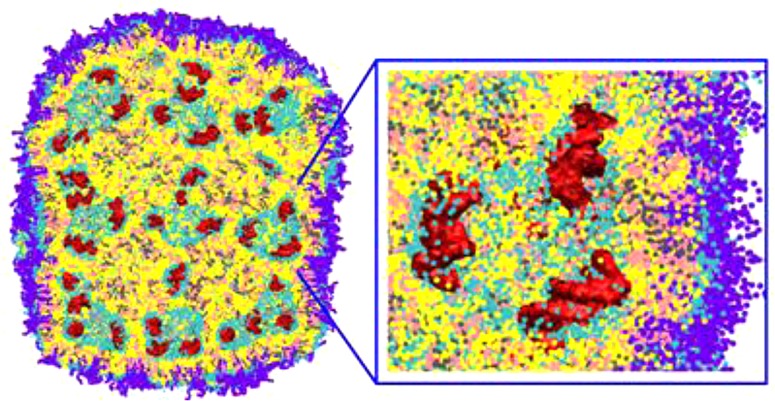
A lipid nanoparticle (LNP) contains irregular water-filled cavities separated by bilayer membranes, with nucleic acids bound to membrane surfaces: side (a), cross-section (b,c), and zoom-in (d) views. Cationic lipid DLinKC2-DMAis shown in yellow, cholesterol in pink, DSPC in gray, lipid polar moiety in cyan, PEG-lipid in violet, and nucleic acids (duplex DNA) in red; water not shown for clarity. The lipid composition was DLinKC2-DMA/DSPC/cholesterol/PEG-lipid (4:1:4:1; mol/mol) and DNA to lipid ratio ∼0.05 wt/wt.
In the LNP core, the numbers of nearest neighbors in the first coordination shell are given in Table 1. There are strong preferential interactions between the DNA phosphates and positively charged groups of DLinKC2-DMAlipids. Increased numbers of DLinKC2-DMA- DSPC neighbors originate from favorable interactions of the positively charged group of the cationic lipid with the negatively charged phosphate groups of DSPC. These interactions lead to formation of molecular pairs which manifest in the radial distribution function (RDF) as a pronounced second peak at a distance of ∼1 nm (data not shown).
Table 1. Simulation Results: Numbers of Nearest Neighbors in the First Coordination Shell for Each Molecule in the LNP Corea.
| no.
of neighbors for molecule |
||||
|---|---|---|---|---|
| DLinKC2-DMA | cholesterol | DSPC | DNA phosphates | |
| DLinKC2-DMA | 0.76 (0.78) | 0.57 (0.78) | 0.43 (0.2) | 0.84 |
| cholesterol | 0.58 (0.51) | 0.43 (0.51) | 0.14 (0.13) | 0.14 |
| DSPC | 1.70 (1.10) | 0.51 (1.10) | 0.27 (0.28) | 0.24 |
| DNA (phosphate) | 2.59 (1.43) | 0.43 (1.43) | 0.19 (0.36) | |
Estimated numbers in the absence of preferential interactions are shown in brackets. Neighbors for DNA are averaged over the entire LNP. The numbers are calculated for DNA phosphates, positively charged moiety of DLinKC2-DMA, the cholesterol polar group, and the DSPC phosphate group (or choline group in the case of DNA neighbors) by integrating the RDFs over the first maximum.
Spatial density distributions for selected groups are shown in Figure 9. The densities of headgroups are higher in the LNP core (Figure 9a) than on the LNP surface (Figure 9b). This is expected from a high negative curvature of the water-filled compartments inside the LNP, and the presence of polymer-grafted lipids on the surface. The distribution of DLinKC2-DMAaround DSPC lipids is shifted towards the DNA phosphates, and cholesterol is distributed somewhat away from DLinKC2-DMA(Figure 9a). Due to a high cationic lipid to DNA ratio, not all cationic lipids in the LNP core are bound to DNA. These free (Figure 9c) and bound (Figure 9d) cationic lipid fractions have a noticeably different 3D neighborhood. A somewhat heterogeneous surrounding of DSPC and cholesterol aligned along the linker of the free lipid becomes for the bound lipid displaced away and to the side by DNA phosphates.
Figure 9.
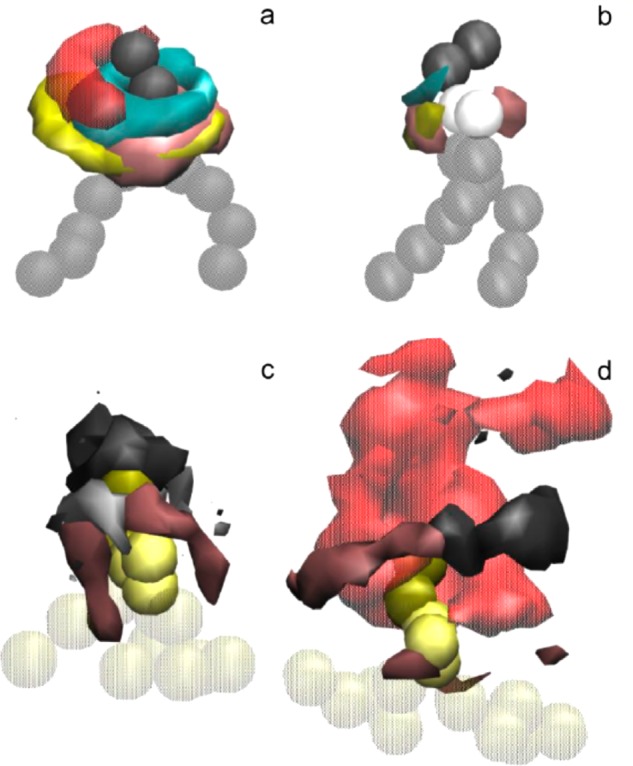
Spatial density distributions for selected molecular groups around DSPC in the LNP core (a) and on the LNP surface (b), and DLinKC2-DMAin the LNP core free (c) and bound to DNA (d). The surfaces of constant number densities are plotted at the values corresponding approximately to the average density in the first coordination shell. In the molecular representations, DSPC headgroup is shown as dark gray, glycerol-ester region as light gray, and hydrocarbon chains as transparent gray beads; DLinKC2-DMAheadgroup is shown as yellow, linker as light yellow, and chains as transparent yellow beads. In the density distributions, DNA phosphates are colored in semitransparent red, DLinKC2-DMAheadgroup in cyan and linker in yellow, cholesterol polar group in pink, DSPC headgroup in dark gray, and glycerol-ester in light gray.
3.9. Both Molecular Simulation and Experiment Support a Nanostructured Core for LNP siRNA Systems
The model for LNP siRNA structure developed by molecular simulation (Figure 8) is fully consistent with the experimental results presented here. The model shows that encapsulated siRNA is located in internalized distorted inverted micelles complexed to cationic lipid, with remaining lipids organized around small internal aqueous compartments. Such organization could account for the increased electron density as compared to bilayer vesicle systems as evidenced by cryo-TEM studies. This organization is also consistent with the ability of particles composed of DLinKC2-DMA and PEG-lipid to adopt structures as small as 14 nm diameter in the absence of siRNA and 23 nm diameter in the presence of siRNA, as well as the lack of external cationic lipid in LNP siRNA systems prepared at high siRNA-to-cationic lipid charge ratios and the essentially complete protection of encapsulated siRNA from external RNase.
The modeling results provide interesting insight into the location and role of DSPC. Previously DSPC has been included to provide increased stability to the preformed vesicles used in the preformed vesicle formulation method30 and has been thought to play a stabilizing role in the LNP structure formed. The results presented here suggest that DSPC could also be forming ion pairs between the DSPC phosphate group and cationic lipid headgroup, leaving the DSPC choline function to associate with siRNA phosphates. Such interactions have been suggested by other investigators.13 A final point is that, as suggested by the molecular modeling results, it is likely that cholesterol is distributed roughly homogeneously in the LNP core and surface, given its ability to partition into both lamellar and inverted lipid structures such as the hexagonal HII phase.31
A major advantage of the model presented in Figure 8 is that it can explain how siRNA encapsulation efficiencies approaching 100% can be achieved during the formulation process. Specifically, during the microfluidic synthesis process, the following steps are achieved: first, rapid mixing between the aqueous phase containing siRNA and the ethanol phase containing cationic lipid, second the association of cationic lipid with siRNA to form hydrophobic nucleating structures, and third, as the polarity of the medium increases further, coating of the nucleating structures by remaining lipids as they reach their solubility limits in the ethanol/water system, forming the final LNP siRNA structure. In this picture, it is clear how essentially all of the siRNA could be incorporated into LNP systems at nonsaturating siRNA loading, leading to complete encapsulation.
There are, however, other structures that could be compatible with high encapsulation efficiencies. In previous work,26,30 we have encapsulated antisense oligonucleotides into LNP systems containing ionizable cationic lipids with encapsulation efficiencies of up to 70% using the PFV method. The LNP formed are small multilamellar vesicles where the oligonucleotide is apparently located at the lamellar interfaces. These systems contained higher levels of the bilayer forming lipid DSPC and lower levels of cationic lipid than employed here. In order to show that the systems we observe here are part of a continuum of structures that are sensitive to the proportions (and types) of lipid components, we examined the cryo-TEM morphology of an LNP siRNA system containing a larger proportion of DSPC and a lower proportion of cationic lipid (DLinKC2-DMA/DSPC/Chol/PEG-lipid in the proportions 20/31.5/47.5/1; mol/mol, as opposed to 40/11.5/47.5/1; mol/mol). This system also exhibited high encapsulation efficiencies above 90% but also showed evidence of multilamellar structure, as shown in Supporting Information Figure 3. It would therefore appear that the internal structure of LNP siRNA systems changes from multilamellar to inverted micellar as the proportion of bilayer-forming species is reduced, as may be expected based on polymorphic phase preferences of component lipids.
It is likely that the results presented here for LNP siRNA systems produced by microfluidic mixing will also extend to LNP siRNA systems composed of the same or similar lipids but prepared by other in-line mixing protocols such as the T-tube mixer, as long as the mixing rates are sufficiently fast. In this regard, it may be noted that LNP siRNA systems containing ionizable cationic lipids prepared by T-tube mixing show the distinctive electron-dense core by cryo-TEM.4
4. Conclusions
In summary, the results presented in this work demonstrate a new type of lipid nanoparticle with a structured core formed by the rapid mixing of cationic lipid-containing ethanol solutions with aqueous solutions of siRNA oligonucleotides. Such structures are consistent with the electron dense core observed by cryo-TEM and the efficient loading characteristics associated with these particles. This work also provides, for the first time, an understanding of the mechanism of formation of LNP siRNA formulations formed by mixing cationic lipids in ethanol with siRNA in aqueous solution. It is anticipated that this understanding will lead to the design of more sophisticated LNP structures.
Acknowledgments
This work was supported by Alnylam Pharmaceuticals and the Canadian Institutes for Health Research (CIHR) through Grant FRN59836. DPT is an Alberta Innovates Health Solutions Scientist and Alberta Innovates Technology Futures Strategic Chair in (Bio)Molecular Simulation; DPT acknowledges support from NSERC. We thank Markus Heller at the Centre of Drug Research and Development (CDRD) for his assistance with the 31P NMR and Bradford Ross at the UBC BioImaging Facility for his assistance with the cryo-TEM.
Supporting Information Available
Supporting Information Figure 1: siRNA encapsulation efficiency of LNP siRNA systems prepared by microfluidic mixing at various siRNA/lipid ratios. Supporting Information Figure 2: Lipid mixing as measured by the FRET assay is a function of the concentration of FRET pairs incorporated into the DOPS vesicles. Supporting Information Figure 3: LNP containing different ratios of cationic lipid and DSPC can exhibit different structures. This material is available free of charge via the Internet at http://pubs.acs.org.
N.M.B., C.L.H. and P.R.C. have a financial interest in Precision NanoSystems; M.J.H. and P.R.C. have a financial interest in AlCana Technologies.
Supplementary Material
References
- Zimmermann T. S.; Lee A. C. H.; Akinc A.; Bramlage B.; Bumcrot D.; Fedoruk M. N.; Harborth J.; Heyes J. A.; Jeffs L. B.; John M.; et al. Nature 2006, 441, 111. [DOI] [PubMed] [Google Scholar]
- Semple S. C.; Akinc A.; Chen J.; Sandhu A. P.; Mui B. L.; Cho C. K.; Sah D. W. Y.; Stebbing D.; Crosley E. J.; Yaworski E.; et al. Nat. Biotechnol. 2010, 28, 172. [DOI] [PubMed] [Google Scholar]
- Jeffs L. B.; Palmer L. R.; Ambegia E. G.; Giesbrecht C.; Ewanick S.; MacLachlan I. Pharm. Res. 2005, 22, 362. [DOI] [PubMed] [Google Scholar]
- Crawford R.; Dogdas B.; Keough E.; Haas R. M.; Wepukhulu W.; Krotzer S.; Burke P. a; Sepp-Lorenzino L.; Bagchi A.; Howell B. J. Int. J. Pharm. 2011, 403, 237. [DOI] [PubMed] [Google Scholar]
- Zhigaltsev I. V.; Maurer N.; Edwards K.; Karlsson G.; Cullis P. R. J. Controlled Release 2006, 110, 378. [DOI] [PubMed] [Google Scholar]
- Abrams M. T.; Koser M. L.; Seitzer J.; Williams S. C.; DiPietro M. A.; Wang W.; Shaw A. W.; Mao X.; Jadhav V.; Davide J. P.; et al. Mol. Ther. 2010, 18, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belliveau N.; Huft J.; Lin P.; Chen S.; Leung A. K. K.; Weaver T. J.; Wild A. W.; Lee J. B.; Taylor R. J.; Tam Y. K.; Hansen C. L.; Cullis P. R. Microfluidic Synthesis of Highly Potent Limit-Size Lipid Nanoparticles for In Vivo Delivery of siRNA”, Molecular Therapy-Nucleic Acids 2012, 10.1038/mtna.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroock A. D.; Dertinger S. K. W.; Ajdari A.; Mezic I.; Stone H. a; Whitesides G. M. Science (New York, N.Y.) 2002, 295, 647–651. [DOI] [PubMed] [Google Scholar]
- Struck D. K.; Hoekstra D.; Pagano R. E. Biochemistry. 1981, 20, 4093. [DOI] [PubMed] [Google Scholar]
- Hafez I. M.; Ansell S.; Cullis P. R. Biophys. J. 2000, 79, 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; de Vries A. H. J. Phys. Chem. B 2007, 111, 7812. [DOI] [PubMed] [Google Scholar]
- Corsi J.; Hawtin R. W.; Ces O.; Attard G. S.; Khalid S. Langmuir 2010, 26, 12119. [DOI] [PubMed] [Google Scholar]
- Lee H.; de Vries A. H.; Marrink S.-J.; Pastor R. W. J. Phys. Chem. B 2009, 113, 13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; Uppsala S.-; Lindahl E. J. Chem. Theory Comput. 2008, 4, 435. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola a.; Haak J. R. J. Chem. Phys. 1984, 81, 3684. [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. J. Chem. Phys. 2007, 126, 014101. [DOI] [PubMed] [Google Scholar]
- Kuntsche J.; Horst J. C.; Bunjes H. Int. J. Pharm. 2011, 417, 120. [DOI] [PubMed] [Google Scholar]
- Hafez I. M.; Maurer N.; Cullis P. R. Gene Ther. 2001, 8, 1188. [DOI] [PubMed] [Google Scholar]
- Hafez I. M.; Cullis P. R. Adv. Drug Delivery Rev. 2001, 47, 139. [DOI] [PubMed] [Google Scholar]
- Tinker D. O.; Pinteric L. Biochemistry 1971, 10, 860. [DOI] [PubMed] [Google Scholar]
- Lafleur M.; Bloom M.; Eikenberry E. F.; Gruner S. M.; Cullis P. R. Biophys. J. 1996, 70, 2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg J. M.; Seeman N. C.; Day R. O.; Rich A. Biochem. Biophys. Res. Commun. 1976, 69, 979. [DOI] [PubMed] [Google Scholar]
- Alwarawrah M.; Dai J.; Huang J. J. Phys. Chem. B 2010, 114, 7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong R.; Macdonald P. M. Biochim. Biophys. Acta 2007, 1768, 1805. [DOI] [PubMed] [Google Scholar]
- DiVerdi J. A.; Opella S. J.; Ma E.-I.; Kallenbach N. R. Biochem. Biophys. Res. Commun. 1981, 102, 885. [DOI] [PubMed] [Google Scholar]
- Maurer N.; Wong K. F.; Stark H.; Louie L.; McIntosh D.; Wong T.; Scherrer P.; Semple S. C.; Cullis P. R. Biophys. J. 2001, 80, 2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein F.; Jovin T. M. Biochemistry 1983, 22, 4546. [DOI] [PubMed] [Google Scholar]
- Hopkins S.; Furman A.; Painter G. R.; Carolina N. Biochem. Biophys. Res. Commun. 1989, 163, 106. [DOI] [PubMed] [Google Scholar]
- Marrink S.-J.; Mark A. E. Biophys. J. 2004, 87, 3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple S. C.; Klimuk S. K.; Harasym T. O.; Dos Santos N.; Ansell S. M.; Wong K. F.; Maurer N.; Stark H.; Cullis P. R.; Hope M. J.; Scherrer P. Biochim. Biophys. Acta 2001, 1510, 152. [DOI] [PubMed] [Google Scholar]
- Cullis P. R.; de Kruijff B. Biochim. Biophys. Acta 1979, 559, 399. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.