

Analysis of human genome variation may focus on one of two possible goals: understanding the genome region under study or solving historical and evolutionary questions specific to the population(s) analyzed. Understanding of variation of a given genome region has a genetic interest because it is a consequence of the dynamics of the genome and thus the evolutionary forces (mutation, selection in its varieties, drift, recombination, … ) may be understood. It is thus a way to understand the mechanisms that produce variation in the genome. On the other hand, when genetic variation is being analyzed, random individuals from specific populations offer the possibility to trace back their origin beyond the limitation of the genome region sampled. The goal then may be the evolutionary reconstitution of specific populations or, for a global sampling, the origin of our species. The underlying idea is very simple: if we are able to trace back the coalescence of genomic regions from an ample worldwide sample, we can infer the phylogeny of humans. The rationale can be sketched as follows:
(i) The evolutionary dynamics of the genome region is known in pattern and tempo.
(ii) From the extant variation the past can be inferred and the coalescence process reconstructed; in some cases just part of the genetic information is being used, and analyses do not fully exploit the information contained in the data.
(iii) Translate the genetic process into a process of individuals that reproduce (the population), and the time and place of the ancestral individual (carrying the ancestral genome) may be recognized.
(iv) If the geographic distribution of derivative genetic stages is known, the expansion process may be dissected as migratory waves and events.
(v) The structure of the genetic variation may reveal demographic characteristics such as population size and subdivision as well as ancient dynamics, such as expansions.
This simple pathway is not easy. Inferences from molecules to populations are not straightforward, and there have been recurrent worries on what was being analyzed, either the genes or genomic regions on one hand or the individuals, populations, or species on the other. There have been worries concerning the accuracy of our knowledge of genome dynamics, worries concerning the ability and power to detect specific processes and disentangle cases where more than one mechanism may have produced similar genetic patterns, and worries concerning the appropriateness of evolutionary models needed for the inference. And finally there have been worries from anthropologists who do not perceive the interface between the evolutionary biology of a species and that of tiny fragments of DNA, usually in noncoding regions, worries surrounding a fast-developing field, heir to classical population genetics, with brilliant novelties but also eager to get headlines.
In this context, two related papers in this issue (1, 2) analyze an ample set of sequences of the Y chromosome to address the issue of human origins. Their main objective is to propose a novelty: the common origin of present humans is not as old as previously believed, but should be much more recent, around 50,000 years ago (ya), the age of coalescence of the Y chromosomes surveyed (2) and in congruence with an onset of expansion of around 30,000 ya (1). Moreover, both studies corroborate the existence of a substantial (or exponential) population growth and an African origin for modern humans, in accord with most other genetic data.
The newly proposed age is much younger than dates consistent with nuclear and mitochondrial DNA (mtDNA) (see references in ref. 2), where the minimum coalescent age was obtained for mtDNA at around 150,000 years and much older for nuclear genes, except for the present case. Similar results have been obtained through other genetic approaches, such as short tandem repeat (STR, also called microsatellite) variation (3), where a figure of 156,000 years for the deepest split in human phylogeny was obtained, or Alu insertions (4), with a proposed age of 137,000 years for the time of separation of African versus non-African populations.
What seems more surprising is the discrepancy with the results of nine diallelic polymorphic sites on the Y chromosome (5), where the analysis with the same methods as those in ref. 2 gave a figure of around 150,000 ya for the coalescence of the variation. The new analysis (2) on the same chromosome gives only one-third of the time. It is thus an interesting new proposal not only in human evolution but also in human evolutionary genetics. It could mean that a population with modern characteristics had to exist in Africa 50,000 ya and spread later to Eurasia and the rest of the world. Besides the concordance of this proposal with archaeological evidence, there are more strictly genetic issues to be discussed, namely the limitations of the theory and of the genetic data, the interpretation of the variation, and possible specific properties unique to the Y chromosome.
New ideas coming from genetics in human evolution have had very different fates, and some cases are remembered for having proposed a paradigm shift strongly attacked by paleoanthropologists but later shown to be correct. This was the case for the time depth of the hominid branch, as a separate group from our close relatives, the chimpanzee and the bonobo. In most cases the process either to acceptance or rejection goes with a lively debate in which scholars from very different disciplines enter the fray with non-mutually intelligible languages. A proposal like the present one (1, 2), stemming from genetics, has implications in several fields, from which specific clarifications may be asked:
(i) Evolutionary genetics: Are conclusions about population fully supported or may there be a bias due to the genomic region under study? Does all of the genome tell the same story?
(ii) Mathematical genetics: May the models used be safely applied to the real world, apart from their theoretical elegance? What are the implications of violation of the theoretical assumptions?
(iii) Human paleontology: Is the evolutionary history of humans as interpreted from the hard evidence (the fossils) compatible with the new genetic proposals?
(iv) Archaeology and Paleodemography: Since cultural innovations are at the base of population expansions, are time and mode of cultural change correlated with their demographic consequences retrieved through genetic data?
To Understand the Genome or the Population?
Take two good papers on evolutionary genetics, one on Drosophila and the other on humans. As an average, there is a striking difference: while the Drosophila paper tends to focus on the understanding of the genomic region and the forces acting on it, keeping demography and history at a second level, the human paper tends to address specific population questions, considering that the mechanisms of genome change are sufficiently well known for the purpose. As a whole, the approaches are very different; perhaps too much so if we consider that the scope of the problem is the same. Human studies have ignored until recently most of the strong development of molecular evolutionary theory, and the incorporation of scholars and methodologies into the study of humans has improved the field and is giving interesting new views.
To what extent do the data support population-related results rather than being a result of genomic properties of the region under analysis? Let us focus on the Y chromosome. Recombination adds complexity to autosomal studies but it does not affect the nonrecombining region of the Y chromosome (NRY). The lack of recombination has a drawback: wherever and however selection acts, it will affect the whole NRY, as a result of positive selection (hitchhiking effect or selective sweep) or of negative selection (background selection). In both cases there is a reduction of nucleotide variation in linked sites, a result that could also be achieved by a reduction of effective population size. It affects the estimation of the coalescent age. In fact, until recently the Y chromosome was thought to be extremely poor in genetic variation, and an easy explanation could be postulated: positive selection in a single gene may have created a dramatic genetic sweep, setting to zero the amount of variation accumulated. The lack of recombination makes the situation easily affected by the action of positive selection.
Exploring population size may help us in recognizing the importance of selection when explaining genetic diversity levels. If selection has not been important in human history, what is at the base of the differentiation of populations is drift. To what extent can drift alone explain levels of differentiation found among Y chromosomes in humans? For STRs (6) it was shown that differentiation between populations (as measured by Fst, a measure of genetic distance) and gene diversity within populations were comparable to those of autosomal STRs when corrections for the smaller effective population size of the Y chromosome were taken into account. Nothing else was needed to explain the 4-fold difference in the value of Fst between Y and autosomes and the 2-fold difference in gene diversity. Thus there was no need to invoke selection, as drift may be a complete explanation for STR variation.
With the new sequence data (1) the situation is similar. Under the infinite-site model and with random mating, it has been shown (7) that the mean of π (nucleotide diversity or average number of nucleotide differences between two sequences randomly chosen from the population) is θ (a function of effective population size), and thus the comparison between θ values should take into consideration the values of N for autosomes, Y chromosomes, and X chromosomes according to
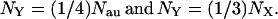 |
Results are given in Table 1.
Table 1.
Observed nucleotide diversity in coding and noncoding regions of autosomes and X and Y chromosomes and values predicted for the Y chromosome by correcting the values for autosomes and the X chromosome for the population size, assuming that male and female effective population sizes are equal
| Regions | Diversity × 10−4
|
||||
|---|---|---|---|---|---|
| Observed
|
Predicted for Y
|
||||
| Autosomes | X | Y | By autosomes | By X | |
| Coding | 2.91 | 1.96 | 0.52 | 0.73 | 0.65 |
| Noncoding | 6.18 | 3.92 | 1.11 | 1.54 | 1.31 |
The differences between the values 0.73 and 0.65 in relation to 0.52 on one hand and between 1.54 and 1.31 in relation to 1.11 on the other may be a footprint of selection, as it may also be the sole fact of having a lower nucleotide diversity for coding regions. Although present results do not allow measuring past selection in the Y chromosome, it seems likely to have been of small importance; explanations based on population structure and history seem to explain better the historical shaping of genetic diversity. Perhaps some background selection will be demonstrated, but it will have little impact on the bulk of the structure of the genetic variation.
A similar reasoning could be done with the distribution of pairwise differences between sequences. While it is mainly assumed that it is a footprint of a population expansion, other phenomena could be involved, such as a selective sweep or an uneven mutation rate along the nucleotides. Thus genomic factors and population factors mimic each other in shaping genetic variation, and only accurate and comparative analyses may solve ambiguities.
There is still another alternative that could account for a discordance of results between Y chromosome age and other genetic results, as the history of the Y chromosome may be different from the history of the autosomes and mtDNA. Conflicting patterns in the structure of genetic variation have been found between Y chromosome and mtDNA, that is, between paternal and maternal lineages (8, 9), which have been interpreted as a sex-specific migratory pattern, with higher mobility for females than males. Could a sex-specific characteristic account for discrepancies in coalescence? A possible explanation could be a lower effective size for Y chromosomes than for mtDNA and for autosomes. The latter is much less likely, as differences should be dramatic to show their effect, as autosomes are in both males and females. But even for mtDNA, differences do not seem to have been important. Known causes of reduced effective number of males, such as polygyny or higher male prereproductive mortality caused by hunting or warfare (ref. 9 and references therein), seem to have had little impact. Thus it does not seem plausible that factors tied to male specificity of the Y chromosome could be important in explaining the data.
Use of Complex Models.
Coalescence theory is providing extremely useful tools for the understanding of molecular variation. Nonetheless, some of the models are very complex, and most geneticists do not feel comfortable with black boxes in which to pour data and extract powerful results, such as precise estimates of ages of each mutation in the coalescent tree, including the age of the most recent common ancestor. This is the case of the interesting approach developed by R. C. Griffiths in the program Genetree (see references in ref. 2), for which the robustness under violation of theoretical assumptions should be better clarified. The problem may not be whether human populations have been of constant size or have followed a fixed exponential model, but heterogeneity in time and space of growth patterns.
The historical demography of humans is much more complex than what all population genetics models assume; this is obvious and it does not add any value to the discussion. What is more difficult to understand is the robustness of the methods to the real demographic history, with likely exponential increases and stasis (or even decreases) in a highly irregular pattern. Undoubtedly both a global and a continental approach to the past are extreme oversimplifications. The well-demonstrated heterogeneity in mtDNA composition of African populations is a clear example of the complex historical patterns. Genomic issues should also be clarified, as should the importance of uneven mutation rates along the nucleotides or, for autosomal loci, the impact of recombination or gene conversion along the gene genealogy.
Another question is related to population subdivision. No doubt that human populations, which for most of the past millennia have had extremely low densities and have occupied vast territories, have experienced extraordinary barriers to gene flow, and subdivision at many levels has been an important genetic force. The distortion that this may produce in coalescence estimations should be analyzed from a theoretical perspective considering what is known or inferred about real past populations.
The discrepancy with the analysis by M. F. Hammer et al. (5) is also puzzling. There is a methodological difference that may be important: whereas in sequence analysis (2) variable positions appear while sequencing, in a survey on polymorphisms defined beforehand there may be a lack of alleles at low frequencies (like singletons), which may affect Genetree behavior. In fact, its use with previously defined polymorphisms may distort the estimates. This should also be a point of clarification.
I would like to call upon theoretical population geneticists to test the robustness of the Genetree estimates in the real genetic and demographic world to allow many users (more comfortable with molecular than with theoretical challenges) to feel more secure in their inferences and interpretations.
Do Bones and Genes Agree?
The answer should be positive, as there is a single human history. But controversy has often arisen between the fossil hard evidence and the genetic evidence. There are still competing explanations for the origin of modern humans, even if the replacement hypothesis with an out of Africa migration is accepted by a wide majority of the scientific community. The paleontological data fit the model, although some cases (especially in Asia) remain debatable. What seems clear is that we should look for the earliest modern morphologies and see whether their place and time fit the new hypothesis of a recent common origin.
The earliest well-characterized anatomically modern humans have been found both in Africa and in the Middle East, and they are significantly older than findings from other parts of the Old World. Ages around 100,000 ya seem well documented for modern morphologies in Africa (Omo Kibish in Ethiopia; Klasies River Mouth in South Africa; Border Cave, South Africa) and in the Middle East (Skhul and Qafzeh). For the rest of Eurasia dates are more recent: less than 65,000 ya in China (Liujiang) and much more recent in Southeast Asia, where Homo erectus seems to have persisted longer. In Europe, the first appearance is dated around 40,000 ya, and modern morphology seems to expand from east to west, with earlier morphologies (i.e., Neanderthal) persisting in southern Iberia until 27,000 ya (ref. 10 and references therein).
In summary, morphological evidence supports the hypothesis of a single and African origin of modern humans, but at a date much older than the proposed 50,000 ya. But it is clear that, in a large part, the spread of modern humans, first to Asia and Australia, later to Europe and America, took place much later than the initial appearance of the morphology. Present morphological data do not exclude a later migration out of Africa (except for the very special case of the Middle East), but they do not support a very recent origin. Perhaps multiple dispersions from Africa took place at different times and from different places (11); complex models, as usual, could better accommodate the existing data.
Biology and Culture in Human Evolution.
The evolutionary framework stemming from biology (be it from genetic or morphological evidence) should fit into the archaeological evidence. Modern human behavior seems to be associated with the shift from the Middle Paleolithic to the Upper Paleolithic (equivalent for most of Africa to the shift from Middle Stone Age to Later Stone Age), which does not seem to have taken place before 40,000 ya. Some archeologists believe that this is a dramatic behavioral shift and may be called a major revolution in human history. This cultural change could account for a demographic expansion, likely related to the spread of modern humans in some regions.
This transition took place in the Middle East at about the same time as in the earliest sites in Europe, around 40,000 ya, but there is controversy about whether they have a common origin and where it could be. The simple model of an origin in the Middle East and a spread to Europe has not been clearly demonstrated, even if it could be true, as a general trend, for the expansion within Europe, mostly in its southern part. Genetic evidence supporting this view was provided by mtDNA analysis (12, 13). The East Asia evidence is too scarce to be considered here.
The evidence for Africa is puzzling. Although the classical view established both a chronological and a technological parallelism between cultural phases, Middle to Later Stone Ages and Middle to Upper Paleolithic, recent evidence points to more elaborate technologies well within the Middle Stone Age with chronologies compatible with the oldest anatomically modern humans. Nonetheless, the correlation between morphology and culture is not found in the earliest modern Africans. We are still far from having a clear picture of the making of modern behavior or even which are the landmarks that would allow recognizing the initial steps.
Cultural evidence tends to show that signs of modernity appeared first in Africa, and later developed in Eurasia (10), with a tempo and mode of dispersion that are clear only for Europe, beginning 40,000 ya and likely to be associated with morphologically modern humans. A recent date for the origin of humans as proposed (2) does not contradict the scarce existing evidence unless the old tool industries in Africa are taken as evidence of modern behavior. The timing of the population expansion (1) at 30,000 ya may fit the archaeological evidence, mainly for Europe.
It has taken years to assemble some pieces of the puzzle of human origins, within the substitution model (with some variants) and an African origin. A consensus between genetic, morphological, and maybe even archaeological sources placed the origin of modern humans at before 100,000 ya. A much more recent date, as proposed (2) does not fit with the present interpretation of the fossil evidence, but it could fit the cultural evidence. The detection of population growth is well supported by the cultural evidence, and the timing of population expansion at 30,000 ya could be related to the Upper Paleolithic transition and, in Europe, to the expansion of modern humans.
Footnotes
References
- 1.Shen P, Wang F, Underhill P A, Franco C, Yang W-H, Roxas A, Sung R, Lin A A, Hyman R W, Vollrath D, et al. Proc Natl Acad Sci USA. 2000;97:7354–7359. doi: 10.1073/pnas.97.13.7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomson R, Pritchard J K, Shen P, Oefner P J, Feldman M W. Proc Natl Acad Sci USA. 2000;97:7360–7365. doi: 10.1073/pnas.97.13.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstein D B, Ruiz Linares A, Cavalli-Sforza L L, Feldman M W. Proc Natl Acad Sci USA. 1995;92:6723–6727. doi: 10.1073/pnas.92.15.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoneking M, Fontius J J, Clifford S L, Soodyall H, Arcot S S, Saha N, Jenkins T, Tahir M A, Deininger P L, Batzer M A. Genome Res. 1997;7:1061–1071. doi: 10.1101/gr.7.11.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammer M F, Karafet T, Rasanayagam A, Wood E T, Altheide T K, Jenkins T, Griffiths R C, Templeton A R, Zegura S L. Mol Biol Evol. 1998;15:427–441. doi: 10.1093/oxfordjournals.molbev.a025939. [DOI] [PubMed] [Google Scholar]
- 6.Pérez-Lezaun A, Calafell F, Seielstad M, Mateu E, Comas D, Bosch E, Bertranpetit J. J Mol Evol. 1997;45:265–270. doi: 10.1007/pl00006229. [DOI] [PubMed] [Google Scholar]
- 7.Watterson G A. Theor Pop Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 8.Seielstad M T, Minch E, Cavalli-Sforza L L. Nat Genet. 1998;20:278–280. doi: 10.1038/3088. [DOI] [PubMed] [Google Scholar]
- 9.Pérez-Lezaun A, Calafell F, Comas D, Mateu E, Bosch E, Martínez-Arias R, Clarimon J, Fiori G, Luiselli D, Facchini F, Pettener D, Bertranpetit J. Am J Hum Genet. 1999;65:208–219. doi: 10.1086/302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewin R. Human Evolution. An Illustrated Introduction. Oxford: Blackwell; 1999. [Google Scholar]
- 11.Lahr M M, Foley R. Evol Anthropol. 1994;3:48–60. doi: 10.1002/evan.21405. [DOI] [PubMed] [Google Scholar]
- 12.Comas D, Calafell F, Mateu E, Pérez-Lezaun A, Bosch E, Bertranpetit J. Hum Genet. 1997;99:443–449. doi: 10.1007/s004390050386. [DOI] [PubMed] [Google Scholar]
- 13.Simoni L, Calafell F, Pettener D, Bertranpetit J, Barbujani G. Am J Hum Genet. 2000;66:262–278. doi: 10.1086/302706. [DOI] [PMC free article] [PubMed] [Google Scholar]