

Summary
Although distinct epigenetic marks correlate with different chromatin states, how they are integrated within single nucleosomes to generate combinatorial signals remains largely unknown. We report the successful implementation of single molecule tools constituting fluorescence correlation spectroscopy (FCS), pulse interleave excitation-based Förster resonance energy transfer (PIE-FRET) and fluorescence lifetime imaging-based FRET (FLIM-FRET) to elucidate the composition of single nucleosomes containing histone variant H2A.Z (Htz1p in yeast) in vitro and in vivo. We demonstrate that yeast nucleosomes containing Htz1p are primarily composed of H4 K12ac and H3 K4me3 but not H3 K36me3 and that these patterns are conserved in mammalian cells. Quantification of epigenetic modifications in nucleosomes will provide a new dimension to epigenetics research and lead to a better understanding of how these patterns contribute to the targeting of chromatin-binding proteins and chromatin structure during gene regulation.
Key words: Histone modification, H2A.Z, Single molecule, Fluorescence correlation spectroscopy (FCS)
Introduction
The simplest structural unit of chromatin, the nucleosome, can exist in a variety of configurations depending on the histone variants and post-translational modifications present. The composition of individual nucleosomes influences chromatin structure and function and provides signals to the cellular machinery to promote gene activation or repression. These signals tend to be dynamic and can vary as a function of the cell cycle or growth conditions and between different cells in a population. Yet, our understanding of the patterns found within individual nucleosomes and presented to the cellular machinery is poorly defined, largely because limited, labor-intensive methodologies have been available to reveal these patterns. Approaches commonly used to characterize chromatin composition at population-based levels include mass spectrometry and chromatin immunoprecipitation (ChIP). Mass spectrometry has been employed to identify and map individual modifications within peptides derived from histones, but, unless coupled to affinity-based purification strategies, cannot describe which of the identified modifications are found within the same nucleosomes. ChIP can identify modifications enriched at chromosomal loci, but provides information averaged from a large starting population of cells. As a powerful complement to these approaches, we provide a toolkit of single molecule strategies to describe and quantify characteristics of nucleosomes containing the histone variant H2A.Z (Htz1p in yeast) both in vitro and in single fixed cells.
Htz1p/H2A.Z has been implicated in the regulation of gene expression through its targeted deposition at the 5′ end of genes in multiple organisms (Raisner et al., 2005; Barski et al., 2007). There, Htz1p/H2A.Z is preferentially assembled into one or two nucleosomes flanking transcriptional start sites (Raisner et al., 2005; Wong et al., 2007) by the ATP-dependent chromatin remodeling complex SWR1-C in Saccharomyces cerevisiae or SRCAP or p400/TIP60 in humans (Kobor et al., 2004; Mizuguchi et al., 2004; Wu et al., 2005; Gévry et al., 2007; Wong et al., 2007). This remodeling event is stimulated by NuA4-dependent acetylation of histone H4 K12ac (Altaf et al., 2010). Htz1p is often enriched at repressed promoters, but can dissociate upon gene activation, leading to a model in which Htz1/H2A.Z functions in creating a state in which genes are poised for activation (Adam et al., 2001; Larochelle and Gaudreau, 2003; Guillemette et al., 2005). Htz1p/H2A.Z promotes the localization of recently repressed genes to the nuclear periphery and facilitates their rapid activation (Brickner et al., 2007). Htz1p/H2A.Z also functions in responses to DNA damage and in maintaining the structural integrity of chromatin (Billon and Côté, 2012).
Like Htz1p, the location of several histone modifications has been mapped throughout the genome in S. cerevisiae (Liu et al., 2005) and mammals (Barski et al., 2007). Similarly to Htz1p, acetylation of several histone residues, including H4 K12ac, is enriched at promoters (Liu et al., 2005). Trimethylation of H3 K4 by the Set1p methyltransferase localizes to the 5′ end of genes and correlates with actively transcribed regions (Santos-Rosa et al., 2002; Ng et al., 2003). Other modifications, including mono- and dimethylation of H3 K4 by Set1p and trimethylation of H3 K36 by Set2p are enriched in the middle or 3′ ends of coding regions, and Set2p-dependent methylation has been linked to transcription elongation (Carrozza et al., 2005; Liu et al., 2005). For most modifications, it is not known which are present simultaneously in individual nucleosomes because both replication-coupled and replication-independent chromatin assembly contributes to continual nucleosome replacement throughout the genome (Mito et al., 2005; Dion et al., 2007; Kaplan et al., 2008).
Histone turnover rates vary as a function of the cell cycle, genomic location and transcriptional activity (Mito et al., 2005; Dion et al., 2007; Kaplan et al., 2008). This dynamic nature of chromatin implies that studies mapping steady-state histone modifications have only provided a partial picture of the composition of nucleosomes. Consistent with this idea, analyses of chromatin dynamics during multiple rounds of transcription using synchronization strategies and ligand-mediated activation of an estrogen receptor-dependent promoter have revealed that chromatin composition varies in a cyclical manner and includes the addition and removal of modifications commonly associated with activation or repression (Métivier et al., 2003).
Limited tools exist for defining the presence of multiple components in a single nucleosome. Strategies for elucidating the modification patterns, or combinatorial code, present at single nucleosome level necessitate a marked departure from conventional methods. We hypothesized that single molecule tools including fluorescence correlation spectroscopy (FCS) and fluorescence cross-correlation spectroscopy (FCCS) could decipher the components in a single nucleosome. FCS tracks the fluctuation in fluorescence intensity using a correlation function to provide information on the diffusion time, molecular size, and number of molecules in a limited focal volume (<1 fl) at single molecule precision (Maiti et al., 1997; Cypionka et al., 2009). Whereas fluctuations in fluorescence intensity of diffusing molecules can be revealed by auto-correlation analysis, interactions between molecules can be determined by FCCS (Schwille et al., 1997; Chen and Irudayaraj, 2010; Chen et al., 2011).
Interaction and distance between two components of a complex can be assessed by Förster resonance energy transfer (FRET), which capitalizes on the energy transfer between a donor and an acceptor fluorophore. Fluorescence lifetime imaging-based FRET (FLIM-FRET) provides a rigorous means of measuring FRET efficiency and is ideal for studying interactions between two targets in vivo because the decrease in fluorescence lifetime of the donor fluorophore in the presence of acceptor can be analyzed independently of the acceptor emission (Wallrabe and Periasamy, 2005). FLIM-FRET is especially attractive because it is independent of fluorophore concentration, photobleaching and spherical aberrations and therefore allows for the monitoring of multiple species in a spatially defined manner in single cells (Vidi et al., 2008).
Although histone modifications and variants in single cells can be assessed by FLIM-FRET, we anticipated that single molecule interactions of mononucleosomes in vitro diffusing in a confocal volume could be better interrogated by pulse-interleave excitation FRET (PIE-FRET), which corrects for artifacts from incomplete FRET pairs with missing or non-fluorescing acceptors, correcting for zero FRET efficiency pairs (Lee et al., 2005; Koopmans et al., 2009). In PIE-FRET, by alternately exciting both donor and acceptor molecules, interacting molecules can be sorted into two-dimensional distributions based on their donor–acceptor energy transfer. The corresponding energy transfer efficiency, distance and donor–acceptor stoichiometry can be estimated from this information (Müller et al., 2005). Application of the above methods provides powerful strategies for evaluating the presence of multiple components within a single complex.
In this work, we report the successful implementation of FCS, FCCS, PIE-FRET and FLIM-FRET to reveal that Htz1p/H2A.Z-containing nucleosomes are primarily comprised of H4 acetylated on K12 (H4 K12ac) and H3 tri-methylated on K4 (H3 K4me3) but not H3 tri-methylated on K36 (H3 K36me3). The approaches presented here set the stage for the development of a quantitative perspective for understanding epigenetics.
Results
Defining post-translational modification patterns in single nucleosomes containing Htz1p
Whether multiple histone modifications and variants identified in population-based studies are present in the same nucleosome is largely unknown. To assess modifications in Htz1p/H2A.Z-containing nucleosomes, we monitored H4 K12ac, H3 K4me3 and H3 K36me3 by PIE-FRET. Acetylation of H4 K12 by NuA4 stimulates nucleosome remodeling by SWR1-C (Altaf et al., 2010). H3 K4me3 and H3 K36me3 are associated with transcriptionally active loci, but differ in their localization within genes. The presence of these modifications within Htz1p/H2A.Z nucleosomes in yeast has not been evaluated previously.
In our analysis, the mean FRET efficiency between labeled antibodies targeting HA–Htz1p and H4 K12ac was 24% for mononucleosomes isolated from yeast expressing HA–Htz1p, whereas no FRET was observed with control mononucleosomes isolated from H4 mutants in which lysine 12 has been mutated to arginine (H4 K12R mutants) expressing HA–Htz1p (Fig. 1C, Fig. 2A). Similarly, the mean FRET efficiency was 21% for HA–Htz1p and H3 K4me3, whereas no FRET was observed in control mononucleosomes isolated from set1Δ mutants expressing HA–Htz1p (Fig. 1D, Fig. 2B). The high FRET efficiency confirms the presence of the above two modifications in nucleosomes containing Htz1p. The distances between the fluorophores on the respective antibodies bound to H4 K12ac or H3 K4me3 and the N-terminal hemagglutinin (HA) tag on Htz1p estimated by PIE-FRET were 6.4 and 6.7 nm, respectively, further confirming their close proximity. Because we employed labeled antibodies to detect targets in mononucleosomes, the antibody (2–3 nm) could also contribute to the distance measurements (Ratcliff and Erie, 2001). Hence, the distances calculated from PIE-FRET data reflect the distances between fluorophores on each antibody, not necessarily between the targets themselves. Nevertheless, they demonstrate the close proximity of the targets. To further validate these findings using a complimentary approach, we employed FCCS to assess the presence of H4 K12ac or H3 K4me3 within HA–Htz1p nucleosomes. H4 K12ac or H3 K4me3 were observed in 11% or 16%, respectively, of nucleosomes containing HA–Htz1p (Fig. 2A,B, right panels).
Fig. 1.

Isolation of mononucleosomes from yeast. (A) Chromatin fractions from wild-type, H3 K4R, htz1Δ or htz1Δ strains expressing HA–Htz1p. Histones in chromatin were analyzed by immunoblots probed with anti-HA antibodies (top), and then stripped and reprobed with anti-H3 antibodies (bottom). (B) Mononucleosome preparation. DNA extracted from nuclear extracts from 1×108 cell equivalents of yeast expressing HA–Htz1p were digested with 0 or 2.0 U MNase/mg of solid and analyzed by agarose gel electrophoresis. (C–E) Analyses of antibody specificity and genotype of yeast used for single molecule studies. Immunoblots of whole cell extracts from the indicated yeast strains were probed with anti-HA antibodies (top panels) to evaluate HA–Hzt1p expression, then stripped and reprobed with anti-H4 K12ac (C), anti-H3 K4me3 (D) or anti-H3 K36me3 (E) antibodies (middle panels) to evaluate histone modifications. Blots were then restripped and reprobed with anti-H3 antibodies because H3 served as a loading control (bottom panels).
Fig. 2.

H4 K12ac and H3 K4me3 are present in Htz1p-containing mononucleosomes, as shown by PIE-FRET and FCS. (A) PIE-FRET efficiency histograms of interactions of Htz1–HA plus H4 K12ac of mononucleosomes isolated from wild-type yeast (left panel) or H4 K12R mutants (middle panel) expressing HA–Htz1p. Fluorescence cross-correlation analysis of Htz1–HA plus H4 K12ac of HA–Htz1p mononucleosomes (right panel). (B) PIE-FRET efficiency histograms of interactions of Htz1–HA plus H3 K4me3 of mononucleosomes isolated from wild-type yeast (left panel) or set1Δ mutants (middle panel) expressing HA–Htz1p. Fluorescence cross-correlation of Htz1–HA plus H3 K4me3 of HA–Htz1p mononucleosomes (right panel). Donor: anti-H4 K12ac–FAM-X (A) or anti-H3 K4me3–Alexa-Fluor-488 (B). Acceptor: anti-HA–Alexa-Fluor-647. (C,D) H3 K36me3 is present in H2A mononucleosomes but undetectable in Htz1p mononucleosomes. PIE-FRET efficiency histograms of interactions of H2A (C) or HA–Htz1p (D) with H3 K36me3 in mononucleosomes isolated from wild-type yeast (left panels) or set2Δ mutants (middle panels) expressing HA–Htz1p. Fluorescence cross-correlation analysis of H3 K36me3 plus H2A of wild-type yeast mononucleosome (C, right panel). Donor: anti-H3 K36me3–Alexa-Fluor-488. Acceptor: anti-H2A–Alexa-Fluor-647 (C) or anti-HA–Alexa-Fluor-647 (D). (E) H4 K12ac and H3 K4me3 are present in the same nucleosome. PIE-FRET efficiency histogram of interactions between H4 K12ac and H3 K4me3 in mononucleosomes isolated from wild-type yeast (left panel) or set1Δ mutants (middle panel). Fluorescence cross-correlation of H4 K12ac plus H3 K4me3 of wild-type yeast mononucleosomes (right panel). Donor: anti-H3 K4me3–Alexa-Fluor-488. Acceptor: anti-H4 K12ac–Alexa-Fluor-647.
Unlike the above modifications, H3 K36me3 was not observed in HA–Htz1p-containing mononucleosomes isolated from either wild-type yeast or set2Δ mutants expressing HA–Htz1p (Fig. 1E), as demonstrated by an insignificant FRET efficiency (Fig. 2D). By contrast, H3 K36me3 and H2A colocalized in wild-type mononucleosomes, but not in control mononucleosomes isolated from set2Δ mutants (Fig. 2C; supplementary material Fig. S1) by PIE-FRET. Together, these results indicate that H3 K36me3 and Htz1p were not likely to be present in the same nucleosome, whereas H3 K36me3 was enriched in nucleosomes containing H2A. This lack of interaction is consistent with previous genome-wide ChIP analyses demonstrating that Htz1p localizes to 5′ ends and H3 K36me3 is enriched in the middle and 3′ ends of genes (Liu et al., 2005; Raisner et al., 2005; Barski et al., 2007) (see Discussion).
Defining the composition of Htz1p-containing nucleosomes in single yeast cells
To confirm that the modification patterns of Htz1p-containing nucleosomes observed in vitro reflected those in vivo, we next examined the composition of Htz1p-containing nucleosomes in single yeast cells. Here, we introduce FLIM-FRET as an elegant technique to monitor interactions between modifications and histone variants in the context of an intact nucleus because a quantitative readout is possible by monitoring the change in the lifetime of the donor fluorophore. Yeast cells expressing or lacking HA–Htz1p were fixed and stained with anti-H4 K12ac–FAM-X, anti-H3 K4me3–Alexa-Fluor-488 or anti-H3 K36me3–Alexa-Fluor-488, along with anti-HA–Alexa-Fluor-647 antibodies and H4 K12ac, H3 K4me3 or H3 K36me3 plus HA–Htz1p were monitored by FLIM-FRET (Fig. 2; Table 1). In our analysis, the lifetime of donor fluorophores (FAM-X or Alexa Fluor 488) targeted to H4 K12ac (Fig. 3A), H3 K4me3 (Fig. 3C), but not H3 K36me3 (Fig. 3E), decreased in the presence of the acceptor (Alexa Fluor 647) targeted to HA–Htz1p in cells expressing HA–Htz1p, but not in cells lacking HA–Htz1p (Fig. 3B,D,F; Table 1). The respective FRET efficiencies of H4 K12ac and H3 K4me3 with HA–Htz1p were ∼16% and ∼14%, respectively, in cells expressing HA–Htz1p. The corresponding distances between the fluorophore pairs were 5.9 and 7.0 nm, respectively, which is less than the diameter of a nucleosome. By contrast, the FRET efficiency of H3 K36me3 with HA–Htz1p was 4% (similar to background) (Table 1), indicating a lack of H3 K36me3 within HA–Htz1p nucleosomes in vivo, which is consistent with our in vitro studies.
Table 1.
FLIM-FRET analysis of histone modifications in yeast and MDA-MB-468 cells
| Cells | Genotype | Antibody targeta | Lifetime (nanoseconds) | FLIM-FRET efficiency | Distance (nm) |
| Yeast | Donor only | H4 K12ac | 4.01±0.0609 | – | – |
| Donor only | H3 K4me3 | 4.07±0.0533 | – | – | |
| Donor only | H3 K36me3 | 4.10±0.0730 | – | – | |
| HTZ1-HA | H4 K12ac + HA | 3.37±0.0812 | 0.16 | 5.93 | |
| HTZ1-HA | H3 K4me3 + HA | 3.51±0.0465 | 0.14 | 7.58 | |
| HTZ1-HA | H3 K36me3 + HA | 3.92±0.0354 | 0.044 | – | |
| HTZ1 | H4 K12ac + HA | 3.91±0.0329 | 0.025 | – | |
| HTZ1 | H3 K4me3 + HA | 3.96±0.0337 | 0.027 | – | |
| HTZ1 | H3 K36me3 + HA | 3.91±0.0372 | 0.046 | – | |
| MDA-MB-468 | H2A.Z | H3 K4me3 (donor only) | 4.01±0.0312 | – | – |
| H2A.Z | H3 K36me3 (donor only) | 4.05±0.0566 | – | – | |
| H2A.Z | H3 K4me3 + H2A.Z | 2.83±0.106 | 0.29 | 6.50 | |
| H2A.Z | H3 K4me3 + HA | 3.97±0.0793 | 0.0099 | – | |
| H2A.Z | H3 K36me3 + H2A.Z | 3.96±0.0129 | 0.022 | – | |
| H2A.Z | H3 K36me3 + HA | 3.98±0.0258 | 0.017 | – |
FAM-X for anti-H4 K12ac antibody; Alexa Fluor 488 for anti-H3 K4me3 and anti-H3 K36me3 antibodies; Alexa Fluor 647 for anti-HA and anti-H2A.Z antibodies. R0 = 5.6 nm for Alexa Fluor 488 and Alexa Fluor 647, and 4.5 nm for FAM-X and Alexa Fluor 647.
Fig. 3.
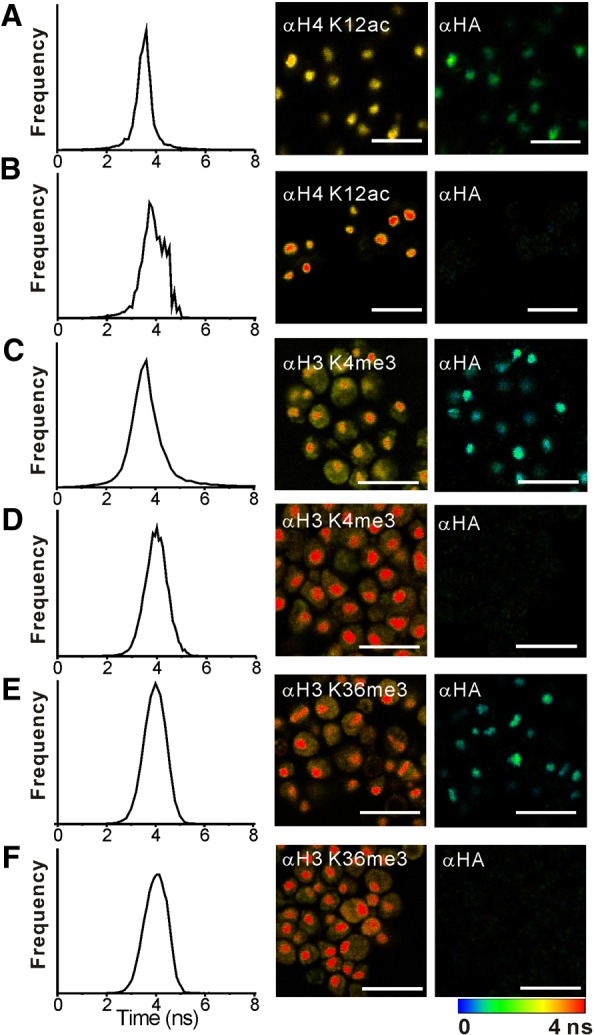
Htz1p is associated with H4 K12ac and H3 K4me3 but not H3 K36me3 in yeast. (A,B) Analysis of H4 K12ac in yeast expressing (A) or lacking (B) HA–Htz1p. (C,D) H3 K4me3 in yeast expressing (C) or lacking (D) HA–Htz1p. (E,F) H3 K36me3 in yeast expressing (E) or lacking (F) HA–Htz1p. Yeast were fixed with methanol:acetic acid and incubated with the indicated fluorescently labeled antibodies and analyzed by FLIM-FRET as described in the Materials and Methods. Left column: Fluorescence lifetime distribution of donor (anti-H4 K12ac–FAM-X, anti-H3 K4me3–Alexa-Fluor-488 or anti-H3 K36me3–Alexa-Fluor-488) in the presence of acceptor (anti-HA–Alexa-Fluor-647). Middle column: FLIM from donor channel. Right column: FLIM from acceptor channel. Scale bars: 10 µm. FLIM scale: 1 nanosecond, blue; 4.5 nanoseconds, red. See also Table 1.
Composition of H2A.Z-containing nucleosomes is evolutionarily conserved in mammals
To evaluate the composition of nucleosomes containing the H2A.Z variant in human cells, we stained fixed MDA-MB-468 cells with anti-H3 K4me3–Alexa-Fluor-488 or anti-H3 K36me3–Alexa-Fluor-488 antibodies alone or in combination with anti-H2A.Z–Alexa-Fluor-647 or negative control anti-HA–Alexa-Fluor-647 antibodies and analyzed samples by FLIM-FRET (Fig. 4; supplementary material Fig. S3). Similar to what we observed in yeast, the fluorescence lifetime of the donor representing H3 K4me3 decreased from 4.01 to 2.83 nanoseconds in the presence of an acceptor targeting H2A.Z and the FRET efficiency was 27%, yielding a distance of 6.5 nm between FRET pairs (Fig. 4A,B,E; Table 1). By contrast, the presence of H3 K36me3 with H2A.Z was not observed from the lifetime measurements and efficiency calculations (Fig. 4C,D,E; Table 1). Together, these results support a model in which the composition of modifications within nucleosomes containing Htz1p/H2A.Z is conserved across phyla.
Fig. 4.

H2A.Z is associated with H3 K4me3 in mammalian cells. (A–D) MDA-MB-468 cells were fixed with methanol:acetic acid and incubated with anti-H3 K4me3–Alexa-Fluor-488 antibodies only (donor) (A), with anti-H3 K4me3–Alexa-Fluor-488 plus anti-H2A.Z–Alexa-Fluor-647 antibodies (donor and acceptor in left and right panels, respectively) (B), with anti-H3 K36me3–Alexa-Fluor-488 antibodies only (donor) (C) or with anti-H3 K36me3–Alexa-Fluor-488 plus anti-H2A.Z–Alexa-Fluor-647 antibodies (donor and acceptor in left and right panels, respectively) (D), and then analyzed by FLIM-FRET. (E) Fluorescence lifetime distribution of samples shown in A–D. (F) Sequence alignments between yeast and human H2A and Hta1p/H2A.Z are shown. Ten amino acid intervals on H2A and Htz1p/H2A.Z are noted by filled and open circles, respectively. Mutation of residues highlighted in red in yeast H2A result in defects in H3 K36me3 by Set2p. The docking domain is shown with a solid line. (G,H) Composition of nucleosomes containing the histone variants. Scale bars: 10 µm. FLIM scale: 0 nanoseconds, blue; 3.9 nanoseconds, red. See also Table 1.
Defining multiple modifications present simultaneously within single nucleosomes
In the above analyses, we observed that H3 K4me3 and H4 K12ac were present in nucleosomes containing Htz1p, implying that both modifications might co-exist within the same nucleosome. To determine whether these modifications were present within a single nucleosome, PIE-FRET experiments were conducted using mononucleosomes isolated from wild-type yeast. The FRET efficiency between anti-H3 K4me3–Alexa-Fluor-488 and anti-H4 K12ac–Alexa-Fluor-647 antibodies targeting the modifications were 14.5% (distance was 7.5 nm) in wild-type and <5% in H4 K12R mutants (Fig. 2E). FCCS analysis indicated that 10% of nucleosomes containing H4 K12ac also contained H3 K4me3 (Fig. 2E, right panel). Together, these experiments implied that both modifications could coexist within Htz1p-containing nucleosomes. Further technology and analysis module development are in progress to address simultaneously whether three targets are in the same nucleosome in a generalized manner at the single molecule level.
Discussion
Here we lay the groundwork for the application of single molecule technologies to epigenetics research and showcase how these methodologies can be tailored to elucidate epigenetic patterns within extracted mononucleosomes and nucleosomes in single cells. By applying fluorescence fluctuation data from FCS and PIE-FRET/FLIM-FRET, we have established that nucleosomes containing Htz1p/H2A.Z have H3 K4me3 and H4 K12ac but not H3 K36me3 (Figs. 2–4; supplementary material Fig. S3). This latter modification was found within H2A-containing nucleosomes (Fig. 2C,D). Moreover, we show that H3 K4me3 and H4 K12ac are present simultaneously within individual nucleosomes (Fig. 2E). The integration of correlation spectroscopy with histogram analysis is invaluable because these approaches could be applied in the future to study the assembly and disassembly of a wide range of specialized nucleosomes or other protein complexes in a variety of cell types.
In our analysis, we observed pairwise coexistence of H3 K4me3 and Htz1p/H2A.Z (Fig. 2B, Fig. 3C,D, Fig. 4A,B,E and Table 1), H4 K12ac and Htz1p (Fig. 2A, Fig. 3A,B and Table 1) and H4 K12ac and H3 K4me3 in nucleosomes (Fig. 2E). H4 K12ac probably facilitates the role of SWR1-C in targeting Htz1p to transcriptional start sites. The bromodomain-containing Bdf1p subunit of SWR1-C binds histone H4 tails with NuA4-dependent acetylation patterns (Allard et al., 1999; Matangkasombut and Buratowski, 2003). Selective binding of Bdf1p to transcription initiation sites and incorporation of Htz1p into chromatin requires NuA4 (Koerber et al., 2009). Consistent with SWR1-C-dependent exchange of H2A for Htz1p being stimulated by NuA4-dependent H4 K12ac (Raisner et al., 2005; Altaf et al., 2010) and this residue remaining acetylated after incorporation of Htz1p, H4 K12ac is present in nucleosomes containing Htz1p in vitro (Fig. 2A) and in single cells (Fig. 3A,B; Table 1, yeast). These results support a model in which the H3/H4 tetramer is not replaced at the time of exchange of H2A for Htz1p by SWR1-C (Luk et al., 2010).
The lack of FRET between H3 K36me3 and Htz1p/H2A.Z both in vitro and in vivo (Fig. 2D, Fig. 3E,F, Fig. 4C–E) might be caused, in part, by a failure of Set2p to bind Htz1p/H2A.Z-containing nucleosomes. Recently, a region on the nucleosomal surface surrounding H3 K36, which is formed by H2A, H3 and H4, was found to be crucial for trimethylation (Du and Briggs, 2010). Mutations in H2A within this proposed Set2p-recognition surface lead to defects in Set2p-dependent trimethylation (Du and Briggs, 2010). This region is diverged between H2A and Htz1p/H2A.Z, especially in humans (Fig. 4F), and is part of the docking domain that interacts with the (H3–H4)2 tetramer and contains a metal ion binding site that is absent in H2A (Suto et al., 2000). Previously, Set2p-dependent methylation events have largely been attributed to interactions between Set2p and RNA polymerase II during transcription elongation (Fuchs et al., 2009). Future investigation of interactions between Set2p and Htz1p/H2A.Z-containing nucleosomes should clarify the role of Htz1p/H2A.Z in ensuring that the hypomethylated form of H3 K36 is maintained at the 5′ end of genes (Liu et al., 2005; Raisner et al., 2005; Barski et al., 2007). The approaches demonstrated here can also be applied in the future to distinguish between modifications present in Htz1p/H2A.Z-containing nucleosomes from those that are in distinct nucleosomal subpopulation(s) arising from cell-to-cell differences due to the cell cycle, transcriptional status or dynamic changes to chromatin during transcription.
Although single molecule FRET has previously been applied to nucleosomes, most of these studies focused on nucleosome remodeling and conformational dynamics using reconstituted nucleosomes labeled in vitro (Li et al., 2005; Blosser et al., 2009; Gansen et al., 2009; Andrews et al., 2010), neither PIE-FRET or FLIM-FRET have been applied previously to resolve histone modifications. In our single cell FRET studies, it is theoretically possible that FRET might occur between fluorophores on antibodies bound to targets on neighboring nucleosomes in intact chromosomes, in addition to targets on the same nucleosomes. However, no such interaction was observed in vivo between H3 K36me3 and Htz1p/H2A.Z (Fig. 3E,F, Fig. 4C,D). Our analyses also clearly confirmed that interactions observed in fixed cells were also present in isolated mononucleosomes in vitro where such interactions in trans are unlikely to occur (Fig. 2, Fig. 4E). In the approach applied here, either an increase or decrease in the measured distance relative to the actual distance between two targets could have occurred due to the size of the antibodies and the position of the fluorophores on these antibodies, which could affect the FRET efficiency. Although paraformaldehyde (PFA) has been widely used for cell fixation in FRET measurement, it is worth noting that fixation might influence FRET efficiency under certain circumstances. Reports suggest that formaldehyde fixation could decease the FRET efficiency of CFP–YFP fusions or of CFP/YFP when fused to endogenous proteins (Anikovsky et al., 2008). However, it is unlikely that fixation could result in a distance change between the donor and acceptor because the donor and acceptor are covalently linked in most cases. It is possible that fixation could reduce the values of the orientation factor (κ2) because immobilization might anchor fluorophores in positions that disfavor energy transfer (Anikovsky et al., 2008). In the case of FLIM-FRET measurement, reports have suggested that PFA fixation is unlikely to alter the fluorescence lifetime of donor and acceptor (Peter et al., 2005; Latz et al., 2007).
Future development of Fab fragments or small molecules such as aptamers to detect modifications will improve the accuracy of distance measurements and decrease the possibility of steric hindrance while evaluating multiple targets.
In summary, the single molecule approaches outlined here represent a powerful set of tools for establishing and quantifying epigenetic modification patterns as well as for tracking chromatin dynamics. We envision that single cell studies will enhance our understanding of how nucleosome composition is affected by the cell cycle and by responses to DNA damage or transcription. Such studies will also aid in defining the order in which modifications are added onto nucleosomes to create patterns and the interdependency between different modifications as well as trans-tail requirements for modifications within individual nucleosomes.
Materials and Methods
Plasmids and yeast strains
Yeast strains used in this study are described in supplementary material Table S1. Strains expressing histone mutants were generated by plasmid shuffling (Adams et al., 1997). Plasmids used in this study are described in supplementary material Table S2. Plasmids expressing histone mutants were generated by site directed mutagenesis as outlined in the Quick Change Site-Directed Mutagenesis Kit protocol (Stratagene) and confirmed by sequencing. Sequences of oligonucleotides used during site-directed mutagenesis are available upon request. Plasmid pAK991 was generated by introducing a HindIII-SalI fragment containing HA–HTZ1 from pTK23 (Santisteban et al., 2000) into HindIII-SalI of pRS316 (Sikorski and Hieter, 1989).
Whole cell extracts of histones
For analysis of histone modifications and HA–Htz1p expression, 5 ml cultures of yeast were grown in complete synthetic media or complete synthetic media lacking uracil to an OD600 of 1. Cells were harvested by centrifugation at 400 g for 5 minutes, resuspended in 250 µl 2 M NaOH, 8% β-mercaptoethanol and incubated on ice for 5 minutes. Samples were pelleted by centrifugation at 16,100 g for 1 minute, washed once with high salt extraction buffer (40 mM HEPES pH 7.5, 350 mM, 0.1% Tween 20 and 10% glycerol), collected by centrifugation at 16,100 g for 1 minute and resuspended in 50 µl 2× SDS-PAGE sample buffer.
Yeast nuclear extractions
Yeast nuclei were isolated from 300 ml cultures grown to an OD600 of 0.8–1.5 in synthetic complete media, synthetic complete media lacking uracil, or YPD as outlined previously (Miller et al., 2008). Nuclei were resuspended in 0.3 ml NP buffer (0.34 M sucrose, 20 mM Tris-HCl pH 7.5, 50 mM KCl, 5.0 mM MgCl2, 1.0 mM PMSF, 1 µg/ml leupeptin and 1 µg/ml pepstatin).
Isolation of chromatin
Approximately 5×108 cell equivalents of yeast nuclei were isolated by centrifugation at 16,100 g at 4°C for 10 minutes. Pellets were resuspended and lysed with RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.5, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) on ice for 5 minutes with intermittent vortexing. The chromatin fraction was isolated by centrifugation at 16,100 g for 10 minutes and enriched for chromatin-associated histones by washing twice each with buffer A (10 mM Tris pH 8.0, 0.5% NP-40 and 75 mM NaCl) and buffer B (10 mM Tris pH 8.0 and 300 mM NaCl). Final pellets were resuspended in 20 µl 2× SDS-PAGE sample buffer (25 mM Tris-HCl pH 6.8, 2.5% SDS, 2.5% glycerol, 0.01% Bromophenol Blue and 1.25% β-mercaptoethanol, final concentration) for protein blot analyses.
Mononucleosome preparation
Approximately 1×109 cells equivalent of yeast nuclei were harvested by centrifugation at 16,100 g for 10 minutes at 4°C. Pellets were resuspended in 0.5 ml RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.5, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) at a final concentration of 0.2×107 cell equivalents/µl. CaCl2 was added to a final concentration of 1 mM to aliquots of 1×108 cell equivalents in RIPA buffer and chromatin was digested with 0, 1.0, 1.75 or 2.0 U micrococcal nuclease (MNase; Sigma, N3755)/mg of solid for 7 minutes at 37°C. Digestions were stopped by adding EGTA to a final concentration of 10 mM. Aliquots were stored at −80°C prior to analysis by PIE-FRET and FCS/FCCS.
DNA extraction from mononucleosomes
To monitor MNase digestions, 50 µl aliquots of approximately 1×108 cell equivalents of MNase-treated mononucleosomes were incubated with 20 µg proteinase K (Sigma, P-2308) at 45°C for 1 hour. Samples were diluted into 0.15 ml TE buffer (10 mM Tris, pH 7.5, 1 mM EDTA), then extracted with phenol/chloroform and precipitated with ethanol. The final pellet was resuspended in 25 µl TE, digested with 20 µg ribonuclease A (Sigma, R-6513) at room temperature for 1 hour prior to analysis by agarose gel electrophoresis.
Immunofluorescence
Yeast cells were prepared for immunofluorescence by growing 5 ml cultures logarithmically to an OD600 of 0.8 in complete synthetic media or complete synthetic media lacking uracil (Q-BIO gene, BIO 101 Systems) with 2% glucose, and fixed with formaldehyde (3.7% final concentration) for 1 hour at room temperature. Cells were collected by centrifugation, washed with 0.1 M potassium phosphate pH 7.5, resuspended in 1 ml 0.1 M potassium phosphate pH 7.5, 50 µg/ml Zymolyase 100T (Seikagaku) and 2 µl/ml β-mercaptoethanol and incubated for 40 minutes at 30°C. No. 1 coverslips (VWR International) were coated with poly L-lysine (Sigma) and cells were deposited onto the coverslips, aspirated and washed 3× with 1× PBS (0.137 M NaCl, 2.7 mM KCl, 5.4 mM Na2HPO4 and 1.8 mM KH2PO4, pH 7.4). The coverslips were immersed in cold (−20°C) methanol for 6 minutes and then in cold (−20°C) acetone for 30 seconds, incubated with 15 µl blocking buffer (1× PBS, 3% BSA) for 1 hour at room temperature in a humid chamber. Coverslips were then incubated with 20 nM fluorescently labeled antibodies in blocking buffer for 1 hour at room temperature and washed 3× with PBS prior to analysis by FLIM-FRET.
Human MDA-MB-468 cells were seeded onto sterilized No. l coverslips and placed in six-well plates in RPMI 1640 containing 10% fetal bovine serum, at 37°C with 5% CO2. After reaching 90% confluency, the cells were fixed with a cold (−20°C) mixture of acetone and methanol (1∶1) for 10 minutes. The slides were washed with 3× PBS and blocked with 1× PBS containing 0.25% Triton X-100 and 1% BSA for 30 minutes. The slides washed with 1× PBS, incubated with the fluorescence labeled antibody overnight at 4°C, and then washed with 3× PBS prior to analysis by FLIM-FRET.
Protein blot analyses
For protein blot analyses (Fig. 1; supplementary material Fig. S1), 2×106 or 5×106 cell equivalents of whole cell extracts or 1×108 cell equivalents of chromatin-associated histones from logarithmically growing cells were separated on 15% polyacrylamide gels, transferred to PVDF membranes (Bio-Rad) and processed as outlined previously (Miller et al., 2008). Membranes were probed with anti-HA (12CA5) (Roche, 11583816001; 1∶2000); anti-histone H3 (tri-methyl K36) (Abcam, ab9050; 1∶10,000), anti-histone H3 (tri-methyl K4) (Abcam, ab1012; 1∶5000); anti-histone H4 (acetyl K12) (Abcam, ab61238; 1∶10,000); anti-H2A (Abcam, ab13923; 1∶5000), anti-histone Htz1 (Active Motif, #39648; 1∶1000) or anti-histone H3, (Abcam, ab1791; 1∶10,000). Secondary antibodies used were Alexa-Fluor-680-conjugated goat anti-rabbit IgG (Molecular Probes, A21109; 1∶5000 or 1∶10,000) or Alexa-Fluor-680-conjugated goat anti-mouse IgG (Molecular Probes, A21058; 1∶20,000). Membranes were stripped as outlined previously before reprobing (Miller et al., 2008). Images were obtained using an Odyssey Infared Imager (Li-Cor Biosciences) by scanning membranes at 169 µm resolution, medium-quality setting using the 700 and 800 nm channels. The intensity was set to 5 for all images obtained. Images were analyzed using Odyssey Software v1.2 (Li-Cor Biosciences).
Antibody labeling
Anti-HA antibodies were pre-labeled with Alexa Fluor 647 (Cell Signaling Technology, 3444). Anti-H4 K12ac (Abcam, ab61238) antibodies were labeled with 6-(fluorescein-5-carboxamido) hexanoic acid, succinimidyl ester (FAM-X) or Alexa Fluor 647 using the SureLINK FAM-X Labeling Kit (KPL) or Alexa Fluor 647 Monoclonal Antibody Labeling Kit (Invitrogen) according to the manufacturer's instructions. Free fluorophores were removed using a spin filter (MWCO 10 KDa, KPL). Anti-histone H3 (tri-methyl K4) (Abcam, ab1012) and anti-histone H3 (tri-methyl K36) (Abcam, ab9050) antibodies were labeled using the APEX Alexa Fluor 488 Antibody Labeling Kit (Invitrogen) according to manufacturer's instructions. Anti-histone H2A (Abcam, ab13923) and anti-histone H2A.Z (Abcam, ab4174) antibodies were labeled with Alexa Fluor 647 using the APEX Alexa Fluor 647 Antibody Labeling Kit (Invitrogen) according to manufacturer's instructions. Labeled antibodies were dialyzed into 1× PBS and stored at −20°C.
Evaluation of antibody labeling by FCS
Antibodies were labeled either with FAM-X or Alexa Fluor 488 for the 465 nm laser excitation or with Alexa Fluor 647 for the 636 nm laser excitation. Labeling was accomplished by following the manufacturer's specification and confirmed through UV-Vis spectroscopy (supplementary material Fig. S2A and data not shown). Two absorbance maxima peaks were noted: the 280 nm peak corresponding to the protein absorbance and the 495/650 nm peak corresponding to FAM-X/Alexa Fluor 488 or Alexa Fluor 647 label, indicating successful labeling. The molar substitution ratio of fluorophore to protein (F/P ratio) was determined by measuring the absorbance at 280 nm and 495/488 nm using the IgG extinction coefficient of 210,000 M−1cm−1 (Bhattacharyya et al., 2008). FCS was applied to confirm the labeling according to changes in diffusion time between free fluorophores and labeled antibody (supplementary material Fig. S2B). The diffusion time was obtained from fitting the autocorrelation curves with the appropriate equation. As expected, the diffusion time of antibodies labeled with fluorophores (∼0.4 milliseconds) increased significantly compared with free fluorescent dyes (∼0.035 milliseconds) and was proportional to the cubic root of the molecular weight. Furthermore, the autocorrelation of antibodies was fitted well with a single component 3D diffusion model, indicating a high degree of purity of the labeled antibodies.
Instrumentation
The Microtime 200 (Picoquant, Berlin, Germany) used for single molecule experiments was fitted with two picosecond pulsing diode lasers with excitation wavelengths of 465 and 636 nm (LDH470, LDH635, PicoQuant) controlled by a laser driver (Sepia PDL 808 Driver) at repetition rate of 40 MHz. The laser beams were focused in the sample volume using apochromatic 60× water immersion objective with 1.2 NA. The emitted fluorescence was collected using the same objective and separated from the excitation beam by a dichroic mirror (see scheme in supplementary material Fig. S4). Fluorescence was detected by two single photon avalanche photodiodes (SPAD, SPCM-AQR-14, Perkin-Elmer) using the time-correlated single photon counting module in the time-tagged time-resolved (TTTR) single photon mode, whereby each photon is tagged with a time stamp that identifies the arrival time after the laser pulse. All of the FCS/FCCS, PIE-FRET, FLIM-FRET and FLIM experiments were conducted using Microtime 200. Details of the instrumentation are provided in the literature (Varghese et al., 2008) and information on the theoretical and experimental basis are provided below.
Fluorescence correlation and cross-correlation spectroscopy
FCS is based on the correlation between fluctuations in fluorescence intensity as molecules diffuse through a small observation volume (less than 1 fl) (Sako and Yanagida, 2003). This highly sensitive technique provides information on the number of interacting molecules in a confocal-limited spot and the binding kinetics of two or more interacting species tagged with appropriate fluorophores (Maiti et al., 1997; Schwille et al., 1999; Bacia and Schwille, 2003; Hwang et al., 2006; Hwang and Wohland, 2007; Varghese et al., 2008).
In FCS, fluorescence fluctuations δI(t) around the average fluorescence <I> was measured in real time and the normalized autocorrelation was calculated as follows (Magde et al., 1974; Maiti et al., 1997; Rigler and Elson, 2001; Müller et al., 2003):
| (1) |
where δI(t) = I(t)−<I(t)>. The autocorrelation curve of fluorophore diffusing in solution was fitted to a 3D diffusion model using one or two components with the SymphoTime software (PicoQuant) and Origin Lab using the equation:
| (2) |
where Ni and τDi are the number of fluorescent molecules in the detection volume and diffusion time of component i, respectively. The parameter κ and the lateral diffusion coefficient D are defined by:
| (3) |
In Eqn 3, κ denotes the ratio of the axial beam size z and radius ω of the laser and τD is the diffusion time of the fluorophore. The effective confocal volumes for 465 and 636 nm excitation were calculated according to Eqn 1 and fitted according to the autocorrelation function (Eqn 2) using aqueous solutions of rhodamine 123 (300 µm2/second) or Atto 655 (390 µm2/second) dyes (Invitrogen Molecular Probes, Eugene, OR) with known diffusion coefficients. Assuming a 3D Gaussian observation volume as approximated by Veff = π3/2ω2z, the confocal volume for the 465 and 636 nm laser was 0.36 and ∼1 fl, respectively.
For monitoring the interaction between two species tagged by two different fluorophores simultaneously excited by the 465 and 636 nm lasers, dual color or FCCS was used. The cross-correlation function for an FCCS experiment is given by (Schwille et al., 1997; Bacia and Schwille, 2007):
 |
(4) |
In Eqn 4, <Ci>, <Cj> and <Cij> are the concentrations of species i, j, and ij (denoting bound species), respectively, diffusing in the confocal volume, and Veff is the effective detection volume for the dual color experiment. With these considerations, the diffusion time and the effective detection volume for cross-correlation analysis are given by:
| (5) |
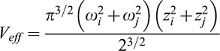 |
(6) |
Here, Dij is the diffusion coefficient of the bound fraction and its concentration Cij is obtained from the cross-correlation analysis using:
| (7) |
In Eqn 7, Gij(0) is the cross-correlation amplitude at time τ = 0, and Gi(0) and Gj(0) are the respective autocorrelation amplitudes of species i and j at time τ = 0. The inverse of the amplitude of the auto-correlation curve (shown in Fig. 2A,C,D,E) gives the apparent number of diffusing molecules in the confocal volume.
The fraction of bound complex was calculated as a percentage of the species (Ci or Cj) according to the following equation:
| (8) |
Under triplet state conditions, the autocorrelation function should compensate for these fractions of fluorescent molecules and can be evaluated using the equation:
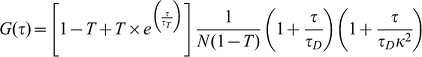 |
(9) |
Here T is the dark or triplet fraction of molecules and τT is the triplet relaxation time.
PIE-FRET
PIE-FRET was established using the combination of time-correlated single-photon counting (TCSPC) techniques and two pulsed picosecond lasers operating at a laser power in the range 50–100 µW to alternately excite both donor and acceptor molecules in a complex (Lee et al., 2005; Rüttinger et al., 2006; Fore et al., 2007). Alternate excitation leads to the generation of two fluorescence lifetime decay curves corresponding to blue and red excitation in the TCSPC histogram window. Time-gating in Symphotime software based on these two decay curves enabled the separation of fluorescence based on the excitation laser source. According to Poisson distribution, most of the photon bursts were composed of fluorescence from single molecules in a sufficiently diluted solution. The FRET efficiency, E, was then determined from the equation:
| (10) |
where IDA denotes the intensity of acceptor fluorescence with donor excitation, ID is the donor fluorescence and γ is a factor correcting for differences in the detection efficiencies. PIE-FRET analysis is presented in Fig. 2. In our application, we have assumed γ to be 1 because the excitation and emission spectra of the FRET dye pair used are well separated and crosstalk and bleed-through can be neglected. In addition to that, two identical single-photon avalanche diodes (SPADs) were employed to detect the emission photons in our instrumentation. The quantum efficiency for different emission wavelengths (Alexa Fluor 488 and Alexa Fluor 647) is similar to that specified by the manufacturer. Similar assumptions with the same detector and FRET dye pair could be found elsewhere (Fore et al., 2007).
PIE-FRET evaluation of mononucleosomes
Antibodies were used at a final concentration of 100 pM to detect single FRET pairs diffusing through the confocal volume. FCS was utilized to monitor the concentration of antibodies from the amplitude [i.e. correlation, G(0)] at time zero, noting that G(0) is inversely proportional to the number of fluorescence molecules (N). A 300-microsecond time bin size was chosen as optimum in our experiments because it was the best compromise between time resolution and the ability to detect a sufficiently large number of photons maintaining a high threshold. A signal-to-background threshold was employed to exclude time bins in which no fluorescence signal was present and only signals that exceeded the threshold were included in the FRET efficiency calculation. PIE-FRET efficiency below 5% was considered to be non-significant (Ruttinger et al., 2006; Fore et al., 2007). First, the 465 nm laser was used, followed by the 636 nm laser, to excite the donor and acceptor molecules alternately. To differentiate and separate the acceptor emissions from the donor energy transfer or direct excitation, time-gating based on these two TCSPC decay curves was used. Subsequently, bins with counts that constituted the sum of mean value of donor emission and acceptor emission in the donor time gate, plus five times the standard deviation of the sum of donor and acceptor channels, were incorporated in the FRET calculations. The FRET efficiency was then calculated to create the respective histograms (Fig. 2).
FLIM-FRET
Fluorescence lifetime is defined as the time in which the intensity decays to 1/e of the initial intensity. FLIM can be used to study FRET interactions because energy transfer from the donor to acceptor (targeting variants and modifications) will result in a decrease in the donor's fluorescence lifetime. FLIM-FRET is an excellent technique to monitor co-presence distances (1–10 nm) between fluorophore pairs because the change in lifetime of the donor can be analyzed independently of acceptor emission (Bastiaens and Squire, 1999; Peter et al., 2005; Suhling et al., 2005; Grant et al., 2008; Vidi et al., 2008). In FLIM-FRET measurements, fluorescence lifetimes were obtained from TCSPC decay curves fitted by an exponential equation using the SymphoTime software (PicoQuant). By characterizing donor lifetimes in the absence and presence of an acceptor, FRET efficiency (E) and distance (R) can be calculated from Eqs 11 and 12:
| (11) |
| (12) |
where τDA and τD are the donor excited state lifetime in the presence and absence of acceptor. R0 is the Förster distance, or distance of 50% energy transfer between donor and acceptor. R0 is given by Eqn 13:
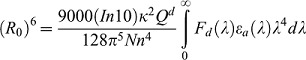 |
(13) |
where Qd is the quantum efficiency of the donor, N is Avogadro's number, n is the index of refraction of the medium between the fluorophores, Fd is the normalized emission spectrum of the donor, εa(λ) is the extinction coefficient of the acceptor at wavelength λ and κ2 is the orientation factor for the interaction between the donor and acceptor.
The Förster distance of FITC–Alexa-Fluor-647 is 4.5 nm (Liu et al., 2009) and that of Alexa-Fluor-488–Alexa-Fluor-647 is 5.6 nm (Huang et al., 2009). The FLIM-FRET analysis of yeast and mammalian cells is presented in Fig. 3, Fig. 4, Table 1 and supplementary material Fig. S3. None of the past efforts have used PIE-FRET or FLIM FRET to resolve histone modifications (Cairns, 2007; Kelbauskas et al., 2009; Llères et al., 2009; Poirier et al., 2009; Rowe and Narlikar, 2010).
Supplementary Material
Acknowledgments
We thank Amy Lossie and Paul Kaufman for helpful discussions, Joe Ogas for critical input to this manuscript and Gabriella Mavaro Velez and Rebecca Funk for technical assistance. We thank Scott Briggs, Paul Kaufman, Mark Parthun, Lorraine Pillus and Mitch Smith for strains and plasmids used in this study. J.C. and A.M. performed the experiments, A.L.K. and J.M.K.I. conceived of and supervised the study, J.C., A.M., A.L.K. and J.M.K.I. wrote the manuscript; all authors discussed the results and approved the manuscript. The authors declare no competing financial interests.
Footnotes
Funding
This work was supported by the National Science Foundation [grant number 0945771 to A.L.K. and J.M.K.I.] and the Purdue University Office of the Vice President of Research (to J.M.K.I., A.L.K. and A.L.). This research was also supported by the National Cancer Institute (NCI) [grant number CCSG CA23168] for data acquired in the Purdue University Center for Cancer Research (PCCR) DNA Sequencing Facility and by a NCI Cancer Center Supplement for Undergraduate Student Research Experiences to the Purdue University Center for Cancer Research. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.101592/-/DC1
References
- Adam M., Robert F., Larochelle M., Gaudreau L. (2001). H2A.Z is required for global chromatin integrity and for recruitment of RNA polymerase II under specific conditions. Mol. Cell. Biol. 21, 6270–6279 10.1128/MCB.21.18.6270-6279.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams A., Gottschling D. E., Kaiser C. A., Stearns T. (1997). Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Allard S., Utley R. T., Savard J., Clarke A., Grant P., Brandl C. J., Pillus L., Workman J. L., Côté J. (1999). NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 18, 5108–5119 10.1093/emboj/18.18.5108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altaf M., Auger A., Monnet–Saksouk J., Brodeur J., Piquet S., Cramet M., Bouchard N., Lacoste N., Utley R. T., Gaudreau L., et al. (2010). NuA4-dependent acetylation of nucleosomal histones H4 and H2A directly stimulates incorporation of H2A.Z by the SWR1 complex. J. Biol. Chem. 285, 15966–15977 10.1074/jbc.M110.117069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews A. J., Chen X., Zevin A., Stargell L. A., Luger K. (2010). The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol. Cell 37, 834–842 10.1016/j.molcel.2010.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anikovsky M., Dale L., Ferguson S., Petersen N. (2008). Resonance energy transfer in cells: a new look at fixation effect and receptor aggregation on cell membrane. Biophys. J. 95, 1349–1359 10.1529/biophysj.107.124313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacia K., Schwille P. (2003). A dynamic view of cellular processes by in vivo fluorescence auto- and cross-correlation spectroscopy. Methods 29, 74–85 10.1016/S1046-2023(02)00291-8 [DOI] [PubMed] [Google Scholar]
- Bacia K., Schwille P. (2007). Practical guidelines for dual-color fluorescence cross-correlation spectroscopy. Nat. Protoc. 2, 2842–2856 10.1038/nprot.2007.410 [DOI] [PubMed] [Google Scholar]
- Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- Bastiaens P. I., Squire A. (1999). Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends Cell Biol. 9, 48–52 10.1016/S0962-8924(98)01410-X [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S., Wang S., Reinecke D., Kiser W., Jr, Kruger R. A., DeGrado T. R. (2008). Synthesis and evaluation of near-infrared (NIR) dye-herceptin conjugates as photoacoustic computed tomography (PCT) probes for HER2 expression in breast cancer. Bioconjug. Chem. 19, 1186–1193 10.1021/bc700482u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billon P., Côté J. (2012). Precise deposition of histone H2A.Z in chromatin for genome expression and maintenance. Biochim. Biophys. Acta 1819, 290–302 [DOI] [PubMed] [Google Scholar]
- Blosser T. R., Yang J. G., Stone M. D., Narlikar G. J., Zhuang X. (2009). Dynamics of nucleosome remodelling by individual ACF complexes. Nature 462, 1022–1027 10.1038/nature08627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner D. G., Cajigas I., Fondufe–Mittendorf Y., Ahmed S., Lee P. C., Widom J., Brickner J. H. (2007). H2A.Z-mediated localization of genes at the nuclear periphery confers epigenetic memory of previous transcriptional state. PLoS Biol. 5, e81 10.1371/journal.pbio.0050081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns B. R. (2007). Chromatin remodeling: insights and intrigue from single-molecule studies. Nat. Struct. Mol. Biol. 14, 989–996 10.1038/nsmb1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza M. J., Li B., Florens L., Suganuma T., Swanson S. K., Lee K. K., Shia W. J., Anderson S., Yates J., Washburn M. P., et al. (2005). Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592 10.1016/j.cell.2005.10.023 [DOI] [PubMed] [Google Scholar]
- Chen J., Irudayaraj J. (2010). Fluorescence lifetime cross correlation spectroscopy resolves EGFR and antagonist interaction in live cells. Anal. Chem. 82, 6415–6421 10.1021/ac101236t [DOI] [PubMed] [Google Scholar]
- Chen J., Nag S., Vidi P. A., Irudayaraj J. (2011). Single molecule in vivo analysis of toll-like receptor 9 and CpG DNA interaction. PLoS ONE 6, e17991 10.1371/journal.pone.0017991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypionka A., Stein A., Hernandez J. M., Hippchen H., Jahn R., Walla P. J. (2009). Discrimination between docking and fusion of liposomes reconstituted with neuronal SNARE-proteins using FCS. Proc. Natl. Acad. Sci. USA 106, 18575–18580 10.1073/pnas.0906677106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion M. F., Kaplan T., Kim M., Buratowski S., Friedman N., Rando O. J. (2007). Dynamics of replication-independent histone turnover in budding yeast. Science 315, 1405–1408 10.1126/science.1134053 [DOI] [PubMed] [Google Scholar]
- Du H. N., Briggs S. D. (2010). A nucleosome surface formed by histone H4, H2A, and H3 residues is needed for proper histone H3 Lys36 methylation, histone acetylation, and repression of cryptic transcription. J. Biol. Chem. 285, 11704–11713 10.1074/jbc.M109.085043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingerman I. M., Li H. C., Briggs S. D. (2007). A charge-based interaction between histone H4 and Dot1 is required for H3K79 methylation and telomere silencing: identification of a new trans-histone pathway. Genes Dev. 21, 2018–2029 10.1101/gad.1560607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fore S., Yuen Y., Hesselink L., Huser T. (2007). Pulsed-interleaved excitation FRET measurements on single duplex DNA molecules inside C-shaped nanoapertures. Nano Lett. 7, 1749–1756 10.1021/nl070822v [DOI] [PubMed] [Google Scholar]
- Fuchs S. M., Laribee R. N., Strahl B. D. (2009). Protein modifications in transcription elongation. Biochim. Biophys. Acta 1789, 26–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansen A., Valeri A., Hauger F., Felekyan S., Kalinin S., Tóth K., Langowski J., Seidel C. A. (2009). Nucleosome disassembly intermediates characterized by single-molecule FRET. Proc. Natl. Acad. Sci. USA 106, 15308–15313 10.1073/pnas.0903005106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gévry N., Chan H. M., Laflamme L., Livingston D. M., Gaudreau L. (2007). p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev. 21, 1869–1881 10.1101/gad.1545707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant D. M., Zhang W., McGhee E. J., Bunney T. D., Talbot C. B., Kumar S., Munro I., Dunsby C., Neil M. A., Katan M., et al. (2008). Multiplexed FRET to image multiple signaling events in live cells. Biophys. J. 95, L69–L71 10.1529/biophysj.108.139204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette B., Bataille A. R., Gévry N., Adam M., Blanchette M., Robert F., Gaudreau L. (2005). Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol. 3, e384 10.1371/journal.pbio.0030384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman M. J., Winston F. (2007). Heme levels switch the function of Hap1 of Saccharomyces cerevisiae between transcriptional activator and transcriptional repressor. Mol. Cell. Biol. 27, 7414–7424 10.1128/MCB.00887-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F., Rajagopalan S., Settanni G., Marsh R. J., Armoogum D. A., Nicolaou N., Bain A. J., Lerner E., Haas E., Ying L., et al. (2009). Multiple conformations of full-length p53 detected with single-molecule fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 106, 20758–20763 10.1073/pnas.0909644106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang L. C., Wohland T. (2007). Recent advances in fluorescence cross-correlation spectroscopy. Cell Biochem. Biophys. 49, 1–13 10.1007/s12013-007-0042-5 [DOI] [PubMed] [Google Scholar]
- Hwang L. C., Gösch M., Lasser T., Wohland T. (2006). Simultaneous multicolor fluorescence cross-correlation spectroscopy to detect higher order molecular interactions using single wavelength laser excitation. Biophys. J. 91, 715–727 10.1529/biophysj.105.074120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan T., Liu C. L., Erkmann J. A., Holik J., Grunstein M., Kaufman P. D., Friedman N., Rando O. J. (2008). Cell cycle- and chaperone-mediated regulation of H3K56ac incorporation in yeast. PLoS Genet. 4, e1000270 10.1371/journal.pgen.1000270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelbauskas L., Woodbury N., Lohr D. (2009). DNA sequence-dependent variation in nucleosome structure, stability, and dynamics detected by a FRET-based analysis. Biochem. Cell Biol. 87, 323–335 10.1139/O08-126 [DOI] [PubMed] [Google Scholar]
- Kelly T. J., Qin S., Gottschling D. E., Parthun M. R. (2000). Type B histone acetyltransferase Hat1p participates in telomeric silencing. Mol. Cell. Biol. 20, 7051–7058 10.1128/MCB.20.19.7051-7058.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobor M. S., Venkatasubrahmanyam S., Meneghini M. D., Gin J. W., Jennings J. L., Link A. J., Madhani H. D., Rine J. (2004). A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin. PLoS Biol. 2, E131 10.1371/journal.pbio.0020131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerber R. T., Rhee H. S., Jiang C., Pugh B. F. (2009). Interaction of transcriptional regulators with specific nucleosomes across the Saccharomyces genome. Mol. Cell 35, 889–902 10.1016/j.molcel.2009.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans W. J., Buning R., Schmidt T., van Noort J. (2009). spFRET using alternating excitation and FCS reveals progressive DNA unwrapping in nucleosomes. Biophys. J. 97, 195–204 10.1016/j.bpj.2009.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle M., Gaudreau L. (2003). H2A.Z has a function reminiscent of an activator required for preferential binding to intergenic DNA. EMBO J. 22, 4512–4522 10.1093/emboj/cdg427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E., Verma A., Visintin A., Gong M., Sirois C. M., Klein D. C., Monks B. G., McKnight C. J., Lamphier M. S., Duprex W. P., et al. (2007). Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat. Immunol. 8, 772–779 10.1038/ni1479 [DOI] [PubMed] [Google Scholar]
- Lee N. K., Kapanidis A. N., Wang Y., Michalet X., Mukhopadhyay J., Ebright R. H., Weiss S. (2005). Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys. J. 88, 2939–2953 10.1529/biophysj.104.054114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Levitus M., Bustamante C., Widom J. (2005). Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 12, 46–53 10.1038/nsmb869 [DOI] [PubMed] [Google Scholar]
- Liu B., Fletcher S., Avadisian M., Gunning P. T., Gradinaru C. C. (2009). A photostable, pH-invariant fluorescein derivative for single-molecule microscopy. J. Fluoresc. 19, 915–920 10.1007/s10895-009-0492-9 [DOI] [PubMed] [Google Scholar]
- Liu C. L., Kaplan T., Kim M., Buratowski S., Schreiber S. L., Friedman N., Rando O. J. (2005). Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 3, e328 10.1371/journal.pbio.0030328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llères D., James J., Swift S., Norman D. G., Lamond A. I. (2009). Quantitative analysis of chromatin compaction in living cells using FLIM-FRET. J. Cell Biol. 187, 481–496 10.1083/jcb.200907029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk E., Ranjan A., Fitzgerald P. C., Mizuguchi G., Huang Y., Wei D., Wu C. (2010). Stepwise histone replacement by SWR1 requires dual activation with histone H2A.Z and canonical nucleosome. Cell 143, 725–736 10.1016/j.cell.2010.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magde D., Elson E. L., Webb W. W. (1974). Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 13, 29–61 10.1002/bip.1974.360130103 [DOI] [PubMed] [Google Scholar]
- Maiti S., Haupts U., Webb W. W. (1997). Fluorescence correlation spectroscopy: diagnostics for sparse molecules. Proc. Natl. Acad. Sci. USA 94, 11753–11757 10.1073/pnas.94.22.11753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matangkasombut O., Buratowski S. (2003). Different sensitivities of bromodomain factors 1 and 2 to histone H4 acetylation. Mol. Cell 11, 353–363 10.1016/S1097-2765(03)00033-9 [DOI] [PubMed] [Google Scholar]
- Métivier R., Penot G., Hübner M. R., Reid G., Brand H., Kos M., Gannon F. (2003). Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115, 751–763 10.1016/S0092-8674(03)00934-6 [DOI] [PubMed] [Google Scholar]
- Miller A., Yang B., Foster T., Kirchmaier A. L. (2008). Proliferating cell nuclear antigen and ASF1 modulate silent chromatin in Saccharomyces cerevisiae via lysine 56 on histone H3. Genetics 179, 793–809 10.1534/genetics.107.084525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito Y., Henikoff J. G., Henikoff S. (2005). Genome-scale profiling of histone H3.3 replacement patterns. Nat. Genet. 37, 1090–1097 10.1038/ng1637 [DOI] [PubMed] [Google Scholar]
- Mizuguchi G., Shen X., Landry J., Wu W. H., Sen S., Wu C. (2004). ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303, 343–348 10.1126/science.1090701 [DOI] [PubMed] [Google Scholar]
- Müller B. K., Zaychikov E., Bräuchle C., Lamb D. C. (2005). Pulsed interleaved excitation. Biophys. J. 89, 3508–3522 10.1529/biophysj.105.064766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J. D., Chen Y., Gratton E. (2003). Fluorescence correlation spectroscopy. Methods Enzymol. 361, 69–92 10.1016/S0076-6879(03)61006-2 [DOI] [PubMed] [Google Scholar]
- Ng H. H., Robert F., Young R. A., Struhl K. (2003). Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell 11, 709–719 10.1016/S1097-2765(03)00092-3 [DOI] [PubMed] [Google Scholar]
- Nislow C., Ray E., Pillus L. (1997). SET1, a yeast member of the trithorax family, functions in transcriptional silencing and diverse cellular processes. Mol. Biol. Cell 8, 2421–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M., Ameer–Beg S. M., Hughes M. K., Keppler M. D., Prag S., Marsh M., Vojnovic B., Ng T. (2005). Multiphoton-FLIM quantification of the EGFP-mRFP1 FRET pair for localization of membrane receptor-kinase interactions. Biophys. J. 88, 1224–1237 10.1529/biophysj.104.050153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier M. G., Oh E., Tims H. S., Widom J. (2009). Dynamics and function of compact nucleosome arrays. Nat. Struct. Mol. Biol. 16, 938–944 10.1038/nsmb.1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisner R. M., Hartley P. D., Meneghini M. D., Bao M. Z., Liu C. L., Schreiber S. L., Rando O. J., Madhani H. D. (2005). Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 123, 233–248 10.1016/j.cell.2005.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliff G. C., Erie D. A. (2001). A novel single-molecule study to determine protein--protein association constants. J. Am. Chem. Soc. 123, 5632–5635 10.1021/ja005750n [DOI] [PubMed] [Google Scholar]
- Rigler R., Elson E. (2001). Fluorescence Correlation Spectroscopy: Theory and Applications. Springer–Verlag, Berlin, Heidelberg [Google Scholar]
- Rowe C. E., Narlikar G. J. (2010). The ATP-dependent remodeler RSC transfers histone dimers and octamers through the rapid formation of an unstable encounter intermediate. Biochemistry 49, 9882–9890 10.1021/bi101491u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüttinger S., Macdonald R., Krämer B., Koberling F., Roos M., Hildt E. (2006). Accurate single-pair Förster resonant energy transfer through combination of pulsed interleaved excitation, time correlated single-photon counting, and fluorescence correlation spectroscopy. J. Biomed. Opt. 11, 024012 10.1117/1.2187425 [DOI] [PubMed] [Google Scholar]
- Sako Y., Yanagida T. (2003). Single-molecule visualization in cell biology. Nat. Rev. Mol. Cell Biol. Suppl. SS1–SS5 [PubMed] [Google Scholar]
- Santisteban M. S., Kalashnikova T., Smith M. M. (2000). Histone H2A.Z regulats transcription and is partially redundant with nucleosome remodeling complexes. Cell 103, 411–422 10.1016/S0092-8674(00)00133-1 [DOI] [PubMed] [Google Scholar]
- Santos–Rosa H., Schneider R., Bannister A. J., Sherriff J., Bernstein B. E., Emre N. C., Schreiber S. L., Mellor J., Kouzarides T. (2002). Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411 10.1038/nature01080 [DOI] [PubMed] [Google Scholar]
- Schwille P., Meyer–Almes F. J., Rigler R. (1997). Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys. J. 72, 1878–1886 10.1016/S0006-3495(97)78833-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille P., Haupts U., Maiti S., Webb W. W. (1999). Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys. J. 77, 2251–2265 10.1016/S0006-3495(99)77065-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R. S., Hieter P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhling K., French P. M., Phillips D. (2005). Time-resolved fluorescence microscopy. Photochem. Photobiol. Sci. 4, 13–22 10.1039/b412924p [DOI] [PubMed] [Google Scholar]
- Suto R. K., Clarkson M. J., Tremethick D. J., Luger K. (2000). Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat. Struct. Biol. 7, 1121–1124 10.1038/81971 [DOI] [PubMed] [Google Scholar]
- Varghese L. T., Sinha R. K., Irudayaraj J. (2008). Single molecule kinetic investigations of protein association and dissociation using fluorescence cross-correlation spectroscopy. Anal. Chim. Acta 625, 103–109 10.1016/j.aca.2008.07.021 [DOI] [PubMed] [Google Scholar]
- Vidi P. A., Chen J., Irudayaraj J. M., Watts V. J. (2008). Adenosine A(2A) receptors assemble into higher-order oligomers at the plasma membrane. FEBS Lett. 582, 3985–3990 10.1016/j.febslet.2008.09.062 [DOI] [PubMed] [Google Scholar]
- Wallrabe H., Periasamy A. (2005). Imaging protein molecules using FRET and FLIM microscopy. Curr. Opin. Biotechnol. 16, 19–27 10.1016/j.copbio.2004.12.002 [DOI] [PubMed] [Google Scholar]
- Winzeler E. A., Shoemaker D. D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J. D., Bussey H., et al. (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 10.1126/science.285.5429.901 [DOI] [PubMed] [Google Scholar]
- Wong M. M., Cox L. K., Chrivia J. C. (2007). The chromatin remodeling protein, SRCAP, is critical for deposition of the histone variant H2A.Z at promoters. J. Biol. Chem. 282, 26132–26139 10.1074/jbc.M703418200 [DOI] [PubMed] [Google Scholar]
- Wu W. H., Alami S., Luk E., Wu C. H., Sen S., Mizuguchi G., Wei D., Wu C. (2005). Swc2 is a widely conserved H2AZ-binding module essential for ATP-dependent histone exchange. Nat. Struct. Mol. Biol. 12, 1064–1071 10.1038/nsmb1023 [DOI] [PubMed] [Google Scholar]
- Yang B., Kirchmaier A. L. (2006). Bypassing the catalytic activity of SIR2 for SIR protein spreading in Saccharomyces cerevisiae. Mol. Biol. Cell 17, 5287–5297 10.1091/mbc.E06-08-0669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B., Britton J., Kirchmaier A. L. (2008). Insights into the impact of histone acetylation and methylation on Sir protein recruitment, spreading, and silencing in Saccharomyces cerevisiae. J. Mol. Biol. 381, 826–844 10.1016/j.jmb.2008.06.059 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.