

Background: Benzo[a]pyrene-7,8-dione is a genotoxic metabolite produced by aldo-keto reductases.
Results: SULT1A1 is identified as the major SULT in lung cells involved in the detoxication of the corresponding catechol.
Conclusion: SULT1A1 may protect lung cells from the genotoxic benzo[a]pyrene-7,8-dione.
Significance: Polymorphisms in SULT1A1 may affect lung cancer susceptibility.
Keywords: Carcinogenesis, Lung Cancer, Reactive Oxygen Species (ROS), Reductase, Sulfotransferase, Aldo-Keto Reductase, Mass Spectrometry, Quinones
Abstract
Polycyclic aromatic hydrocarbons (PAH) are environmental and tobacco carcinogens. Human aldo-keto reductases catalyze the metabolic activation of proximate carcinogenic PAH trans-dihydrodiols to yield electrophilic and redox-active o-quinones. Benzo[a]pyrene-7,8-dione a representative PAH o-quinone is reduced back to the corresponding catechol to generate a futile redox-cycle. We investigated whether sulfonation of PAH catechols by human sulfotransferases (SULT) could intercept the catechol in human lung cells. RT-PCR identified SULT1A1, -1A3, and -1E1 as the isozymes expressed in four human lung cell lines. The corresponding recombinant SULTs were examined for their substrate specificity. Benzo[a]pyrene-7,8-dione was reduced to benzo[a]pyrene-7,8-catechol by dithiothreitol under anaerobic conditions and then further sulfonated by the SULTs in the presence of 3′-[35S]phosphoadenosine 5′-phosphosulfate as the sulfonate group donor. The human SULTs catalyzed the sulfonation of benzo[a]pyrene-7,8-catechol and generated two isomeric benzo[a]pyrene-7,8-catechol O-monosulfate products that were identified by reversed phase HPLC and by LC-MS/MS. The various SULT isoforms produced the two isomers in different proportions. Two-dimensional 1H and 13C NMR assigned the two regioisomers of benzo[a]pyrene-7,8-catechol monosulfate as 8-hydroxy-benzo[a]pyrene-7-O-sulfate (M1) and 7-hydroxy-benzo[a]pyrene-8-O-sulfate (M2), respectively. The kinetic profiles of three SULTs were different. SULT1A1 gave the highest catalytic efficiency (kcat/Km) and yielded a single isomeric product corresponding to M1. By contrast, SULT1E1 showed distinct substrate inhibition and formed both M1 and M2. Based on expression levels, catalytic efficiency, and the fact that the lung cells only produce M1, it is concluded that the major isoform that can intercept benzo[a]pyrene-7,8-catechol is SULT1A1.
Introduction
Polycyclic aromatic hydrocarbons (PAH)2 are ubiquitous environmental pollutants that originate from fossil fuel combustion and tobacco smoke and are suspect lung carcinogens (1, 2). PAH are not biologically reactive and require metabolic activation to elicit their deleterious effects; thus they are procarcinogens (3). Benzo[a]pyrene (B[a]P) is a representative PAH and is widely used to study the metabolic activation of PAH (3, 4).
There are three pathways for the activation of B[a]P. In the first pathway, called the radical cation pathway, B[a]P is activated by one-electron oxidation catalyzed by either P450 monooxygenases or peroxidases to yield a radical cation at C6 of B[a]P, which leads to the formation of depurinating adducts and results in G to T transversions (5, 6). The second pathway is known as the diol-epoxide pathway catalyzed by cytochrome P450s. In this pathway B[a]P is initially converted to (−)trans-7,8-dihydroxy-7,8-dihydro-benzo[a]pyrene (B[a]P-7,8-trans-dihydrodiol), a proximate carcinogen intermediate by the sequential actions of P4501A1/1B1 and epoxide hydrolase. B[a]P-7,8-trans-dihydrodiol is then further monooxygenated by P450 1A1/1B1 to yield the ultimate carcinogen (+)-anti-7,8-dihydroxy-9α,10α-epoxy-7,8,9,10-tetra-hydrobenzo[a]pyrene (anti-B[a]PDE) (4, 7, 8). Anti-B[a]PDE is electrophilic and reacts with dGuo to form (+)-trans-anti-BPDE-N2-dGuo adducts (9, 10), which are believed to be the cause of mutagenicity in bacterial and mammalian cells as well as tumorigenicity in pulmonary adenomas of mice (11, 12). The third pathway is termed the o-quinone pathway catalyzed by aldo-keto reductases (AKR). In this pathway, B[a]P-7,8-trans-dihydrodiol is oxidized by AKR1A1 and AKR1C1–1C4 to yield a ketol that spontaneously rearranges to form B[a]P-7,8-catechol, which is not stable and undergoes autooxidation to yield benzo[a]pyrene-7,8-dione (B[a]P-7,8-dione) and the generation of reactive oxygen species (ROS) (13–15) (Fig. 1).
FIGURE 1.

Metabolic activation of B[a]P by AKRs and detoxication of B[a]P-7,8-dione by SULTs.
B[a]P-7,8-dione is electrophilic and highly reactive with endogenous nucleophiles. It readily forms thioether conjugates with l-cysteine, N-acetyl-l-cysteine, and GSH (16, 17). B[a]P-7,8-dione can also react with DNA to form both stable and depurinating adducts (18–20). B[a]P-7,8-dione is also redox-active, and in the presence of NADPH it is reduced back to the catechol for subsequent rounds of autooxidation thereby amplifying ROS in futile redox-cycles (21). Recently, we found that this process was exacerbated enzymatically by NQO1 and by AKRs themselves (21). Using a yeast-based p53 mutagenesis assay, we find that B[a]P-7,8-dione was more mutagenic than diol-epoxides provided that the o-quinone was allowed to redox cycle and that the mutagenic efficiency of B[a]P-7,8-dione showed a linear correlation with the presence of 8-oxo-dGuo in the p53 DNA (22–24). In human lung adenocarcinoma (A549) cells with high constitutive expression of AKRs, the metabolic activation of B[a]P-7,8-trans-dihydrodiol to B[a]P-7,8-dione by AKRs led to the formation ROS and 8-oxo-dGuo adducts in cellular DNA, which were exacerbated by the presence of a catechol-O-methyl transferase (COMT) inhibitor (25). The contribution of COMT to detoxication of PAH o-quinone was confirmed by demonstrating that human recombinant COMT detoxified structurally diverse PAH o-quinones, including B[a]P-7,8-dione, via O-methylation of PAH catechols (26).
It is still unknown whether sulfonation of B[a]P-7,8-catechol by human sulfotransferases (SULT) is a feasible detoxication route for B[a]P-7,8-dione. SULTs are a group of cytosolic enzymes responsible for transfer of a sulfonate group from a 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to either a hydroxyl moiety or an amine group (27, 28). On the basis of sequence identity, human SULTs are divided into two major families, SULT1 and SULT2, which are also termed the phenol sulfotransferase and the hydroxysteroid sulfotransferase family, respectively. The SULT1 family is further classified into four subfamilies, phenol sulfotransferases (SULT1A), thyroid hormone sulfotransferases (SULT1B), hydroxyarylamine sulfotransferases (SULT1C), and estrogen sulfotransferases (SULT1E) (29). The SULT2 family consists of SULT2A and -2B subfamilies and catalyze sulfonation of the hydroxyl group of steroids including both 3α- and 3β-hydroxysteroids (30). Estrogen o-quinones are structurally related to the PAH o-quinones, and multiple SULTs catalyze the sulfonation of estrogen catechols such as 2-hydroxyestradiol, 4-hydroxyestradiol, 2-hydroxyestrone, and 4-hydroxyestrone (31–33). SULT enzymes are also widely expressed and found in human lung (34). Therefore, it is imperative to investigate whether sulfonation of B[a]P-7,8-catechol by SULTs is a detoxication pathway for B[a]P-7,8-dione that will limit its ability to redox cycle.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
p-Nitrophenol and PAPS, were purchased from Sigma. 3′-[35S]Phosphoadenosine 5′-phosphosulfate (1–3 Ci/mmol, 0.5 mCi/ml) was obtained from PerkinElmer Life Sciences. B[a]P-7,8-dione was synthesized according to published procedures (35). All solvents were HPLC grade, and all other chemicals used were of the highest grade available.
Cell Lines and Culture Conditions
A549 (human lung adenocarcinoma cells) were obtained from American Type Culture Collection (ATCC number CCL-185) and maintained in F-12K nutrient mixture (Kaighn's modification) with 10% heat-inactivated FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. H358 (human bronchoalveolar cells) were obtained from American Type Culture Collection (ATCC number CRL-5807) and maintained in RPMI 1640 nutrient mixture with 10% heat-inactivated FBS, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. HBEC-KT (immortalized human bronchial epithelial cells) originated from a patient without lung cancer were a gift from Dr. John Minna at University of Texas Southwestern Medical Center and maintained in keratinocyte-serum free medium with 0.1–0.2 ng/ml recombinant EGF, 20–30 μg/ml bovine pituitary extract, and 2 mm l-glutamine. BEAS-2B (normal human bronchial epithelium cells) cells from American Type Culture Collection (ATCC number CRL-9609) were cultured in BEGM Bronchial Epithelium Medium (Cambrex CC-3170). Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2 and were passaged every 3 days at a 1:6 dilution. Cultured cells with a passage number of 10–20 were used in the experiments to reduce variability during cell culture.
RT-PCR Analysis of SULT mRNA Expression in Human Lung Cells
Total RNA from each cell line was extracted using RNeasy Kits (Qiagen, Valencia, CA). cDNA was synthesized with GeneAmp® RNA PCR Core kit according to the manufacturer's protocol (Applied Biosystems, Carlsbad, CA). An aliquot of 1 μl of the reverse transcriptase reaction mixture was used for PCR. In 25 μl of PCR system there was 1× PCR buffer (10 mm Tris-HCl buffer (pH 8.3), 50 mm KCl), 2.5 mm MgCl2, 250 μm dNTPs, 0.2 μm primers and 0.5 units of TaqDNA polymerase (Applied Biosystems). The sequences of forward and reverse primer pairs for SULT1A1, -1A3, -1B1, -1C2, -1E1, and -2A1, β-actin were taken from previous studies (36, 37). PCR amplification was carried out with PerkinElmer Life Sciences GeneAmp PCR System 2400 (Waltham, MA) using the following protocol. After an initial denaturation step at 95 °C for 45 s, amplification was conducted by denaturation at 95 °C for 15 s, annealing at 68 °C (for SULT1E1) or 66 °C (for other SULT isoforms) for 30 s, and extension at 72 °C for 45 s for 30 cycles. The final extension reaction was performed at 72 °C for 7 min. Plasmids corresponding to pGEX-2TK-SULT1A1*3, -1A3, -1B1, -1C2, -1E1, and -2A1 were kindly provided by Prof. Ming-Cheh Liu (Department of Pharmacology, College of Pharmacy, University of Toledo, OH) and were used as positive controls, whereas H2O was used as negative control. The control samples were amplified using the same conditions as described above. An aliquot of 10 μl of PCR products was analyzed by electrophoresis in a 2% agarose gel containing ethidium bromide and visualized under UV light.
Preparation of pGEX-2TK-SULT1A1*1 Wild Type Expression Vectors
The pGEX-2TK-SULT1A1*3 (Met223Val) was used as a template to prepare a wild type construct by conducting site-directed mutagenesis using the QuikChange method following the manufacturer's protocol. The following forward and reverse primers were used (where the underlined nucleotides indicate the mutation introduced): 5′-dGAGACCGTGGACTTCATGGTTCAGCACACGTCG-3′ and 5′-dCGACGTGTGCTGAACCATGAAGTCCACGGTCTC-3′. The introduction of the wild type sequence of SULT1A1*1 into the construct was verified by dideoxysequencing.
Standard Radiometric Assay for SULT Activity
SULT enzyme assays were conducted as described by Foldes and Meek (38). Briefly, the reaction system contained 10 mm KPO4 buffer of pH 7.4, 1.0 mm dithiothreitol, 5.0 mm MgCl2, 20 μm [35S]PAPS (100 cpm/pmol), 10 μm (for SULT1A1) or 500 μm (for SULT1A3 and -1E1) of p-nitrophenol, and human recombinant SULTs in a final volume of 100 μl. Reactions were initiated by addition of [35S]PAPS. After incubation at 37 °C for 15 min, the reactions were terminated by the sequential addition of 50 μl each of 0.1 m barium acetate, 0.1 m barium hydroxide, and 0.1 m zinc sulfate. The precipitate was pelleted by centrifugation at 16,000 × g for 10 min. An aliquot of 50 μl supernatant was pipetted and mixed with scintillation fluid. The radioactivity was measured on a Tri-Carb 2100TR scintillation counter (machine efficiency for tritium is 65%). Control reactions were performed in the absence of p-nitrophenol. The trace amount of [35S]PAPS unprecipitated by the barium treatment in the controls served as a background value that was subtracted from the values obtained from reactions containing p-nitrophenol. The volume-corrected cpm were converted to pmol of p-nitrophenol sulfate formed using the specific radioactivity of [35S]PAPS. It was also confirmed that a single product of p-nitrophenol-sulfate was formed by comparing the retention time of authentic p-nitrophenyl sulfate (Sigma) by reversed phase HPLC.
Expression and Purification of SULT
The SULTs expressed in human lung cells were purified as recombinant enzymes following the reported method with slight modification (39). The pGEX-2TK-SULT1A1, -1A3, and -1E1 expression vectors were transformed respectively into competent Escherichia coli C41(DE3) cells. The cells were grown in 750-ml cultures of Luria-Bertani medium at 37 °C (containing 100 μg/ml ampicillin). Upon reaching A600 0.6, 1 mm isopropyl-1-thio-β-d-galactopyranoside was added to induce enzyme expression, and cells were cultured overnight. The culture was centrifuged for 10 min at 10,000 × g at 4 °C. Harvested cells were washed with saline, and the pellets were resuspended in buffer A (20 mm Tris-HCl (pH 7.5) containing 10 mm NaCl and 1 mm EDTA). Resuspensions were lysed by sonication and centrifuged twice for 10 min at 10,000 × g at 4 °C. The lysate was loaded onto a glutathione-Sepharose column (GE Healthcare) equilibrated with buffer A. GST-SULT fusion proteins were eluted with buffer consisting 10 mm GSH, 20 mm Tris-HCl (pH 7.5), 10 mm NaCl, 1 mm EDTA. The collected fractions were digested with 40 units of thrombin for 1 h to cleave the GST tag from the fusion protein and release native SULTs, and the thrombin was inactivated by adding 20 μg of aprotinin. The resulting cleavage products were dialyzed overnight in buffer. The cleaved GST tags were removed by loading the dialysate onto a glutathione-Sepharose column equilibrated with buffer A. The eluent containing native SULTs was concentrated into 2 ml using a Centricon (Millipore, Billerica, MA) and further purified with HiPrep 16/60 gel filtration column (GE Healthcare). The active fractions containing SULTs were identified using the standard SULT assay conditions, measurement of A280 nm, and visualization of the protein content of each fraction by SDS-PAGE. The homogeneous human recombinant SULT1A1, -1A3, and -1E1 with an Mr 35 kDa were obtained by this procedure, as judged by SDS-polyacrylamide gel electrophoresis. The specific activities of SULT1A1, -1A3, and -1E1 were 25, 33, and 27 nmol of p-nitrophenol sulfate formed/min/mg, respectively, and compare favorably with published values for these enzymes (39–41). The purified enzymes were stored in 20 mm Tris-HCl (pH 7.5) containing 1 mm EDTA, 10 mm DTT, 20% glycerol at −80 °C for future use.
Kinetic Studies on the Formation of B[a]P-7,8-Catechol Sulfates
Experiments were conducted anaerobically in a glove box purged with argon. All the solvent and aqueous solutions were degassed by freeze-pump-thaw cycling five times and stored in sealed containers filled with argon. The reactions were performed in 1.5-ml amber glass vials with polytetrafluoroethylene/silicone septa closures. The reaction system contained 10 mm KPO4 buffer of pH 7.4, 1.0 mm dithiothreitol, 5.0 mm MgCl2, 20 μm [35S]PAPS (100 cpm/pmol), 0–10 μm B[a]P-7,8-dione in a final volume of 0.2 ml. The reactions were initiated by the addition of 0.48–0.96 μg of human recombinant SULTs at 25 °C. The amount of enzyme used was always in the linear range as determined by plots of initial velocity versus enzyme concentrations. The reactions were quenched by the addition of 50 μl of ice-cold 1% formic acid and were placed on ice. The reaction mixtures were then extracted twice with 0.5 ml aliquots of ethyl acetate twice by vortex mixing and centrifuged at 16,000 × g to help phase separation. The combined ethyl acetate layer was backwashed with 0.2 ml of 1% formic acid by vigorous vortexing and centrifuged at 16,000 × g. The organic phase was then dried by a SpeedVac concentrator (Thermo Scientific). The residue was dissolved in 100 μl methanol, and an aliquot of 50 μl was analyzed by scintillation counting or by HPLC analysis. The initial velocity was estimated by the slope of the linear portion of the progress curve over 10 min. Kinetic analyses using nonlinear regression were performed by fitting the Michaelis-Menten equation to the data with the program Grafit,
When substrate inhibition was observed, the following equation was fitted to the initial velocity data in a similar manner,
where v is the initial velocity of the reaction, [S] is the molar concentration of the substrate, and Km is the Michaelis-Menten constant for the substrate. Because of the iterative fits of the equations to each data set, each fit provided estimates of the kinetic parameters as mean ± S.E. Dividing Vmax by the molar concentration of the enzyme gave kcat. Ki is the dissociation constant for the substrate to dissociate from the enzyme inhibitor complex.
Metabolism of B[a]P-7,8-dione in Human Lung Cells
The A549, HBEC-KT, and H358 cells (5 × 106) at confluency were treated with B[a]P-7,8-dione (2 μm, 0.2% DMSO) in HBSS buffer containing 1 mm sodium pyruvate. The culture media were collected at 0 and 24 h, respectively, and subsequently acidified with 0.1% formic acid before extraction with a 2× 1.5-fold volume of cold H2O-saturated ethyl acetate. The organic phases of culture media were combined and dried under a vacuum. The residue was dissolved in 100 μl of methanol. A 20-μl aliquot was analyzed by LC-MS/MS.
Identification of B[a]P-7,8-catechol Sulfates by HPLC-RAM-UV and LC-MS/MS
The B[a]P-7,8-catechol sulfates generated with [35S]PAPS were analyzed by a Waters Alliance 2695 chromatographic system (Waters Corp., Milford, MA) in tandem with a Waters 996 photodiode array detector and a β-RAM inline radiometric detector (IN/US Systems Inc., Tampa, FL). Chromatographic separation was achieved on a reversed phase column (Zorbax-ODS C18, 5 μm, 4.6 × 250 mm, DuPont) eluted with the following linear gradient of H2O (0.1% trifluoroacetic acid and 5 mm ammonium acetate; solvent A)/MeOH (0.1% trifluoroacetic acid and 5 mm ammonium acetate; solvent B) at a flow rate of 0.5 ml/min. Solvent B was changed from 65 to 80% (v/v) over 45 min, kept at 80% over 5 min, changed from 80 to 65% over 1 min, and kept at 65% for 9 min. Eluates from the column were introduced into the inline radiometric detector following mixture of the scintillant with the HPLC effluent (IN/US Systems Inc.) at a flow rate 1.5 ml/min.
LC-MS/MS identification of the O-sulfated catechols was conducted with a Finnigan TSQ Quantum Ultra spectrometer (Thermo Fisher, San Jose, CA) equipped with a heated electrospray ionization probe as ionization source. The mass spectrometer was operated in the positive ion mode or negative ion mode with the following parameters: spray voltage (4500 V at positive ion mode or −2000 V at negative ion mode), vaporizer temperature (400 °C), sheath gas pressure (35 arbitrary units), auxiliary gas pressure (10 arbitrary units), capillary temperature (350 °C), and collision energy (20 V). The masses of the metabolites were obtained by detecting the molecular ion from Q1 full scan, and the corresponding mass spectrum of each metabolite was obtained from a Q3 full scan of the product ions of the molecular ion. The chromatography conditions for the metabolites were identical to those used for HPLC-RAM-UV detection.
Identification of the B[a]P-7,8-catechol Sulfates by 1H and 13C NMR Analysis
To generate sufficient B[a]P-7,8-catechol-O-monosulfates for characterization, the sulfonation reactions were performed on a large scale. Metabolites were synthesized in a 100-ml system containing 10 mm KPO4 buffer (pH 7.4), 1.0 mm dithiothreitol, 5.0 mm MgCl2, 20 μm PAPS, and 10 μm B[a]P-7,8-dione under anaerobic conditions. The reaction was started by the addition of 1.2 mg of human recombinant SULT1A3 and incubated for 90 min at 37 °C. The reaction was quenched by the addition of 250 μl of formic acid. Metabolites were extracted with 1× 100 ml of ethyl acetate followed by 1 × 50 ml of ethyl acetate. The pooled extracts were evaporated to complete dryness. The residues were dissolved in 2 ml of methanol and subjected to reversed phase HPLC (Zorbax-ODS C18, 5 μm, 4.6 × 250 mm, DuPont). The column was eluted with the linear gradient of H2O (0.1% trifluoroacetic acid solvent A)/MeOH (0.1% trifluoroacetic acid; solvent B) at a flow rate of 0.5 ml/min. Solvent B started with 50% for 5 min and was changed from 50 to 70% (v/v) in 3 min and changed afterward from 70 to 73% over 27 min and then further changed from 73 to 80% in 5 min, kept at 80% for 5 min, changed from 80 to 50% over 1 min, and kept at 50% for 14 min. The B[a]P-7,8-catechol-O-monosulfates were collected on the basis of their characteristic UV spectra. The pooled elutes were evaporated to complete dryness. One- and two-dimensional 1H and 13C NMR data were collected on a Bruker AVIII (cryo500) NMR spectrometer operating at 500 MHz using methanol-D4 as the solvent (Sigma).
RESULTS
Gene Expression of SULTs in Human Lung Cells
To determine the expression level of SULTs in four human lung cells, RT-PCR was employed. Among the six different SULT isoforms, SULT1A1, -1A3, and -1E1 were expressed at the mRNA level (Fig. 2). Although semiquantitative, this screen showed that SULT1A3 was the most abundant at the mRNA level in human lung cells followed by SULT1A1. By contrast. SULT1E1 mRNA showed only a low level of expression in human lung cells. Therefore, the following studies focused on the sulfonation of B[a]P-7,8-catechol by recombinant SULT1A1, -1A3, and -1E1.
FIGURE 2.

Gene expression of SULTs in human lung cells is shown (A). A549, human lung adenocarcinoma cell; H358, human bronchoalveolar cell; HBEC-KT, immortalized human bronchial epithelial cell; BEAS-2B, normal human bronchial epithelial cell. SDS-PAGE of purified human recombinant SULTs is shown (B).
Identification of B[a]P-7,8-catechol-O-Monosulfates Produced by Human Recombinant SULT Isoforms
Recombinant human SULT1A1, -1A3, and -1E1 were expressed as GST-fusion proteins, and upon removal of the tag and subsequent chromatography they were obtained in homogeneous form with appropriate specific activities for the sulfonation of p-nitrophenol. Discontinuous assays were performed with [35S]PAPS as the sulfonate group donor, and the products were profiled by reversed phase HPLC with online radiometric detection. Reactions were subsequently performed with unlabeled PAPS for product identification by LC-MS/MS. Recombinant SULT1A1 catalyzed the formation of a single metabolite (M1), whereas SULT1A3 and -1E1 catalyzed the formation of two metabolites (M1, M2) of B[a]P-7,8-catechol, which were detected by HPLC-UV-RAM (Fig. 3). The HPLC-UV-RAM gave information on the ratios of the two [35S]B[a]P-7,8-catechol sulfates generated with [35S]PAPS. SULT1A3 and -1E1 formed both M1 and M2 in the ratios of 82:18 and 37:63 (M1:M2), respectively. The UV spectra of two metabolites were different. LC-MS analysis demonstrated that the molecular ions of both metabolites were the same. In the positive ion mode, they gave an m/z 365, and in the negative ion mode they gave an m/z 363. MS/MS analysis provided additional structural information for the two metabolites. In the positive ion mode, the product ion spectra of the two metabolites were identical and gave the characteristic cleavage at -SO3H with the loss of 81 atomic mass units from the molecular ion (Fig. 4), indicating that they were sulfate conjugates. Cleavage at one of the C-OH bonds resulted in a daughter ion of m/z 267 representing the loss of -OH. Rearrangement at the remaining phenolic group likely resulted in a change from a -C-OH to -C O bond. The-CO group was then lost, resulting in fragment ion at m/z 239. In the negative ion mode, product ion spectra of two metabolites also demonstrated characteristic cleavage at -SO3− resulting in daughter ions at m/z 283. LC-MS/MS analyses in both the positive and negative ion modes confirmed that the metabolites formed in the reaction system were B[a]P-7,8-catechol-O-monosulfates. However, because the mass spectra of M1 and M2 were identical, LC-MS/MS could not distinguish the position of sulfate group in the two isomers.
O bond. The-CO group was then lost, resulting in fragment ion at m/z 239. In the negative ion mode, product ion spectra of two metabolites also demonstrated characteristic cleavage at -SO3− resulting in daughter ions at m/z 283. LC-MS/MS analyses in both the positive and negative ion modes confirmed that the metabolites formed in the reaction system were B[a]P-7,8-catechol-O-monosulfates. However, because the mass spectra of M1 and M2 were identical, LC-MS/MS could not distinguish the position of sulfate group in the two isomers.
FIGURE 3.

HPLC/UV/RAM identification of sulfated metabolites (M1 and M2) of the B[a]P-7,8-catechol. B[a]P-7,8-catechol (10 μm) was generated in situ under anaerobic conditions in the presence of 1 mm dithiothreitol. The B[a]P-7,8-catechol was converted to the O-sulfate(s) in the presence of [35S]PAPS and SULTs in the incubation buffer. A, B, and C, shown are HPLC-UV chromatograms of B[a]P-7,8-catechol sulfates formed by SULT1A1, -1A3, and -1E1, respectively; D, E, and F, shown are HPLC-RAM chromatograms of B[a]P-7,8-catechol sulfates formed by SULT1A1, -1A3, and -1E1, respectively. The red triangles indicate the beginning and end of peak integration in the chromatograms. AU, absorbance units.
FIGURE 4.

LC/MS/MS identification of sulfated metabolites (M1 and M2) of the B[a]P-7,8-catechol. B[a]P-7,8-catechol (10 μm) was generated in situ under anaerobic conditions in the presence of 1 mm dithiothreitol. The B[a]P-7,8-catechol was converted to the O-sulfate(s) in the presence of PAPS and SULTs in the incubation buffer. The reactions were quenched by 1% formic acid and extracted with of ethyl acetate. The organic phase was then dried and dissolved in MeOH for LC-MS/MS analysis. A, B, and C, shown are positive ion chromatograms of B[a]P-7,8-catechol sulfates by monitoring the reaction transition m/z 365→284 ([M+H]+→ [M+H-SO3H]+). D, shown is a positive ion MS2 of isomer 1 and MS2 of isomer 2; E, shown is a negative ion MS2 of isomer 1 and MS2 of isomer 2.
Kinetic Studies on the Formation of B[a]P-7,8-catechol Sulfates
To obtain steady state kinetic parameters for the formation of the B[a]P-7,8-catechol-O-monosulfates, we conducted discontinuous assays using the recombinant SULT isoforms. We used an assay based on ethyl acetate extraction of the products under acidic conditions, which was favorable for the partition of metabolites in the organic phase. Reverse phase-HPLC analysis confirmed that when M1 and M2 were generated, they were formed concurrently in reactions containing SULT1A3 and -1E1. Only M1 was formed in reactions containing SULT 1A1. We find that the kinetic profiles for the formation of B[a]P-7,8-catechol sulfates catalyzed by the three SULTs were quite different (Fig. 5 and Table 1). Both the Michaelis-Menten equation and the equation for substrate inhibition were fitted to kinetic data with nonlinear regression analysis. Goodness of fit clearly distinguished between these two kinetic patterns. B[a]P-7,8-catechol sulfonation catalyzed by SULT1A3 followed normal Michaelis-Menten kinetics. By contrast, B[a]P-7,8-catechol sulfonation catalyzed by SULT1A1*1 and -1E1 showed modest to potent substrate inhibition yielding Ki values of 30.0 and 0.08 μm, respectively. kcat/Km values showed that SULT1A1 was the most catalytically efficient isozyme followed by SULT1E1 and -1A3, respectively. The Km value for SULT1A1*1 was the lowest, yielding a value in the submicromolar range that was 63- and 15-fold lower than those seen with SULT1A3 and -1E1. B[a]P-7,8-catechol sulfonation by the thermostable SULT1A1*3 variant followed normal Michaelis-Menten kinetics. The Km value of SULT1A1*3 was close to that of SULT1A1*1, whereas the kcat/Km value of SULT1A1*3 was about 50% that of the wild type enzyme (Table 1). Interestingly, the catalytic efficiency of SULT1A1*1 and COMT to form B[a]P-7,8-catechol monosulfate and O-methylated B[a]P-7,8-catechol were similar.
FIGURE 5.
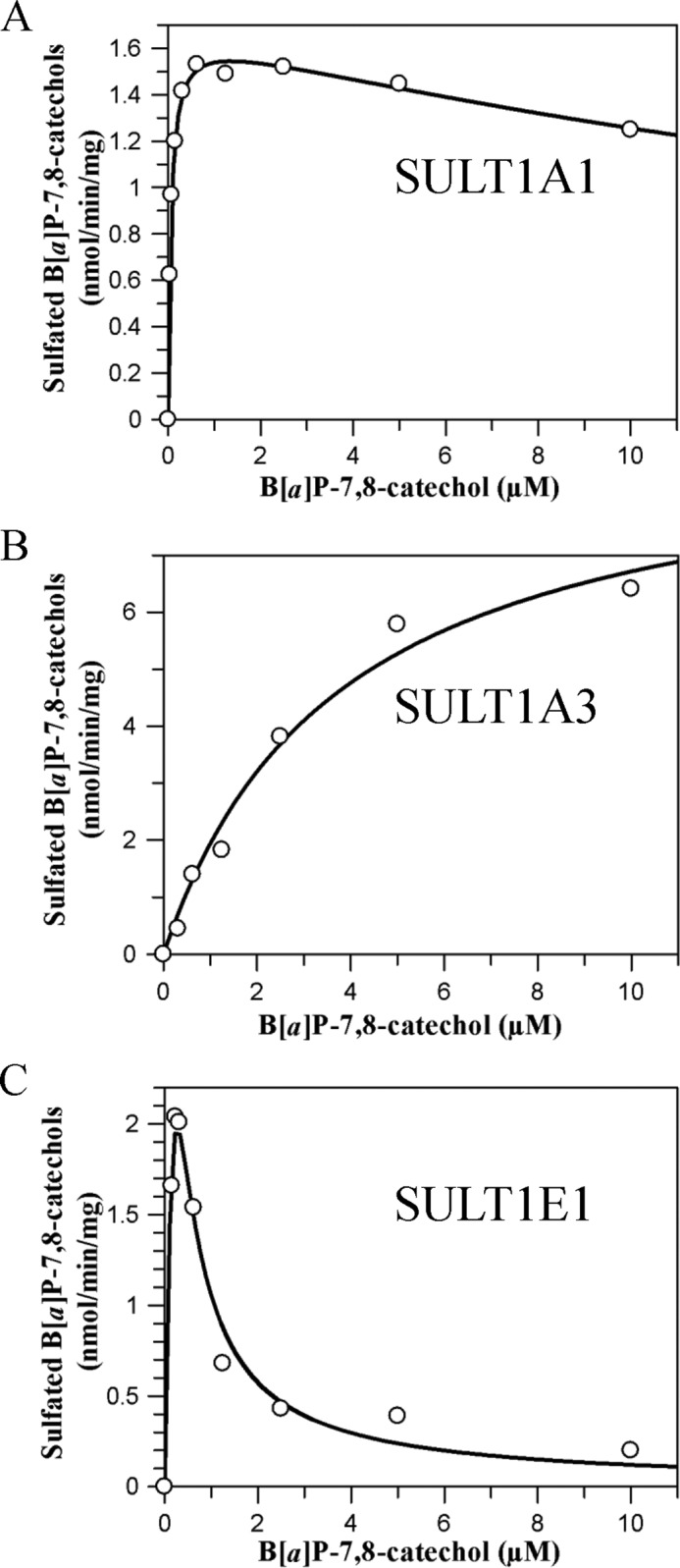
Kinetic characterization of sulfonation of B[a]P-7,8-catechol by SULTs. Initial velocities were estimated by the slope of a linear portion of the progress curve over 10 min. Kinetic analyses were performed by fitting the Michaelis-Menten equation or the substrate inhibition equation to the data. Reactions contained 10 mm KPO4 buffer of pH 7.4, 1.0 mm dithiothreitol, 5.0 mm MgCl2, 20 μm [35S]PAPS, 0–10 μm B[a]P-7,8-dione, and 0.48–0.96 μg of human recombinant SULTs at 25 °C. A, B, and C, shown is velocity versus S curve for sulfonation of B[a]P-7,8-catechol by SULT1A1, -1A3, and -1E1, respectively.
TABLE 1.
Kinetic constants of o-sulfonation of B[a]P-7,8-catechol
| Enzymes | Vmax | kcat | Km | Ki | kcat/Km |
|---|---|---|---|---|---|
| nmol/min/mg | min−1 | μm | μm | min−1 μm−1 | |
| SULT1A1*1 | 1.68 ± 0.07 | 0.059 ± 0.002 | 0.061 ± 0.004 | 29.9 ± 3.5 | 0.97 |
| SULT1A1*3 | 0.84 ± 0.02 | 0.030 ± 0.001 | 0.08 ± 0.009 | NA | 0.38 |
| SULT1A3 | 9.2 ± 0.9 | 0.32 ± 0.03 | 3.8 ± 0.8 | NA | 0.084 |
| SULT1E1 | 15.4 ± 3.7 | 0.54 ± 0.13 | 0.90 ± 0.26 | 0.08 ± 0.02 | 0.60 |
| COMTa | 54.2 | 1.48 | 2.10 | NA | 0.70 |
a Ref. 26.
Formation of B[a]P-7,8-catechol Sulfates in Human Lung Cells Treated with 2 μm B[a]P-7,8-dione
One B[a]P-7,8-catechol-O-monosulfate isomer was detected in A549, H358, and HBEC-KT cell lines after incubation of the cells with 2 μm B[a]P-7,8-dione for 24 h and was detected by LC-MS/MS (Fig. 6). Similar experiments were not performed on BEAS-2B cells as they are AKR null by real-time PCR.3 Using the negative ion mode by monitoring the reaction transition m/z 363→283, the metabolite in the human lung cells had the same retention time as M1. No significant amount of the other isomeric B[a]P-7,8-catechol-O-monosulfate was detected in the cells.
FIGURE 6.

LC-MS/MS detection of sulfated metabolites (M1 and M2) of the B[a]P-7,8-catechol formed by recombinant SULT1A3 (A) and by A549 (B), H358 (C), and HBEC-KT cells (D) at 24 h. B[a]P-7,8-dione (2 μm, 0.2% DMSO) in HBSS buffer were incubated with lung cells and collected at 0 and 24 h, respectively. The culture media were extracted with ethyl acetate. The organic phases were dried under vacuum and redissolved in methanol. B[a]P-7,8-catechol sulfates were analyzed with LC-MS/MS in the negative ion mode by monitoring the reaction transition m/z 363→283 ([M-H]− → [M-H-SO3]−). The peak M1 had the same retention time and mass transition as authentic 8-hydroxy-B[a]P-7-O-sulfate.
NMR Analysis of B[a]P-7,8-catechol Monosulfates
To identify the M1 and M2 B[a]P-7,8-catechol-O-monosulfates unequivocally, they were enzymatically generated using SULT1A3 on a preparative scale and purified by reversed phase-HPLC. Two-dimensional 1H NMR and 14C NMR of M1 and one-dimensional 1H NMR of M2 were obtained. Two-dimensional 1H and 13C NMR of M2 could not be obtained due to its poor yield. By comparing previous published two-dimensional 1H NMR of B[a]P-7,8-catechol, the protons in the spectra of B[a]P-7,8-catechol sulfates could be assigned (17). Generally, the 1H NMR spectra of catechol and catechol sulfates are quite similar. All the protons were aromatic and were located between 7.5 and 9.1 ppm. H-6 in both M1 and M2 was a singlet due to a lack of coupling with other protons. The cross-peaks at two-dimensional 1H NMR of M1 indicated the coupling between H-9 and H-10, H-11 and H-12, H-1 and H-2, and H-2 and H-3 as well as H-4 and H-5 (Fig. 7). A complete list of the connectivities obtained from the cross-peaks is provided in Table 2. To determinate the position of the sulfate group, it was necessary to examine the influence of the -SO3 group on the chemical shift of the related protons. Based on diagnostic peaks of H-6, H-9, and H-10, M1 was assigned as 8-hydroxy-B[a]P-7-O-sulfate, and M2 was 7-hydroxy-B[a]P-8-O-sulfate. 13C NMR spectrum of M1 was also acquired (see supplemental Fig. S1). The chemical shifts were as follow: δ = 146.72, 131.36, 130.99, 130.07, 128.20, 127.94, 127.46, 127.30, 127.18, 125.49, 125.15, 125.09, 124.52, 123.69, 121.97, 121.81, 121.36, 119.18, 118.50 ppm. The carbon at the most downfield position was modified by -O-SO3H.
FIGURE 7.

500 MHz 1H COSY 8-hydroxy-B[a]P-7-O-sulfate (M1) and 1H NMR spectra of 7-hydroxy-B[a]P-8-O-sulfate (M2).
TABLE 2.
Proton assignments for 8-hydroxy-benzo[a]pyrene-7-O-sulfate
m, multiplet; d, doublet; and s, singlet.
| Proton | Chemical shift, multiplicity | Coupling constants (Hz) | Connectivities |
|---|---|---|---|
| ppm | J | ||
| H-1 or H3 | 8.08, d; 8.22, d | 7.7 or 7.8 | H-2 with H-1/H-3 |
| H-2 | 7.95, m | H-1/H-3 with H-2 | |
| H-4 | 7.92, m | H-4 with H-5 | |
| H-5 | 8.05, d | 9.1 | H-5 with H-4 |
| H-6 | 8.88, s | ||
| H-9 | 7.52, d | 9.3 | H-9 with H-10 |
| H-10 | 8.88, d | 9.1 | H-10 with H-9 |
| H-11 | 9.02, d | 9.1 | H-11 with H-12 |
| H-12 | 8.31, d | 9.1 | H-12 with H-11 |
It was found that the 1H NMR spectra of M1 and M2 were different. H-6, H-9, and H-10, are closest to the 7- and 8-hydroxyl groups, and their chemical shift values are most influenced by conjugation of the -SO3 group. Chemical shift values were used as diagnostic peaks to assign the position of the -SO3 group, which was accomplished by comparing their values in the spectra of M1 and M2 with those previously reported for B[a]P-7,8-catechol (17). The -SO3 moiety is an electron withdrawing group that makes carbons at the ortho and para positions carry a partial positive charge that results in the deshielding of protons at these positions. When the -SO3 group is at C7, this would place H-10 at the para position with respect to the -SO3 group, and its resonance would be shifted to a more downfield position. This is observed because H-10 and H-6 now overlap in the spectrum of the M1 isomer. By contrast the chemical shift of H-9 would be unaltered because the -SO3 group would be in a meta position relative to this proton, and this is observed in the spectrum of M1. When the SO3 group is at the C8 position, H-10 and H-6 are clearly separated, but H-9 would now be in the ortho position, and its resonance would be expected to be shifted downfield. For M2, the -SO3 group shifted the H-9 resonance downfield when it was compared with the chemical shifts of H-9 in the spectra of either B[a]P-7,8-catechol or M1. This indicated that in M2, H-9 should be in the ortho position with respect to the -SO3 group. The chemical shift values of the other aromatic protons in M1 and M2 were quite similar because they are far away for the -SO3 group and thus there was a minimal effect on their chemical environment. This analysis shows that M1 is 8-hydroxy-B[a]P-7-O-sulfate and M2 is 7-hydroxy-B[a]P-8-O-sulfate. Using these compounds as standards, the isomer produced in human lung cells was found to be M1 or 8-hydroxy-B[a]P-7-O-sulfate.
DISCUSSION
We are conducting a systematic study to identify the conjugating enzymes that can intercept PAH-catechols and prevent their redox cycling to the corresponding PAH o-quinones. These studies will document which enzymes attenuate the ability of AKR products to form ROS that can lead to the mutagenic lesion 8-oxo-dGuo. Previously we have shown that PAH o-quinones are enzymatically redox-cycled at a robust rate by recombinant NQO1 and by AKRs themselves and that human COMT can O-methylate a series of PAH-catechols (21, 26). In this study we identify SULT1A1 as the major isoform responsible for the formation of B[a]P-7,8-catechol-O-monosulfate based on catalytic efficiency and enzyme expression level in human lung cells. We also identify the major metabolite formed as 8-hydroxy-benzo[a]pyrene-7-O-sulfate based on NMR data.
Three SULT isoforms, SULT1A1, -1A3, and -1E1, were detected in human lung cells and were recombinantly expressed for this study. SULT1A1 and -1A3 share 95.6% similarity in amino acid sequence, whereas SULT1A1 and -1E1 share only 65.4% similarity. SULT1A1 formed only the M1 metabolite, 8-hydroxy-benzo[a]pyrene-7-O-sulfate, whereas SULT1A3 showed a distinct preference for the formation of M1. By contrast SULT1E1 formed M1 and M2 in approximately equal amounts. It is probable the high amino acid sequence similarity and, therefore, similar three-dimensional structure of SULT1A1 and -1A3 accounts for their similar regioselectivity of sulfonation of B[a]P-7,8-catechol. SULTs perform sulfonation via a SN2 mechanism without going through a sulfonated enzyme intermediate. To determine the structural basis for the formation of M1 and M2 at the same active site will require an x-ray crystallographic approach.
SULT1A1 was the most catalytically efficient isoform among the three SULTs in sulfonating B[a]P-7,8-catechol. The Km value of SULT1A1 was in the nanomolar range, which indicates that it can detoxify B[a]P-7,8-catechol at very low substrate concentrations but would also be easily saturated. Even though there is 95.6% similarity in amino acid sequence between SULT1A1 and -1A3, their kinetic profiles were different. Although the kcat value of SULT1A3 was higher than that of SULT1A1, the Km of SULT1A3 is orders of magnitude greater that of SULT1A1; thus, SULT1A3 is less efficient at detoxication of B[a]P-7,8-catehcol than SULT1A1. It was also found that B[a]P-7,8-catechol caused significant substrate inhibition of SULT1E1, which would compromise its detoxication ability. Moreover, the mRNA level of SULT1E1 was relatively low in human lung cells. When the kinetic profiles and expression levels of the SULTs are considered, SULT1E1 was not as important as SULT1A1 and -1A3 in the sulfonation of B[a]P-7,8-catechol. Interestingly, SULT1E1 is the most efficient SULT isoform for sulfonation of 17β-estradiol and estrogen-3,4-catechol, which prevents formation of estrogen o-quinones (31). Estrogen o-quinones are regarded as endogenous tumor initiators and are able to attack DNA to form potentially carcinogenic stable or depurinating DNA adducts (42). It is possible that inhibition to SULT1E1 by B[a]P-7,8-catechol and other PAH catechols may increase the risk factor for estrogen-dependent genotoxicity.
We also demonstrated that B[a]P-7,8-dione was metabolized to B[a]P-7,8-catechol sulfate in three human lung cell-based models. At 2 μm B[a]P-7,8-dione, only 8-hydroxy-B[a]P-7-O-sulfate (M1) was detected in the cells. In the metabolism study using recombinant SULTs, the ratios of M1/M2 formation for SULT1A1, -1A3, and -1E1 were 100:0, 82:18, 36:63, respectively. This ratio could be used as an index to reflect in part the contribution of SULTs in cellular detoxication of B[a]P-7,8-dione. The predominant metabolite found in the cells (M1) implied that SULT1A1 makes the most significant contribution to O-sulfonation. This is consistent with the analysis based on the kinetic profiles and expression levels of each SULT.
We found that in A549, H358, and HBEC-KT human lung cells, the O-monosulfate conjugate of B[a]P-7,8-catechol accounts for 12.2, 15, and 3.5% of the metabolites derived from B[a]P-7,8-dione (43). The amount of O-monosulfated B[a]P-7,8-catechol formed was about twice that of the O-methylated B[a]P-7,8-catechol. This indicated that detoxication of B[a]P-7,8-dione through the sulfation pathway is preferred over O-methylation. Inhibition of COMT in A549 cells increased DNA strand breaks and formation of 8-oxo-dGuo in cells treated with B[a]P-7,8-dione, which supports the contention that O-methylation of B[a]P-7,8-catechol contributes to the detoxication of B[a]P-7,8-dione by preventing its ability to redox cycle (25). Because of the higher level of O-sulfonation in these cells, we predict that SULTs will play a more important role than COMT in intercepting B[a]P-7,8-dione to prevent the generation of reactive oxygen species. The expression of SULT and COMT in lung tissue has been established by others (34, 44) and suggests that is reasonable to infer from our cell-based study that both enzymes will play a significant role in the detoxication of B[a]P-7,8-dione.
SULT1A1 polymorphism has been associated with an increased lung cancer risk (45), and polymorphic variants of SULT1A1 may affect the detoxication of PAH o-quinones. The common SULT1A1 allozymes mainly include *1 (wild type), *2 variant (R213H), and *3 variant (M223V) (46). The allelic frequencies for SULT1A1*1, -*2, and *3 in Caucasian were 0.656, 0.332, 0.012, respectively. Despite the low frequency of SULT1A1*3 in Caucasians, it has an allelic frequency of 0.229 in African Americans (46). It has been reported that SULT1A1 recombinant allozymes have variable thermal stability and specific activity toward p-nitrophenol, catechol estrogens, and dietary flavonoids (31, 47, 48). The SULT1A1*2 variant was associated with low enzyme activity and thermal stability (45, 47, 48). Although SULT1A1*3 had compatible thermal stability to wild type enzyme, its specific activities for SULT1A1 substrates were usually lower than that of wild type (48).
Although the Km value of SULT1A1*3 was comparable to its wild type counterpart, its kcat was less. As a result the catalytic efficiency of SULT1A1*3 for sulfonation of B[a]P-7,8-catechol was about half that observed for the wild type SULT1A1. Because SULT1A1 plays an important role in detoxication of B[a]P-7,8-dione, this study provides some evidence that the polymorphic variants that reduce stability or catalytic efficiency of SULT1A1 in sulfonation of PAH catechols may increase susceptibility to lung cancer caused by smoking and air pollution. Unlike high allele frequencies of SULT1A1 variants, SULT1A3 and SULT1E1 variants were found to be very rare, which indicates that genetic polymorphism of these two enzymes may have less influence on detoxication of B[a]P-7,8-dione (49, 50).
Sulfonation of PAH catechols by SULTs described in this study may be one important mechanism for detoxication of PAH o-quinones. First, SULTs could terminate the futile redox cycling of PAH o-quinones by intercepting o-quinones that cause ROS generation and subsequent oxidative DNA damage. Second SULTs could eliminate the electrophilicity of PAH o-quinones as the formation of catechol O-sulfates prevents the formation of covalent PAH o-quinone adducts with protein or DNA. Third, sulfation usually results in more polar metabolites and enhances renal or biliary excretion of xenobiotics or drugs; thus, sulfate conjugation of PAH catechols may facilitate the elimination of PAH o-quinones.
Supplementary Material
Acknowledgments
We thank Dr. George Furst for conducting NMR analysis, Drs. Adegoke Adeniji and Ding Lu for the advice on NMR assignments, Dr. Mo Chen for help in recombinant SULT purification, and Dr. Xiaojing Liu for advice on LC/MS method development.
This work was supported, in whole or in part, by National Institutes of Health Grants P30-ES-013508, R01-CA39504, and PA-DOH4100038714 (to T. M. P.).

This article contains supplemental Fig. S1.
M. E. Kushman and T. M. Penning, unpublished information.
- PAH
- polycyclic aromatic hydrocarbon
- B[a]P
- benzo[a]pyrene
- B[a]P-7,8-trans-dihydrodiol
- (−)trans-7,8-dihydroxy-7,8-dihydro-benzo[a]pyrene
- AKR
- aldo-keto reductase
- B[a]P-7,8-catechol
- benzo[a]pyrene-7,8-catechol
- B[a]P-7,8-dione
- benzo[a]pyrene-7,8-dione
- COMT
- catechol-O-methyl transferase
- PAPS
- 3′-phosphoadenosine 5′-phosphosulfate
- SULT
- sulfotransferase
- ROS
- reactive oxygen species
- RAM
- radioactive monitor.
REFERENCES
- 1. Grimmer G., Bohnke H. (1975) Polycyclic aromatic hydrocarbon profile analysis of high-protein foods, oils, and fats by gas chromatography. J. Assoc. Anal. Chem. 58, 725–733 [PubMed] [Google Scholar]
- 2. Burczynski M. E., Lin H. K., Penning T. M. (1999) Isoform-specific induction of a human aldo-keto reductase by polycyclic aromatic hydrocarbons (PAH), electrophiles, and oxidative stress. Implications for the alternative pathway of PAH activation catalyzed by human dihydrodiol dehydrogenase. Cancer Res. 59, 607–614 [PubMed] [Google Scholar]
- 3. Gelboin H. V. (1980) Benzo[a]pyrene metabolism, activation, and carcinogenesis. Role and regulation of mixed-function oxidases and related enzymes. Physiol. Rev. 60, 1107–1166 [DOI] [PubMed] [Google Scholar]
- 4. Conney A. H. (1982) Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons. G. H. A. Clowes Memorial Lecture. Cancer Res. 42, 4875–4917 [PubMed] [Google Scholar]
- 5. Cavalieri E. L., Rogan E. G. (1995) Central role of radical cations in metabolic activation of polycyclic aromatic hydrocarbons. Xenobiotica 25, 677–688 [DOI] [PubMed] [Google Scholar]
- 6. Sagher D., Strauss B. (1983) Insertion of nucleotides opposite apurinic/apyrimidinic sites in deoxyribonucleic acid during in vitro synthesis. Uniqueness of adenine nucleotides. Biochemistry 22, 4518–4526 [DOI] [PubMed] [Google Scholar]
- 7. Shimada T., Hayes C. L., Yamazaki H., Amin S., Hecht S. S., Guengerich F. P., Sutter T. R. (1996) Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1. Cancer Res. 56, 2979–2984 [PubMed] [Google Scholar]
- 8. Shimada T., Gillam E. M., Oda Y., Tsumura F., Sutter T. R., Guengerich F. P., Inoue K. (1999) Metabolism of benzo[a]pyrene to trans-7,8-dihydroxy-7, 8-dihydrobenzo[a]pyrene by recombinant human cytochrome P450 1B1 and purified liver epoxide hydrolase. Chem. Res. Toxicol. 12, 623–629 [DOI] [PubMed] [Google Scholar]
- 9. Kim J. H., Stansbury K. H., Walker N. J., Trush M. A., Strickland P. T., Sutter T. R. (1998) Metabolism of benzo[a]pyrene and benzo[a]pyrene-7,8-diol by human cytochrome P450 1B1. Carcinogenesis 19, 1847–1853 [DOI] [PubMed] [Google Scholar]
- 10. Koreeda M., Moore P. D., Wislocki P. G., Levin W., Yagi H., Jerina D. M. (1978) Binding of benzo[a]pyrene 7,8-diol-9,10-epoxides to DNA, RNA, and protein of mouse skin occurs with high stereoselectivity. Science 199, 778–781 [DOI] [PubMed] [Google Scholar]
- 11. Malaveille C., Kuroki T., Sims P., Grover P. L., Bartsch H. (1977) Mutagenicity of isomeric diol-epoxides of benzo[a]pyrene and benz[a]anthracene in S. typhimurium TA98 and TA100 and in V79 Chinese hamster cells. Mutat. Res. 44, 313–326 [DOI] [PubMed] [Google Scholar]
- 12. Nesnow S., Ross J. A., Stoner G. D., Mass M. J. (1995) Mechanistic linkage between DNA adducts, mutations in oncogenes, and tumorigenesis of carcinogenic environmental polycyclic aromatic hydrocarbons in strain A/J mice. Toxicology 105, 403–413 [DOI] [PubMed] [Google Scholar]
- 13. Burczynski M. E., Harvey R. G., Penning T. M. (1998) Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms. Oxidation of trans-7, 8-dihydroxy-7,8-dihydrobenzo[a]pyrene to the activated o-quinone metabolite benzo[a]pyrene-7,8-dione. Biochemistry 37, 6781–6790 [DOI] [PubMed] [Google Scholar]
- 14. Palackal N. T., Burczynski M. E., Harvey R. G., Penning T. M. (2001) The ubiquitous aldehyde reductase (AKR1A1) oxidizes proximate carcinogen trans-dihydrodiols to o-quinones. Potential role in polycyclic aromatic hydrocarbon activation. Biochemistry 40, 10901–10910 [DOI] [PubMed] [Google Scholar]
- 15. Palackal N. T., Lee S. H., Harvey R. G., Blair I. A., Penning T. M. (2002) Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional overexpression in human lung carcinoma (A549) cells. J. Biol. Chem. 277, 24799–24808 [DOI] [PubMed] [Google Scholar]
- 16. Murty V. S., Penning T. M. (1992) Polycyclic aromatic hydrocarbon (PAH) ortho-quinone conjugate chemistry. Kinetics of thiol addition to PAH ortho-quinones and structures of thioether adducts of naphthalene-1,2-dione. Chem. Biol. Interact. 84, 169–188 [DOI] [PubMed] [Google Scholar]
- 17. Murty V. S., Penning T. M. (1992) Characterization of mercapturic acid and glutathionyl conjugates of benzo[a]pyrene-7,8-dione by two-dimensional NMR. Bioconjug. Chem. 3, 218–224 [DOI] [PubMed] [Google Scholar]
- 18. Shou M., Harvey R. G., Penning T. M. (1993) Reactivity of benzo[a]pyrene-7,8-dione with DNA. Evidence for the formation of deoxyguanosine adducts. Carcinogenesis 14, 475–482 [DOI] [PubMed] [Google Scholar]
- 19. McCoull K. D., Rindgen D., Blair I. A., Penning T. M. (1999) Synthesis and characterization of polycyclic aromatic hydrocarbon o-quinone depurinating N7-guanine adducts. Chem. Res. Toxicol 12, 237–246 [DOI] [PubMed] [Google Scholar]
- 20. Balu N., Padgett W. T., Nelson G. B., Lambert G. R., Ross J. A., Nesnow S. (2006) Benzo[a]pyrene-7,8-quinone-3′-mononucleotide adduct standards for 32P postlabeling analyses. Detection of benzo[a]pyrene-7,8-quinone-calf thymus DNA adducts. Anal. Biochem. 355, 213–223 [DOI] [PubMed] [Google Scholar]
- 21. Shultz C. A., Quinn A. M., Park J. H., Harvey R. G., Bolton J. L., Maser E., Penning T. M. (2011) Specificity of human aldo-keto reductases, NAD(P)H:quinone oxidoreductase, and carbonyl reductases to redox-cycle polycyclic aromatic hydrocarbon diones and 4-hydroxyequilenin-o-quinone. Chem. Res. Toxicol. 24, 2153–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu D., Berlin J. A., Penning T. M., Field J. (2002) Reactive oxygen species generated by PAH o-quinones cause change-in-function mutations in p53. Chem. Res. Toxicol. 15, 832–842 [DOI] [PubMed] [Google Scholar]
- 23. Park J. H., Troxel A. B., Harvey R. G., Penning T. M. (2006) Polycyclic aromatic hydrocarbon (PAH) o-quinones produced by the aldo-keto-reductases (AKRs) generate abasic sites, oxidized pyrimidines, and 8-oxo-dGuo via reactive oxygen species. Chem. Res. Toxicol. 19, 719–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shen Y. M., Troxel A. B., Vedantam S., Penning T. M., Field J. (2006) Comparison of p53 mutations induced by PAH o-quinones with those caused by anti-benzo[a]pyrene diol epoxide in vitro. Role of reactive oxygen and biological selection. Chem. Res. Toxicol. 19, 1441–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park J. H., Mangal D., Tacka K. A., Quinn A. M., Harvey R. G., Blair I. A., Penning T. M. (2008) Evidence for the aldo-keto reductase pathway of polycyclic aromatic trans-dihydrodiol activation in human lung A549 cells. Proc. Natl. Acad. Sci. U.S.A. 105, 6846–6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang L., Jin Y., Chen M., Huang M., Harvey R. G., Blair I. A., Penning T. M. (2011) Detoxication of structurally diverse polycyclic aromatic hydrocarbon (PAH) o-quinones by human recombinant catechol-O-methyltransferase (COMT) via O-methylation of PAH catechols. J. Biol. Chem. 286, 25644–25654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Negishi M., Pedersen L. G., Petrotchenko E., Shevtsov S., Gorokhov A., Kakuta Y., Pedersen L. C. (2001) Structure and function of sulfotransferases. Arch. Biochem. Biophys. 390, 149–157 [DOI] [PubMed] [Google Scholar]
- 28. Coughtrie M. W., Sharp S., Maxwell K., Innes N. P. (1998) Biology and function of the reversible sulfation pathway catalysed by human sulfotransferases and sulfatases. Chem. Biol. Interact. 109, 3–27 [DOI] [PubMed] [Google Scholar]
- 29. Weinshilboum R. M., Otterness D. M., Aksoy I. A., Wood T. C., Her C., Raftogianis R. B. (1997) Sulfation and sulfotransferases 1. Sulfotransferase molecular biology. cDNAs and genes. FASEB J. 11, 3–14 [PubMed] [Google Scholar]
- 30. Falany C. N., Comer K. A., Dooley T. P., Glatt H. (1995) Human dehydroepiandrosterone sulfotransferase. Purification, molecular cloning, and characterization. Ann. N.Y. Acad. Sci. 774, 59–72 [DOI] [PubMed] [Google Scholar]
- 31. Adjei A. A., Weinshilboum R. M. (2002) Catechol estrogen sulfation. Possible role in carcinogenesis. Biochem. Biophys. Res. Commun. 292, 402–408 [DOI] [PubMed] [Google Scholar]
- 32. Taskinen J., Ethell B. T., Pihlavisto P., Hood A. M., Burchell B., Coughtrie M. W. (2003) Conjugation of catechols by recombinant human sulfotransferases, UDP-glucuronosyltransferases, and soluble catechol O-methyltransferase. Structure-conjugation relationships and predictive models. Drug Metab. Dispos. 31, 1187–1197 [DOI] [PubMed] [Google Scholar]
- 33. Hui Y., Yasuda S., Liu M. Y., Wu Y. Y., Liu M. C. (2008) On the sulfation and methylation of catechol estrogens in human mammary epithelial cells and breast cancer cells. Biol. Pharm. Bull. 31, 769–773 [DOI] [PubMed] [Google Scholar]
- 34. Cappiello M., Franchi M., Giuliani L., Pacifici G. M. (1989) Distribution of 2-naphthol sulfotransferase and its endogenous substrate adenosine 3′-phosphate 5′-phosphosulfate in human tissues. Eur. J. Clin. Pharmacol. 37, 317–320 [DOI] [PubMed] [Google Scholar]
- 35. Harvey R. G., Dai Q., Ran C., Penning T. M. (2004) Synthesis of the o-quinones and other oxidized metabolites of polycyclic aromatic hydrocarbons implicated in carcinogenesis. J. Org. Chem. 69, 2024–2032 [DOI] [PubMed] [Google Scholar]
- 36. Ebmeier C. C., Anderson R. J. (2004) Human thyroid phenol sulfotransferase enzymes 1A1 and 1A3. Activities in normal and diseased thyroid glands and inhibition by thyroid hormones and phytoestrogens. J. Clin. Endocrinol. Metab. 89, 5597–5605 [DOI] [PubMed] [Google Scholar]
- 37. Stanley E. L., Hume R., Coughtrie M. W. (2005) Expression profiling of human fetal cytosolic sulfotransferases involved in steroid and thyroid hormone metabolism and in detoxification. Mol. Cell. Endocrinol. 240, 32–42 [DOI] [PubMed] [Google Scholar]
- 38. Foldes A., Meek J. L. (1973) Rat brain phenolsulfotransferase. Partial purification and some properties. Biochim. Biophys. Acta 327, 365–374 [DOI] [PubMed] [Google Scholar]
- 39. Liu M. C., Suiko M., Sakakibara Y. (2000) Mutational analysis of the substrate binding/catalytic domains of human M form and P form phenol sulfotransferases. J. Biol. Chem. 275, 13460–13464 [DOI] [PubMed] [Google Scholar]
- 40. Sakakibara Y., Takami Y., Nakayama T., Suiko M., Liu M. C. (1998) Localization and functional analysis of the substrate specificity/catalytic domains of human M-form and P-form phenol sulfotransferases. J. Biol. Chem. 273, 6242–6247 [DOI] [PubMed] [Google Scholar]
- 41. Pai T. G., Sugahara T., Suiko M., Sakakibara Y., Xu F., Liu M. C. (2002) Differential xenoestrogen-sulfating activities of the human cytosolic sulfotransferases. Molecular cloning, expression, and purification of human SULT2B1a and SULT2B1b sulfotransferases. Biochim. Biophys. Acta 1573, 165–170 [DOI] [PubMed] [Google Scholar]
- 42. Cavalieri E. L., Stack D. E., Devanesan P. D., Todorovic R., Dwivedy I., Higginbotham S., Johansson S. L., Patil K. D., Gross M. L., Gooden J. K., Ramanathan R., Cerny R. L., Rogan E. G. (1997) Molecular origin of cancer. Catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. Sci. U.S.A. 94, 10937–10942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang M., Liu X., Basu S. S., Zhang L., Kushman M. E., Harvey R. G., Blair I. A., Penning T. M. (2012) Metabolism and disposition of benzo[a]pyrene-7,8-dione (B[a]P-7,8-dione) in human lung cells by liquid chromatography tandem mass spectrometry. Detection of N7 adenine B[a]P-7,8-dione adduct. Chem. Res. Toxicol. 25, 993–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Somers G. I., Lindsay N., Lowdon B. M., Jones A. E., Freathy C., Ho S., Woodrooffe A. J., Bayliss M. K., Manchee G. R. (2007) A comparison of the expression and metabolizing activities of phase I and II enzymes in freshly isolated human lung parenchymal cells and cryopreserved human hepatocytes. Drug. Metab. Dispos. 35, 1797–1805 [DOI] [PubMed] [Google Scholar]
- 45. Wang Y., Spitz M. R., Tsou A. M., Zhang K., Makan N., Wu X. (2002) Sulfotransferase (SULT) 1A1 polymorphism as a predisposition factor for lung cancer. A case-control analysis. Lung Cancer 35, 137–142 [DOI] [PubMed] [Google Scholar]
- 46. Carlini E. J., Raftogianis R. B., Wood T. C., Jin F., Zheng W., Rebbeck T. R., Weinshilboum R. M. (2001) Sulfation pharmacogenetics. SULT1A1 and SULT1A2 allele frequencies in Caucasian, Chinese, and African-American subjects. Pharmacogenetics 11, 57–68 [DOI] [PubMed] [Google Scholar]
- 47. Raftogianis R. B., Wood T. C., Weinshilboum R. M. (1999) Human phenol sulfotransferases SULT1A2 and SULT1A1. Genetic polymorphisms, allozyme properties, and human liver genotype-phenotype correlations. Biochem. Pharmacol. 58, 605–616 [DOI] [PubMed] [Google Scholar]
- 48. Nagar S., Walther S., Blanchard R. L. (2006) Sulfotransferase (SULT) 1A1 polymorphic variants *1, *2, and *3 are associated with altered enzymatic activity, cellular phenotype, and protein degradation. Mol. Pharmacol. 69, 2084–2092 [DOI] [PubMed] [Google Scholar]
- 49. Hildebrandt M. A., Salavaggione O. E., Martin Y. N., Flynn H. C., Jalal S., Wieben E. D., Weinshilboum R. M. (2004) Human SULT1A3 pharmacogenetics. Gene duplication and functional genomic studies. Biochem. Biophys. Res. Commun. 321, 870–878 [DOI] [PubMed] [Google Scholar]
- 50. Glatt H., Meinl W. (2004) Pharmacogenetics of soluble sulfotransferases (SULTs). Naunyn. Schmiedebergs. Arch. Pharmacol. 369, 55–68 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.