

Abstract
Recent comparisons between anatomically modern humans and ancient genomes of other hominins have raised the tantalizing, and hotly debated, possibility of hybridization. Although several tests of hybridization have been devised, they all rely on the degree to which different modern populations share genetic polymorphisms with the ancient genomes of other hominins. However, spatial population structure is expected to generate genetic patterns similar to those that might be attributed to hybridization. To investigate this problem, we take Neanderthals as a case study, and build a spatially explicit model of the shared history of anatomically modern humans and this hominin. We show that the excess polymorphism shared between Eurasians and Neanderthals is compatible with scenarios in which no hybridization occurred, and is strongly linked to the strength of population structure in ancient populations. Thus, we recommend caution in inferring admixture from geographic patterns of shared polymorphisms, and argue that future attempts to investigate ancient hybridization between humans and other hominins should explicitly account for population structure.
Keywords: ABBA-BABA, D statistic, Neanderthal introgression, stepping stone model, out-of-Africa
The possibility of hybridization between anatomically modern humans and other hominins has been the focus of a heated debate spanning several decades (1–5). Four main alternative models have been put forward describing the origin of anatomically modern humans and their relationship with other hominins (6, 7). The Recent African Origin model argues that anatomically modern humans are the descendants of a recent expansion out of Africa that replaced older hominin populations without significant hybridization (8). The African Hybridization and Replacement model is similar to the Recent African Origin, but it allows for some hybridization with indigenous hominins during the expansion (9). Multiregional Evolution denies a recent African origin of modern humans, and argues that most hominins over the past 2 Myr are regional forms of a single species, with some gene flow occurring among contemporaneous populations in different areas throughout their existence (10, 11). Finally, the Assimilation model takes elements of the other models, because it accepts an important contribution of a recent African expansion, but it emphasizes assimilation rather than replacement of indigenous hominins during the expansion (7).
A pioneering study by Cann et al. (12) showed that the phylogeny of a global sample of mitochondrial sequences had the deepest divergences in Africa, and that most African and all non-African sequences clustered together in a single, more recent branch. This result was interpreted as supporting a replacement without hybridization scenario, and later work on the Y chromosome provided very similar results (13). Several studies on overall genomic diversity have also shown that African populations are more diverse than non-African ones, and that diversity declines progressively with increasing distance from Africa (14–16), as would be expected from the sequential bottlenecks experienced during an expansion without hybridization (15, 17). Though all these lines of evidence point strongly to a recent out-of-Africa (OOA) expansion providing the largest contribution to the gene pool of anatomically modern humans, they cannot rule out small levels of hybridization with other hominins, as the signal left behind in modern populations would be very difficult to detect (18). Some attempts to detect hybridization based on patterns of linkage disequilibrium have found a possible signal (19, 20), but these approaches rely on simplistic demographic models with assumptions that are not testable (21).
Our ability to directly assess the possibility of hybridization between anatomically modern humans and other hominins has been recently revolutionized by the development of techniques allowing the recovery of reliable genetic information from ancient specimens. In principle, if hybridization with another hominin only occurred within part of the range of anatomically modern humans, we would expect a recovered genome of the hybridizing hominin to share more polymorphisms with modern populations in that area compared with modern populations in other areas. Indeed, a first analysis of the Neanderthal genome revealed intriguing patterns in terms of shared polymorphisms with modern-day humans: Neanderthals are genetically more similar to present-day Eurasians than to present-day Africans (22). This asymmetry has been interpreted as evidence for hybridization between Neanderthals and anatomically modern humans during the latter’s exit out of Africa (22). Given that there is no difference between Europeans and Asians in their similarity to Neanderthals, it has been argued that such hybridization would have had to happen at the very beginning of the OOA expansion, before the split between these two groups (22). Similarly, the genome from a newly discovered hominin from the Denisova caves in Siberia has been shown to share unusual polymorphisms with Australians and Melanesians (23–25), again suggesting hybridization. However, our lack of knowledge of both the geographic range of Denisovans, and of their exact taxonomic affinity to modern humans, makes it difficult to identify the exact scenario.
A possible complication in interpreting spatially heterogeneous patterns of similarity between any ancient hominin and modern human populations is that, though such differences might arise through recent hybridization, they could also, in principle, be the consequence of population structure in early humans (22), i.e., a case of incomplete lineage sorting (26). Despite both archaeological (27, 28) and genetic evidence (28–32) for ancient population structure in Africa, none of the genetic models used so far to study the geographic patterns of similarity has incorporated structure in Africa. Currat and Excoffier (5) used a spatial framework to investigate admixture with Neanderthals, but concentrated on the role of gene surfing during the expansion wave in affecting the distribution of polymorphisms unique to Neanderthals across Eurasia, provided admixture did happen. Their model showed that gene surfing leads to overestimates of admixture in nonspatially structured models (5); however, they assumed the ancestors of African modern humans to be unstructured and ignored the common ancestry between Neanderthals and modern humans. To fill this gap, we build a spatially explicit framework of the shared history of anatomically modern humans and Neanderthals (Fig. 1 and Materials and Methods), which allows us to investigate the extent to which ancient population structure might drive spatial differences in genetic overlap between an ancient hominin and modern-day populations. We restrict our analysis to Neanderthals, because we have a reasonably clear idea of their ancient distribution and ancestral relationship with anatomically modern humans, but the conclusions of our study apply generally to any hominin contemporary with early anatomically modern humans in Eurasia.
Fig. 1.

Schematic representation of the spatially structured model representing the history of Neanderthals and anatomically modern humans. We start with a connected string of demes representing the common ancestor (in light blue), spanning Africa and Eurasia (A). Following the split (B), the northern part of the range becomes Neanderthals (purple), and the African part of the range eventually becomes modern humans (in orange). From this African range, modern humans later expand to colonize Eurasia (in red) (C). This figure only provides the logic of the model; for details, including the actual number of demes, see Materials and Methods.
Results
In our model, Africa and Eurasia are represented as a linear string of identical populations (demes), each of which can exchange migrants only with its adjacent neighbors. The simulation starts at 500 kya, with all demes occupied by the common ancestor of Neanderthals and anatomically modern humans (Fig. 1A). The connection between the African and Eurasian branches was truncated at 320 kya to represent the speciation of Neanderthals outside Africa (Fig. 1B). The populations in the African branch, which eventually turns into modern humans, were allowed to reexpand, eventually colonizing a set of Eurasian demes parallel to the ones occupied by Neanderthals (but without any admixture; Fig. 1C). As in Gronau et al. (33), the timings of the transition to modern demography, and of the subsequent expansion out of Africa, were free parameters in the model. We explored dates ranging from 100 to 200 kya for the transition to modern demography, and from 40 to 80 kya for the expansion. The population dynamics of modern humans (with parameters accounting for population size, growth rate, and colonization and migration rates to adjacent demes) was allowed to differ between this expansion and the ancestral phase. This part of the model is analogous to previous spatial models (14, 34–36) used to reconstruct the OOA expansion of anatomically modern humans. Our model differs from models previously used to investigate the similarity between Neanderthal and present-day humans in that it explicitly includes population structure within Africa and Eurasia, whereas previous models considered continents as unstructured compartments within which individuals mix freely.
We parameterized our model by comparing the patterns of genetic variability in modern populations predicted by the model with the observed quantities in the Human Genetic Diversity Project of the Centre d'Etude du Polymorphisme Humain (HGDP-CEPH) (37, 38), which comprises 51 populations covering the whole globe and typed for a large number of neutral microsatellites. This dataset has been used extensively to reconstruct the expansion out of Africa of anatomically modern humans (14, 15, 34, 39), and it provides a sensible yardstick to define the range of possible ancient demographics that would be compatible with the global distribution of genetic diversity in present-day humans. We used an approximate Bayesian computation (ABC) framework (40) to assess the likelihood of combinations of demographic parameters over a broad range of values, using time to most recent common ancestor (TMRCA) between major continents as summary statistics (see Materials and Methods for details and Fig. S1 for a schematic representation of the analysis). The demographic parameters describing the OOA expansion (Fig. S2) were compatible to those found in earlier studies using similar spatial models (14, 35): a rapid expansion with relatively small founding populations in each deme, and high migration rate between neighboring demes after the initial colonization. The ancient population demography was characterized by small carrying capacities and high migration rate. The time of transition to modern demography was compatible to recent estimates based on whole-genome sequences (33). Times of expansion in the explored range between 40 and 80 kya provided generally good fits, without any particular time being favored by the model.
To explore how different measures of the similarity between Neanderthal and different populations of present-day humans were affected by realistic levels of population structure, we used the best 0.05 percentile of demographic parameter combinations (referred to as demographic scenarios in the rest of the text), weighted by their likelihood as estimated by ABC. We considered two measures originally developed in the paper reporting the draft of the Neanderthal Genome (22). Because both approaches lead to similar conclusions, we present the results from the D statistics [supplementary online material (SOM) 15 in ref. 22] in the main text, whereas the results from the degree of matching between ancient and derived SNPs in candidate regions (SOM 17 in ref. 22) are reported in the supplementary text. The D statistics follow the following rationale. Consider four sequenced genomes: one each from two modern populations (P1 and P2); one from an ancient hominin, such as Neanderthal (N); and one from an outgroup, such as a chimpanzee (O). We confine our analysis to a subset of the genome that we can align perfectly across the four samples. We now focus on positions where P1 and P2 differ from each other, and for which N and O also differ. We call allele “A” the variant found in O, and allele “B” the other variant found in N. For the ordered set {P1,P2,N,O}, we can then have either pattern ABBA or BABA. Pattern ABBA represents a situation where the first modern population is more similar to the ancestral individual than the second population, whereas BABA represents the opposite case. The D statistic is the difference in the number of sites with either pattern (no. of ABBA − no. of BABA) divided by the total number of sites being considered (no. of ABBA + no. of BABA). Under a scenario of no admixture between the ancestral population and the two modern populations (and assuming unstructured populations), we expect D to be equal to zero. Conversely, if admixture occurred between the ancient hominin and one of the modern populations, D will deviate from zero.
When the Neanderthal genome was compared with genomes from modern populations, a significant deviation from zero was detected for D when comparing Africans to Europeans and Africans to Asians, but not when comparing Europeans to Asians (22). This pattern could imply admixture between Neanderthals and the ancestor of both Europeans and Asians before their split. Population structure can confound results from the D statistic (21), but the key question is whether realistic levels of population structure could generate the patterns observed in the Neanderthal genome. When we used our demographic scenarios (described above) to generate synthetic genomes for both Neanderthal and modern human populations, we found that the predicted levels of D for the different pairwise comparisons fitted very well the observed patterns. For Africans vs. Europeans and Africans vs. Asians, the distributions of predicted D values are tightly centered around the corresponding observed values; for European vs. Asians, we recover the expected negative values, even though the predicted distribution is centered around values slightly closer to zero than the observed one (Fig. 2). The same conclusion holds for any choice of African and OOA populations (Fig. S3), which is agreement with the finding that the Aborigines and Eurasian genomes show similar overlap with the Neanderthal genome (25). Similar conclusions were reached using the alternative measure presented in SOM 17 of Green et al. (22) (SI Text and Fig. S4).
Fig. 2.

Observed and simulated D statistics. D statistics for Europe vs. Africa (red), Asia vs. Africa (blue), and Europe vs. Asia (orange) in the original data (22) (triangles, with a 95% confidence interval) and as predicted by our spatial model (lines, representing the distributions of possible values).
Discussion
For both measures of shared polymorphisms between Neanderthals and modern humans, the patterns observed in the data could be generated by our spatial model that included ancient population structure of a strength that is compatible with the modern distribution of genetic diversity, even in the complete absence of hybridization. For the D statistics, the patterns produced by our spatial model were virtually identical to the ones observed in the data. For the candidate region approach, we also obtained a very good fit to the data; indeed, our spatial model provided a more realistic description of the data than the original nonspatial model that included hybridization in Green et al. (22), which failed to produce the correct level of matching for ancestral tag SNPs (see SI Text for details).
Currat and Excoffier (5) had already shown that models lacking spatial realism tend to overestimate the level of hybridization by ignoring gene surfing during the human expansion into Eurasia; including ancient structure in Africa further undermines the case for hybridization. Our conclusion was robust to the use of different measures of shared polymorphism, clearly indicating the limitation of looking for polymorphisms that are only shared between a single ancient genome and a restricted group of modern populations. In addition, our results show the extent to which ignoring spatial population structure causes models to underestimate the geographic differences expected in the absence of hybridization. Therefore, the issues raised in this paper should also apply to claims of admixture based on the distribution of linkage disequilibrium (19, 41).
Since the Neanderthal genome was published, another ancient genome has been recovered from a previously undescribed hominin from the Denisova caves (23). Comparisons with a wide range of modern populations revealed an excess of shared polymorphisms between this hominin and Australians and Melanesians, which has been interpreted as a sign of recent admixture (23, 24). Lack of knowledge on the past distribution and exact taxonomic affinity of Denisovans prevents us from quantifying the role ancient population structure played in creating similarities between modern humans and this hominin. However, our Neanderthal results suggest that ancient population structure could be an important determinant of spatial patterns of genetic overlap in this case also.
In conclusion, we urge caution in inferring recent admixture from geographic differences in genetic overlap between ancient hominins and modern-day populations. Though we do not claim that anatomically modern humans never admixed with other hominins, our results imply that current evidence for such admixture events is inconclusive at best. Future tests, to be convincing, will need to show that the genetic patterns used to invoke hybridization cannot be explained by population structure, for which there is both genetic (28–31) and archaeological evidence (27, 28). A key step toward such evidence will be the addition of ancient genomes of modern humans and other hominins from multiple locations, because they will allow reconstruction of ancient population structure and its possible effects on the genetic patterns in modern humans.
Materials and Methods
Demographic Model.
We represented the joint history of anatomically modern humans and Neanderthals using a spatially explicit stepping-stone model (Fig. 1). Before the modern human/Neanderthal split, the common ancestor of the two hominins occupied a linear string of demes (separated by 100 km) stretching from south sub-Saharan Africa into Eurasia (Fig. 1A; 130 demes in total). These demes contained K0 individuals and exchanged m0K0 migrants with each neighboring deme each generation. At 320 kya, future modern humans and Neanderthal split and evolved separately; this was represented by introducing a barrier, at deme 70 (in North Africa), to migration between Africa and the Eurasian branch of the stepping-stone model (Fig. 1B).
At time tmodern, while modern humans were still confined to Africa, the demographic parameters were allowed to change from ancestral to modern: carrying capacities changed from K0 to K, and migration rates from m0 to m. At texit, the barrier preventing the exit out of Africa was removed, and modern humans spread to colonize Eurasia. This process was represented by connecting the African branch of the stepping-stone model to a new branch with initially empty demes, stretching across Eurasia and the American continent (Fig. 1C, 260 demes). During the spread, full demes sent a fraction c of colonizers to adjacent empty demes. Population size grew linearly at the rate rK per generation, until it reached the new carrying capacity K. Each pair of adjacent occupied demes exchanged mNmin migrants per generation, where Nmin represents the smaller of the two population sizes. Thus, when the new branch was connected to Africa, humans spread across it, taking approximately (1 − c)/r generations to fill up a deme and send colonizers to the next empty deme. Once all demes had reached carrying capacity, population size stayed constant (at K) until present.
Parameterizing the Model.
To parameterize the model, we fitted the parameters (m, c, r, K, m0, K0, tmodern, and texit) to estimates of average TMRCA within and between populations (see Fig. S1 for a schematic representation). TMRCAs were calculated from the mean square difference of repeat counts in di- and trinucleotide microsatellite markers (41), genotyped in over 1,000 individuals from 51 populations in the HGDP-CEPH panel (37, 38). Dinucleotide markers where calibrated using the mutation rate of Dib et al. (42), μ = 1.52 × 10−3 single-step mutations per 27 y TMRCA of trinucleotide markers were scaled to match the average TMRCA of the dinucleotide markers.
The predicted TMRCA for a given parameter combination was calculated as follows: we first ran the demographic model described in the previous section, and then generated 100 gene genealogies for 10 individuals in each of the 51 populations corresponding to the HGDP-CEPH populations in our data (placed according to the deme corresponding to the distance from a location in sub-Saharan Africa, calculated using shortest distances on land, as in refs. 15 and 43). We traced gene genealogies backward in time, generation by generation, assuming diploid, random mating within each colonized deme, and with migration probabilities to neighboring demes given by the demographic model.
We fitted our model in the ABC framework, using the ABC-GLM algorithm implemented in the ABCtoolbox software (44). We generated six summary statistics from the average TMRCA between continents. We treated Europe and Central Asia as one continent (Eurasia), and East Asia as a separate continent. Because Oceania only has two populations (both in Papua New Guinea), we included these populations in the East Asian set. Our summary statistics are thus TAfrica,Eurasia, TAfrica,EastAsia, TAfrica,America, TEurasia,EastAsia, TEurasia,America, and TAmerica,America.
We started by randomly sampling 2.2 million parameter values from the following ranges: 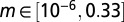 ,
, 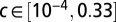 ,
, 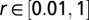 ,
, 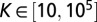 ,
, 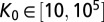 ,
, 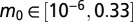 ,
, 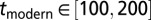 (kya), and
(kya), and 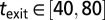 (kya). All parameters (with the exception of tmodern and texit) were log-transformed to ensure an adequate exploration of the large ranges of possible values. We further imposed (through rejection sampling) the constraint
(kya). All parameters (with the exception of tmodern and texit) were log-transformed to ensure an adequate exploration of the large ranges of possible values. We further imposed (through rejection sampling) the constraint 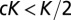 (cannot send out more colonists than individuals). Finally, we used ABC to estimate the likelihood of the 0.05% best-fitting scenarios (corresponding to 1,115 scenarios) and to generate parameter posterior distributions (Fig. S2).
(cannot send out more colonists than individuals). Finally, we used ABC to estimate the likelihood of the 0.05% best-fitting scenarios (corresponding to 1,115 scenarios) and to generate parameter posterior distributions (Fig. S2).
Analysis of the D Statistic for Allele Sharing Between Humans and Neanderthals.
We attempted to match the sample design and analysis of SOM 15 of Green et al. (22) as closely as possible within our framework. For each of the parameter combinations selected by the rejection step of ABC, we generated 10 million unlinked SNPs. When generating samples from our models, we took 10 individuals each from deme 10 (Africa), deme 90 (Europe), and deme 130 (East Asia). We placed the Neanderthal on the northern branch of the stepping-stone model (which became separated from the African at the split between humans and Neanderthals) at location that matches the distance between the origin in sub-Saharan Africa and the Vindija cave in Croatia [where the Neanderthal sample used in Green et al.’s (22) analysis was found]. For each SNP, the gene genealogy of the sample was generated as described in the previous section. For each of the scenarios selected by ABC, we then generated mutations on the gene genealogy according to the Jukes–Cantor model (assuming a split with chimpanzees 6 Mya) with mutation rate 2.5 × 10−8 per gamete per generation (45), and calculated the D statistic for each pair of populations. For each of the three comparisons (Europe vs. Africa, Asia vs. Africa, and Europe vs. Asia), we estimated the corresponding distributions of D values from 10,000 bootstrap samples. Finally, to see how the D statistic depends on the distance from sub-Saharan Africa, we repeated the analysis with 10 individuals from every 10th deme (every 1,000 km). Fig. S3 shows the average D between all pairs of populations, as a function of distance from the origin of the stepping-stone model.
Analyses of Candidate Regions for Gene Flow from Neanderthals.
To be able to directly compare our model to the real data and simulations in Green et al. (22), we closely followed the analyses described in SOM 17 of that paper. See SI Text for details.
Supplementary Material
Acknowledgments
We thank S. Lycett, A. Vail, and three anonymous referees for useful comments on the manuscript. This work was supported by the Leverhulme Trust and the Biotechnology and Biological Sciences Research Council.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1200567109/-/DCSupplemental.
References
- 1.Herrera KJ, Somarelli JA, Lowery RK, Herrera RJ. To what extent did Neanderthals and modern humans interact? Biol Rev Camb Philos Soc. 2009;84:245–257. doi: 10.1111/j.1469-185x.2008.00071.x. [DOI] [PubMed] [Google Scholar]
- 2.Hofreiter M. Drafting human ancestry: What does the Neanderthal genome tell us about hominid evolution? Commentary on Green et al. (2010) Hum Biol. 2011;83:1–11. doi: 10.3378/027.083.0101. [DOI] [PubMed] [Google Scholar]
- 3.Currat M, Excoffier L. Modern humans did not admix with Neanderthals during their range expansion into Europe. PLoS Biol. 2004;2:e421. doi: 10.1371/journal.pbio.0020421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trinkaus E, et al. An early modern human from the Peştera cu Oase, Romania. Proc Natl Acad Sci USA. 2003;100:11231–11236. doi: 10.1073/pnas.2035108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Currat M, Excoffier L. Strong reproductive isolation between humans and Neanderthals inferred from observed patterns of introgression. Proc Natl Acad Sci USA. 2011;108:15129–15134. doi: 10.1073/pnas.1107450108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stringer C. Modern human origins: Progress and prospects. Philos Trans R Soc B Biol Sci. 2002;357:563–579. doi: 10.1098/rstb.2001.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aiello LC. The fossil evidence for modern human origins in Africa—a revised view. Am Anthropol. 1993;95:73–96. [Google Scholar]
- 8.Stringer CB, Andrews P. Genetic and fossil evidence for the origin of modern humans. Science. 1988;239:1263–1268. doi: 10.1126/science.3125610. [DOI] [PubMed] [Google Scholar]
- 9.Brauer G. The evolution of modern humans: A comparison of the African and non-African evidence. In: Mellars P, Stringer CB, editors. The Origins and Dispersal of Modern Humans: Behavioural and Biological Perspectives. Edinburgh: Edinburgh Univ Press; 1989. [Google Scholar]
- 10.Hawks J, et al. An Australasian test of the recent African origin theory using the WLH-50 calvarium. J Hum Evol. 2000;39:1–22. doi: 10.1006/jhev.1999.0384. [DOI] [PubMed] [Google Scholar]
- 11.Wolpoff MH, Hawks J, Frayer DW, Hunley K. Modern human ancestry at the peripheries: A test of the replacement theory. Science. 2001;291:293–297. doi: 10.1126/science.291.5502.293. [DOI] [PubMed] [Google Scholar]
- 12.Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- 13.Underhill PA, Kivisild T. Use of y chromosome and mitochondrial DNA population structure in tracing human migrations. Annu Rev Genet. 2007;41:539–564. doi: 10.1146/annurev.genet.41.110306.130407. [DOI] [PubMed] [Google Scholar]
- 14.Liu H, Prugnolle F, Manica A, Balloux F. A geographically explicit genetic model of worldwide human-settlement history. Am J Hum Genet. 2006;79:230–237. doi: 10.1086/505436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prugnolle F, Manica A, Balloux F. Geography predicts neutral genetic diversity of human populations. Curr Biol. 2005;15:R159–R160. doi: 10.1016/j.cub.2005.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li JZ, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 17.Manica A, Amos W, Balloux F, Hanihara T. The effect of ancient population bottlenecks on human phenotypic variation. Nature. 2007;448:346–348. doi: 10.1038/nature05951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Relethford JH. Genetic evidence and the modern human origins debate. Heredity (Edinb) 2008;100:555–563. doi: 10.1038/hdy.2008.14. [DOI] [PubMed] [Google Scholar]
- 19.Plagnol V, Wall JD. Possible ancestral structure in human populations. PLoS Genet. 2006;2:e105. doi: 10.1371/journal.pgen.0020105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammer MF, Woerner AE, Mendez FL, Watkins JC, Wall JD. Genetic evidence for archaic admixture in Africa. Proc Natl Acad Sci USA. 2011;108:15123–15128. doi: 10.1073/pnas.1109300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28:2239–2252. doi: 10.1093/molbev/msr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reich D, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reich D, et al. Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. Am J Hum Genet. 2011;89:516–528. doi: 10.1016/j.ajhg.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rasmussen M, et al. An Aboriginal Australian genome reveals separate human dispersals into Asia. Science. 2011;334:94–98. doi: 10.1126/science.1211177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pamilo P, Nei M. Relationships between gene trees and species trees. Mol Biol Evol. 1988;5:568–583. doi: 10.1093/oxfordjournals.molbev.a040517. [DOI] [PubMed] [Google Scholar]
- 27.Gunz P, et al. Early modern human diversity suggests subdivided population structure and a complex out-of-Africa scenario. Proc Natl Acad Sci USA. 2009;106:6094–6098. doi: 10.1073/pnas.0808160106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheinfeldt LB, Soi S, Tishkoff SA. Colloquium paper: Working toward a synthesis of archaeological, linguistic, and genetic data for inferring African population history. Proc Natl Acad Sci USA. 2010;107(Suppl 2):8931–8938. doi: 10.1073/pnas.1002563107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell MC, Tishkoff SA. The evolution of human genetic and phenotypic variation in Africa. Curr Biol. 2010;20:R166–R173. doi: 10.1016/j.cub.2009.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henn BM, et al. Hunter-gatherer genomic diversity suggests a southern African origin for modern humans. Proc Natl Acad Sci USA. 2011;108:5154–5162. doi: 10.1073/pnas.1017511108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harding RM, McVean G. A structured ancestral population for the evolution of modern humans. Curr Opin Genet Dev. 2004;14:667–674. doi: 10.1016/j.gde.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 32.Blum MGB, Jakobsson M. Deep divergences of human gene trees and models of human origins. Mol Biol Evol. 2011;28:889–898. doi: 10.1093/molbev/msq265. [DOI] [PubMed] [Google Scholar]
- 33.Gronau I, Hubisz MJ, Gulko B, Danko CG, Siepel A. Bayesian inference of ancient human demography from individual genome sequences. Nat Genet. 2011;43:1031–1034. doi: 10.1038/ng.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramachandran S, et al. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci USA. 2005;102:15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deshpande O, Batzoglou S, Feldman MW, Cavalli-Sforza LL. A serial founder effect model for human settlement out of Africa. Proc Biol Sci. 2009;276:291–300. doi: 10.1098/rspb.2008.0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeGiorgio M, Jakobsson M, Rosenberg NA. Out of Africa: Modern human origins special feature. Explaining worldwide patterns of human genetic variation using a coalescent-based serial founder model of migration outward from Africa. Proc Natl Acad Sci USA. 2009;106:16057–16062. doi: 10.1073/pnas.0903341106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cann HM, et al. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- 38.Rosenberg NA, et al. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 39.Fagundes NJR, et al. Statistical evaluation of alternative models of human evolution. Proc Natl Acad Sci USA. 2007;104:17614–17619. doi: 10.1073/pnas.0708280104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beaumont MA, Zhang WY, Balding DJ. Approximate Bayesian computation in population genetics. Genetics. 2002;162:2025–2035. doi: 10.1093/genetics/162.4.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eriksson A, Manica A. 2011. Detecting and removing ascertainment bias in microsatellites from the HGDP-CEPH Panel. G3 (Bethesda) 1:479–488. [DOI] [PMC free article] [PubMed]
- 42.Dib C, et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 43.Manica A, Prugnolle F, Balloux F. Geography is a better determinant of human genetic differentiation than ethnicity. Hum Genet. 2005;118:366–371. doi: 10.1007/s00439-005-0039-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wegmann D, Leuenberger C, Neuenschwander S, Excoffier L. ABCtoolbox: A versatile toolkit for approximate Bayesian computations. BMC Bioinformatics. 2010;11:116. doi: 10.1186/1471-2105-11-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.