

Background: Microglial activation plays an important role in the pathogenesis of neurodegenerative disorders.
Results: Taylor-Couette-Poiseuille flow-modified saline (RNS60) inhibits microglial inflammation via type 1A phosphatidylinositol 3-kinase-Akt-CREB-mediated up-regulation of IκBα and inhibition of NF-κB activation.
Conclusion: These results delineate a novel biological function of a physically modified saline.
Significance: RNS60 may be of therapeutic benefit in neurodegenerative disorders.
Keywords: CREB, Inflammation, Microglia, NF-κB, Nitric-oxide Synthase, PI 3-Kinase, Glial Activation, IκBα, Modified Saline, Nanobubbles
Abstract
Chronic inflammation involving activated microglia and astroglia is becoming a hallmark of many human diseases, including neurodegenerative disorders. Although NF-κB is a multifunctional transcription factor, it is an important target for controlling inflammation as the transcription of many proinflammatory molecules depends on the activation of NF-κB. Here, we have undertaken a novel approach to attenuate NF-κB activation and associated inflammation in activated glial cells. RNS60 is a 0.9% saline solution containing charge-stabilized nanostructures that are generated by subjecting normal saline to Taylor-Couette-Poiseuille (TCP) flow under elevated oxygen pressure. RNS60, but not normal saline, RNS10.3 (TCP-modified saline without excess oxygen), and PNS60 (saline containing excess oxygen without TCP modification) were found to inhibit the production of nitric oxide (NO) and the expression of inducible NO synthase in activated microglia. Similarly, RNS60 also inhibited the expression of inducible NO synthase in activated astroglia. Inhibition of NF-κB activation by RNS60 suggests that RNS60 exerts its anti-inflammatory effect through the inhibition of NF-κB. Interestingly, RNS60 induced the activation of type IA phosphatidylinositol (PI) 3-kinase and Akt and rapidly up-regulated IκBα, a specific endogenous inhibitor of NF-κB. Inhibition of PI 3-kinase and Akt by either chemical inhibitors or dominant-negative mutants abrogated the RNS60-mediated up-regulation of IκBα. Furthermore, we demonstrate that RNS60 induced the activation of cAMP-response element-binding protein (CREB) via the PI 3-kinase-Akt pathway and that RNS60 up-regulated IκBα via CREB. These results describe a novel anti-inflammatory property of RNS60 via type IA PI 3-kinase-Akt-CREB-mediated up-regulation of IκBα, which may be of therapeutic benefit in neurodegenerative disorders.
Introduction
Although inflammation is a protective cellular response aimed at removing injurious stimuli and initiating the healing process, prolonged inflammation, known as chronic inflammation, goes beyond physiological control, and eventually destructive effects override the beneficial effects. Nowadays, persistent inflammation is considered as an underlying contributor to virtually every chronic disease. Recent studies demonstrate that chronic inflammation in the central nervous system (CNS) is also a hallmark of various neurodegenerative disorders in which progressive loss of structure and function of neurons and neuronal death are observed (1–5). For example, the concentration of NO2− (nitrite), a metabolite of nitric oxide (NO), increases in the cerebrospinal fluid of patients with Parkinson disease and Alzheimer disease in comparison with age-matched controls (6). Consistently, the ablation of inducible nitric-oxide synthase (iNOS)2 in mutant mice significantly protects dopaminergic neurons from MPTP neurotoxicity, indicating that iNOS is essential in MPTP-induced substantia nigra pars compacta dopaminergic neurodegeneration (7). A variety of pro-inflammatory cytokines, including tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), IL-6, eicosanoids, and other immune neurotoxins, are found in either cerebrospinal fluid or affected brain regions of patients with neurodegenerative disorders (8). Finally, NF-κB, a transcription factor required for the transcription of most proinflammatory molecules, is activated in the substantia nigra pars compacta of Parkinson disease patients and MPTP-intoxicated mice and monkeys, and selective inhibition of NF-κB in mice and monkeys by NF-κB essential modifier-binding domain peptides protects dopaminergic neurons from MPTP toxicity (9, 10). Therefore, inflammation is an important target for neuronal protection in neurodegenerative disorders.
RNS60 is a physically modified saline that is generated by subjecting normal saline to Taylor-Couette-Poiseuille (TCP) flow under elevated oxygen pressure. TCP flow combines intense local mixing with limited axial dispersion (11), providing large turbulent energy dissipation rates and surface-to-volume mixing ratios (12). Gas/liquid interface and energy transfer were maximized by using a rotor with numerous cavities and by employing rotational speeds above 3,000 rpm. These conditions generate a strong shear layer at the interface between the vapor and liquid phases near the rotor cavities, which correlates with the generation of small bubbles from cavitation (13).
Although nanobubbles are used as contrast agents in biomedical imaging and have been explored for drug delivery, they have not been shown to have direct biological effects. Here, we delineate new beneficial properties of RNS60. RNS60 suppressed the expression of proinflammatory molecules and the activation of NF-κB in activated glial cells via type 1A phosphatidylinositol-3 (PI-3) kinase-Akt-CREB-mediated up-regulation of IκBα, a specific endogenous inhibitor of the classical NF-κB heterodimer p65:p50 (14, 15). These results establish a novel mode of action of RNS60 and show promise for developing a new class of anti-inflammatory agents as primary or adjunct therapy for neuroinflammatory diseases.
EXPERIMENTAL PROCEDURES
Isolation of Primary Mouse Microglia and Astroglia
Microglia were isolated from mixed glial cultures according to the procedure of Giulian and Baker (16) with modifications as described previously by us (17). Briefly, on day 9, the mixed glial cultures were washed three times with Dulbecco's modified Eagle's medium/F-12 and subjected to shaking at 240 rpm for 2 h at 37 °C on a rotary shaker. The floating cells were washed, seeded onto plastic tissue culture flasks, and incubated at 37 °C for 1 h. The attached cells were seeded onto new plates for further studies. Ninety to 95% of cells were found to be positive for Mac-1. On day 11, mixed glial cultures were washed and shaken again on a rotary shaker at 180 rpm for 18 h. Floating cells were removed, and the remaining cells were trypsinized and seeded onto new plates for experiments. More than 96% of cells were positive for GFAP. Mouse BV-2 microglial cells (kind gift from Virginia Bocchini, University of Perugia) were also maintained and induced as indicated above.
Preparation of RNS60
RNS60 was generated at Revalesio (Tacoma, WA) using a rotor/stator device (18), which incorporates controlled turbulence and TCP flow. Briefly, sodium chloride (0.9%) for irrigation, USP (pH 5.6) (4.5–7.0, Hospira), was processed at 4 °C, and a flow rate of 32 ml/s under 1 atm of oxygen back-pressure (7.8 ml/s gas flow rate) while maintaining a rotor speed of 3,450 rpm. The resulting fluid was immediately placed into glass bottles (KG-33 borosilicate glass, Kimble-Chase) and sealed using gray chlorobutyl rubber stoppers (USP class 6, West Pharmaceuticals) to maintain pressure and minimize leachables. When tested after 24 h, the oxygen content was 55 ± 5 ppm. Chemically, RNS60 contains water, sodium chloride, 50–60 ppm oxygen but no active pharmaceutical ingredients.
The following controls for RNS60 were also used in this study: (a) normal saline (NS) from the same manufacturing batch. This saline contacted the same device surfaces as RNS60 and was bottled in the same way; (b) RNS10.3, saline that was processed through the same device without adding excess oxygen, and (c) PNS60, saline with the same oxygen content (55 ± 5 ppm) that was prepared inside the same device but was not processed with TCP flow. Careful analysis demonstrated that all three fluids were chemically identical.
Analysis of RNS60, NS, RNS10.3, and PNS60 by Liquid Chromatography Quadrupole Time-of-Flight Mass Spectrometry (LC-Q-TOF)
To test for compositional differences in RNS60, NS, and PNS60, the LC-Q-TOF system was configured with an electrospray ionization interface, and the analysis was performed in both positive and negative modes. The samples were introduced in triplicate via flow injection analysis into the system, and data acquisition was performed in TOF mode (MS only, scan) in the 100–1,000 m/z mass range.
Atomic Force Microscopy (AFM)
Tapping mode AFM was performed at ambient temperature (20 ± 1 °C) and at pH 5.6 with a D3000 microscope, a DTFML-DD liquid cell, and DNPS silicon nitride cantilevers (all Bruker Instruments) with a quoted stiffness of 0.35 newtons/m, and height images were obtained. A scan rate of 1 Hz and amplitude set point of 90% were used for all images. The substrate is a silicon wafer spin-coated with polystyrene to give a root mean square roughness of 0.44 nm and a static contact angle of 93°.
Antibodies
Rabbit anti-mouse iNOS antibody was obtained from Calbiochem. FITC-conjugated anti-PIP3 antibody was purchased from Echelon Biosciences, Salt Lake City, UT. Rabbit and goat anti-NF-κB p65 and goat anti-glial fibrillary acidic protein (GFAP) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-mouse phospho-Akt (Ser-473) and rabbit anti-mouse phospho-CREB (Ser-133) antibodies were purchased from Cell Signaling (Danvers, MA). Rat anti-mouse CD11b was purchased from Abcam (Cambridge, MA). Cy2- and Cy5-conjugated antibodies were obtained from Jackson ImmunoResearch (West Grove, PA).
Assay for NO Synthesis
Synthesis of NO was determined by assay of culture supernatant for nitrite, a stable reaction product of NO with molecular oxygen, using “Griess” reagent as described earlier (17, 19, 20).
Assay of Type 1A and Type 1B PI 3-Kinases
After stimulation, cells were lysed with ice-cold lysis buffer containing 1% v/v Nonidet P-40, 100 mm NaCl, 20 mm Tris (pH 7.4), 10 mm iodoacetamide, 10 mm NaF, 1 mm sodium orthovanadate, 1 mm phenylmethylsulfonyl chloride, 1 μg/ml leupeptin, 1 μg/ml antipain, 1 μg/ml aprotinin, and 1 μg/ml pepstatin A. Lysates were incubated at 4 °C for 15 min followed by centrifugation at 13,000 × g for 15 min. The supernatant was precleared with protein G-Sepharose beads (Bio-Rad) for 1 h at 4 °C followed by the addition of 1 μg/ml p85α, p101, or p84 monoclonal antibodies. After a 2-h incubation at 4 °C, protein G-Sepharose beads were added, and the resulting mixture was further incubated for 1 h at 4 °C. The immunoprecipitates were washed twice with lysis buffer, once with phosphate-buffered saline, once with 0.5 m LiCl and 100 mm Tris (pH 7.6), once in water, and once in kinase buffer (5 mm MgCl2, 0.25 mm EDTA, 20 mm HEPES (pH 7.4)). PI 3-kinase activity was determined as described earlier (21) using a lipid mixture of 100 μl of 0.1 mg/ml phosphatidylinositol and 0.1 mg/ml phosphatidylserine dispersed by sonication in 20 mm HEPES (pH 7.0) and 1 mm EDTA. The reaction was initiated by the addition of 20 μCi of [γ-32P]ATP (3,000 Ci/mmol; PerkinElmer Life Sciences) and 100 μm ATP and terminated after 15 min by the addition of 80 μl of 1 n HCl and 200 μl of chloroform/methanol (1:1). Phospholipids were separated by TLC and visualized by exposure to iodine vapor and autoradiography. Similarly, to monitor p110α-, p110β-, and p110δ-associated PI 3-kinase activity, supernatants were immunoprecipitated with antibodies against p110α, p110β, and p110δ followed by immunocomplex lipid kinase assay as described above.
Expression of Different Mutant Constructs of PI 3-Kinase
Class IA PI 3-kinase consists of a catalytic subunit (p110) of 110 kDa and a regulatory subunit (p85) of 85 kDa. In the dominant-negative form of p85α, 35 amino acids in the inter-Src homology 2 region from residues 479 to 513 of wild type p85α, important for binding the p110α/β subunit of PI 3-kinase, were deleted, and two other amino acids (Ser-Arg) were inserted in this deleted position (22). In the constitutively active mutant of p110α/β (p110*), the inter-Src homology 2 domain of p85 is ligated to the NH2 terminus of p110, whereas in the kinase-deficient mutant of p110α/β (p110-kd), the ATP-binding site was mutated (23). Microglial cells plated in 12-well plates were transfected with 0.2–0.25 μg of different plasmids using Lipofectamine-Plus (Invitrogen) following the manufacturer's protocol as described previously (21).
Electrophoretic Mobility Shift Assay (EMSA) of NF-κB and CREB
Nuclear extracts were prepared, and EMSA was carried out as described previously (20, 24, 25) using IRDyeTM infrared dye end-labeled oligonucleotides containing the consensus binding sequence for NF-κB and CREB (LI-COR Biosciences, Lincoln, NE). Six micrograms of nuclear extract were incubated with binding buffer and with IR-labeled probe for 20 min. Subsequently, samples were separated on a 6% polyacrylamide gel in 0.25× TBE buffer (Tris borate/EDTA) and analyzed by Odyssey Infrared Imaging System (LI-COR Biosciences).
Assay of Transcriptional Activities of NF-κB and CREB
Transcriptional activity was assayed as described previously (20, 21, 24). Briefly, cells plated at 50–60% confluence in 12-well plates were transfected with 0.25 μg of either pBIIX-Luc (a NF-κB-dependent reporter construct) or pCRE-Luc (a CREB-dependent reporter construct) and 12.5 ng of pRL-TK (a plasmid encoding Renilla luciferase, used as transfection efficiency control; Promega, Madison, WI) using Lipofectamine Plus (Invitrogen). After 24 h of transfection, cells were stimulated with MPP+ for an additional 6 h, and firefly and Renilla luciferase activities were recorded in a TD-20/20 Luminometer (Turner Designs, Sunnyvale, CA) by analyzing total cell extract according to standard instructions provided in the Dual-Luciferase kit (Promega). Relative luciferase activity of cell extracts was typically represented as (firefly luciferase value/Renilla luciferase value) × 10−3.
Immunoblot Analysis
Immunoblot analysis for IκBα, p65, p50, and iNOS was carried out as described earlier (24, 26). Briefly, cell homogenates were electrophoresed, proteins were transferred onto a nitrocellulose membrane, and bands were visualized with an Odyssey infrared scanner after immunolabeling with respective primary antibodies followed by infrared fluorophore-tagged secondary antibody (Invitrogen).
Densitometric Analysis
Protein blots were analyzed using ImageJ (National Institutes of Health, Bethesda, MD), and bands were normalized to their respective β-actin loading controls. Data are representative of the average fold change with respect to control for three independent experiments.
Real Time PCR Analysis
DNase-digested RNA was analyzed by real time PCR in the ABI-Prism7700 sequence detection system (Applied Biosystems, Foster City, CA) as described earlier (20, 21, 27) using TaqMan universal master mix and optimized concentrations of FAM-labeled probes and primers. Data were processed using the ABI Sequence Detection System 1.6 software.
Statistics
All values are expressed as means ± S.E. One-way analysis of variance was performed while analyzing dose-dependent effect of RNS60 on mRNA expression of IκBα in activated microglial cells.
Immunofluorescence Analysis
It was performed as described earlier (9, 17, 24). Briefly, coverslips containing 100–200 cells/mm2 were fixed with 4% paraformaldehyde followed by treatment with cold ethanol and two rinses in PBS. Samples were blocked with 3% BSA in PBS/Tween 20 (PBST) for 30 min and incubated in PBST containing 1% BSA and goat anti-CD11b (1:50), rabbit anti-iNOS (1:200), or goat anti-GFAP (1:50). After three washes in PBST (15 min each), slides were further incubated with Cy2 (Jackson ImmunoResearch). For negative controls, a set of culture slides was incubated under similar conditions without the primary antibodies. The samples were mounted and observed under a Bio-Rad MRC1024ES confocal laser-scanning microscope.
RESULTS
RNS60 Is Chemically Identical to Control Saline Solutions
To detect any chemical contaminants, RNS60 was carefully compared with two control solutions as follows: (a) NS, unprocessed normal saline from the same manufacturing batch that contacted the same device surfaces and was bottled in the same way as RNS60; (b) PNS60, normal saline from the same manufacturing batch that was prepared inside the same device and bottled in the same way as RNS60, but not processed with the TCP flow. Oxygen pressure was applied above the solution until PNS60 contained a level of oxygen comparable with RNS60 (50–60 ppm).
Careful ICP-MS testing for 26 metals and total organic carbon analysis revealed no differences between RNS60 and the control solutions within detection limits (supplemental Table 1). Similarly, triplicate MS-TOF analysis with and without chromatographic pretreatment, using either negative or positive ion modes, did not reveal chemical composition differences in the 100–1000 m/e range (only a part of it has been presented in supplemental Fig. 1). In all cases, samples for RNS60 and saline source solution were found to be identical within ∼1 ppb.
RNS60 Has a Nanobubble Composition Different from That of Control Saline
Small bubble formation occurs through hydrodynamic cavitation (28), and nanobubbles are produced within various types of mixing devices when sharp pressure variations are generated (29). Measurements of nanobubble-based structures in bulk solution are controversial (30), although recent molecular dynamics studies suggest that bulk nanobubbles exist and are stable for much longer periods of time than previously expected (31). These relationships are tied to size and charge and the distribution of the bubble populations within the solution. Observations on surfaces using AFM, however, are increasingly accepted (32) and may represent characteristics of the bubbles within the solution (33). Therefore, we studied the effects of RNS60 on nanobubble nucleation using AFM. We acquired AFM images of polystyrene-coated silicon wafers immersed in RNS60, PNS60, NS, and distilled water. As evident from AFM images, deionized water (Fig. 1A), NS (Fig. 1B), PNS60 (Fig. 1C), and RNS60 (Fig. 1D) displayed readily nucleated nanobubbles. Compared with control solutions (NS and PNS60), the AFM images for RNS60 reveal approximately half the number of nanobubbles, with an average diameter that increased from 26 to 33 nm. This same relative pattern of nanobubble number and size was observed when positive potentials were applied to AFM surfaces with the same control solutions, suggesting that charge is involved. These data suggest that RNS60 has a different bulk nanobubble composition than its control solutions.
FIGURE 1.

RNS60-derived nanobubbles on polystyrene film show distinct differences from control solutions. Nanobubbles were formed on polystyrene film in distilled water (A), isotonic saline NS (B), PNS60 (C), and RNS60 (D). AFM images obtained after 15 scanning cycles are essentially identical to images generated after 2 h of scanning (data not shown), demonstrating that there are no time- and scan load-dependent artifacts.
RNS60 Attenuates the Expression of Proinflammatory Molecules in Activated Mouse Microglia and Astroglia
Activated microglia and astroglia are known to produce excessive amounts of NO and proinflammatory cytokines, which have the potential of damaging neurons in neurodegenerative disorders (2, 4). LPS is a prototype inducer of various proinflammatory molecules in different cell types, including mouse microglia. Therefore, mouse BV-2 microglial cells, preincubated with different doses of RNS60 for 2 h, were stimulated with LPS under serum-free conditions. Although at lower concentrations (1 and 2% v/v), RNS60 was not effective in inhibiting the production of NO (data not shown), and at higher concentrations, RNS60 markedly suppressed LPS-induced production of NO in microglial cells as evident from the estimation of nitrite (Fig. 2A). Conversely, normal saline (NS), TCP-modified saline without excess oxygen (RNS10.3), and saline containing excess oxygen in the absence of TCP modification (PNS60) had no such inhibitory effect on the production of NO (Fig. 2A). To understand the mechanism further, we investigated the effect of RNS60 on protein and mRNA levels of iNOS in microglial cells. It is clearly evident from Western blot in Fig. 2B and real time PCR in Fig. 2C that RNS60, but not NS, RNS10.3, and PNS60, inhibited LPS-induced protein and mRNA expression of iNOS in microglial cells.
FIGURE 2.
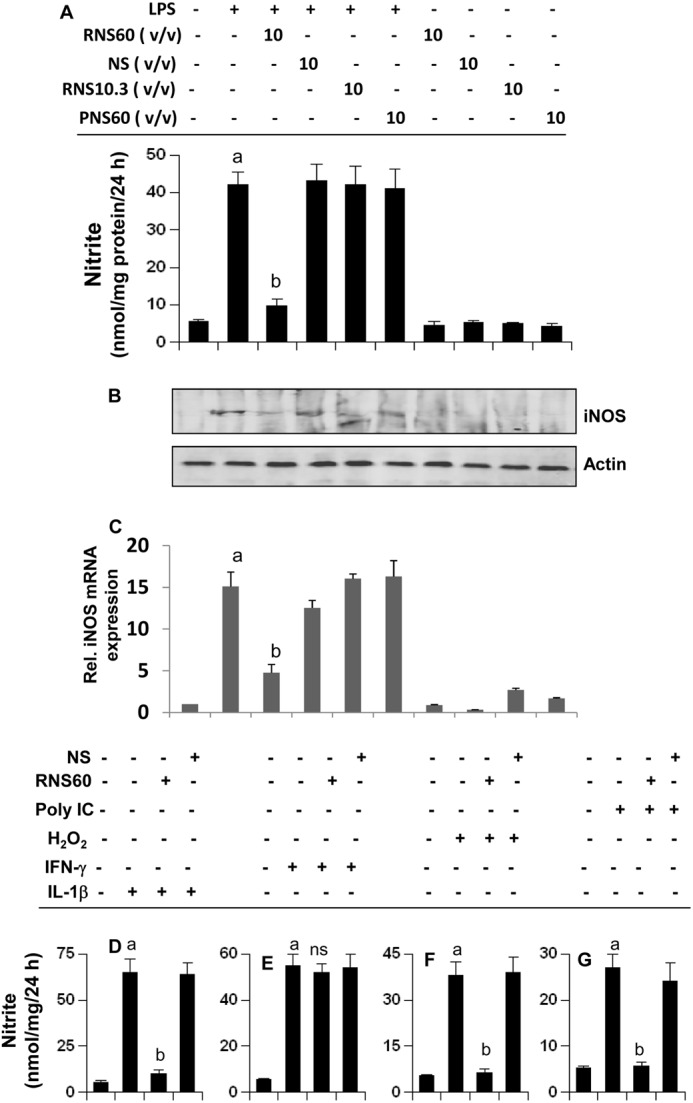
RNS60 inhibits the production of NO and the expression of iNOS in activated mouse BV-2 microglial cells. A, microglial cells preincubated with RNS60, NS, RNS10.3, or PNS60 in serum-free media for 1 h were stimulated with LPS (1 μg/ml). After 24 h of stimulation, concentration of nitrite was measured in supernatant by “Griess” reagent (A), and the level of iNOS protein was monitored in cells by Western blot (B). After 6 h of stimulation, total RNA was isolated, and the mRNA expression of iNOS was analyzed by quantitative real time PCR (C). Data are mean ± S.D. of three different experiments. a, p < 0.001 versus control; b, p < 0.001 versus LPS. Cells preincubated with 10% (v/v) RNS60 or NS for 1 h were stimulated with 50 ng/ml IL-1β (D), 25 milliunits/ml IFN-γ (E), 200 μm H2O2 (F), and 100 μg/ml poly(IC) (G). After 24 h of stimulation, concentration of nitrite was measured in supernatants. Data are mean ± S.D. of three different experiments. a, p < 0.001 versus control; a, p < 0.001 versus stimuli.
Next, to investigate whether RNS60 suppresses the expression of iNOS in primary cells, primary mouse microglia preincubated with RNS60 or NS for 2 h were stimulated with LPS. As expected, LPS induced the expression of iNOS protein in primary microglia. RNS60, but not NS, markedly suppressed LPS-induced expression of iNOS in microglia (Fig. 3A). In addition to microglia, there are other glial cells in the CNS. Earlier we have demonstrated that IL-1β is a potent inducer of iNOS in astroglia (34). Therefore, we examined the effect of RNS60 on the expression of iNOS in IL-1β-stimulated primary astroglia. Similar to its effect on microglia, RNS60 markedly inhibited IL-1β-stimulated iNOS in primary mouse astroglia, although NS had no such effect (Fig. 3B).
FIGURE 3.

RNS60 inhibits the expression of iNOS protein in primary mouse microglia and astroglia. A, primary mouse microglia preincubated with RNS60 or NS (10% v/v) in serum-free media for 1 h were stimulated with 1 μg/ml LPS. After 18 h of stimulation, the level of iNOS was monitored by double-label immunofluorescence using CD11b as a microglial marker. B, primary mouse astroglia preincubated with RNS60 or NS (10% v/v) in serum-free media for 1 h were stimulated with 20 ng/ml IL-1β. After 18 h of stimulation, the level of iNOS was monitored by double-label immunofluorescence using GFAP as an astroglial marker. Results represent three independent experiments.
RNS60 Inhibits IL-1β-, H2O2-, and Double-stranded RNA (Poly(IC))-induced Production of NO in Microglial Cells
Activated microglia are considered to play an important role in different neurodegenerative disorders (2, 35). Because RNS60 inhibited LPS-induced production of NO and the expression of iNOS in microglia, we were prompted to investigate whether RNS60 was also capable of suppressing the production of NO in microglial cells stimulated with etiological reagents of various neurological disorders. Oxidative stress is a hallmark of many human disorders, including neurodegenerative disorders. BV-2 microglial cells were challenged with H2O2 (for oxidative stress), double-stranded RNA in the form of poly(IC) (one of the etiological reagents for viral encephalopathy), and IL-1β (one of the etiological reagents for different neurodegenerative disorders). It is clearly evident from Fig. 2, D, F, and G, that IL-1β, H2O2, and poly(IC) induced the production of NO in microglial cells. However, RNS60 markedly knocked down IL-1β-, poly(IC)-, and H2O2-induced production of NO in microglial cells (Fig. 2, D, F, and G). However, NS had no such inhibitory effect (Fig. 2, D, F, and G), suggesting the specificity of the effect. In contrast, under the same experimental condition, RNS60 had no effect on IFN-γ-induced microglial production of NO (Fig. 2E), suggesting that IFN-γ induces NO production in microglia via a RNS60-insensitive pathway.
RNS60 Inhibits Microglial Activation of NF-κB
LPS and other inflammatory stimuli are known to induce iNOS expression via activation of NF-κB (17, 36–39). Because RNS60 attenuated the expression of iNOS in glial cells, we examined its effect on NF-κB activation. Activation of NF-κB was monitored by both DNA binding and transcriptional activities of NF-κB. Both IL-1β (Fig. 4, A and C) and LPS (Fig. 4, B and D) induced the DNA binding (Fig. 4, A and B) and transcriptional (Fig. 4, C and D) activities of NF-κB in BV-2 microglial cells. However, RNS60, but not NS, inhibited IL-1β- and LPS-induced DNA binding and transcriptional activities of NF-κB in microglial cells (Fig. 4, A–D). These results suggest that RNS60 attenuates the expression of iNOS by suppressing the activation of NF-κB.
FIGURE 4.

RNS60 attenuates activation of NF-κB and up-regulates IκBα in mouse microglia. A, BV-2 microglial cells were treated with RNS60 or NS for 1 h followed by stimulation with 20 ng/ml IL-1β (A) or 1 μg/ml LPS (B) under serum-free conditions. After 30 min of stimulation, the DNA binding activity of NF-κB was monitored. BV-2 cells plated in 12-well plates were co-transfected with 0.25 μg of PBIIX-Luc (an NF-κB-dependent reporter construct) and 12.5 ng of pRL-TK. Twenty four hours after transfection, cells received different concentrations of RNS60 or NS. After 2 h of incubation, cells were stimulated with IL-1β (C) or LPS (D) for 4 h under serum-free conditions. Firefly and Renilla luciferase activities were obtained by analyzing the total cell extract. Results are mean ± S.D. of three different experiments. Rel Luc, relative luciferase. a, p < 0.001 versus control; b, p < 0.001 versus IL-1β; c, p < 0.001 versus LPS. BV-2 cells were treated with RNS60 or NS (10% v/v) under serum-free conditions for different minute intervals followed by monitoring protein levels of IκBα and p65 by Western blot (E) and mRNA levels of IκBα by real time PCR (F). a, p < 0.001 versus control. G, BV-2 cells were treated with different concentrations of RNS60 for 1 h followed by monitoring protein levels of IκBα, p65, and p50 by Western blot. Actin was run as a loading control. H, bands were scanned and results presented as protein expression relative to actin. a, p < 0.001 versus control. I, primary mouse microglia were treated with RNS60 or NS for 1 h followed by monitoring IκBα protein by double-label immunofluorescence using CD11b as a marker for microglia. J, BV-2 cells were treated with 10% (v/v) RNS60, RNS10.3, or PNS60 for 1 h followed by monitoring the protein level of IκBα by Western blot. K, bands were scanned, and results are presented as protein expression relative to actin. a, p < 0.001 versus control.
RNS60 Up-regulates IκBα in Glial Cells
Although there are multiple mechanisms by which an anti-inflammatory molecule may suppress the activation of NF-κB, in resting cells the classical p65:p50 heterodimer is arrested in the cytoplasm as an inactive complex by IκBα (15). It has also been demonstrated that newly synthesized IκBα proteins accumulate in the cytoplasm as well as in the nucleus, where it reduces NF-κB binding (40). Therefore, we hypothesized that RNS60 might up-regulate IκBα to suppress the activation of classical NF-κB heterodimer. Indeed, RNS60, but not NS, markedly up-regulated IκBα protein in microglial cells in a time-dependent fashion (Fig. 4E). Significant increase in IκBα protein was observed within only 30 min of RNS60 treatment (Fig. 4E). This effect was specific as RNS60 did not up-regulate the expression of RelA p65 (Fig. 4E). Real time PCR data also show marked time-dependent increase in IκBα mRNA by RNS60 but not NS (Fig. 4F). Similarly, RNS60 also dose-dependently increased the expression of IκBα, but not of p65 or p50 (Fig. 4, G and H). In fact, we observed down-regulation of p65 and p50 by RNS60 (Fig. 4, E and G). To confirm this IκBα up-regulation, we also performed double-label immunofluorescence. As evident from Fig. 4I, RNS60, but not NS, markedly increased the expression of IκBα in CD11b-positive primary microglia. To confirm that the effect of RNS60 on IκBα was specific, we used RNS10.3 and PNS60. In contrast to RNS60, RNS10.3 and PNS60 remained unable to up-regulate IκBα in microglia (Fig. 4, J and K).
To understand whether this IκBα up-regulation was cell type-specific, we included primary astroglia. RNS60, but not NS, also up-regulated IκBα in GFAP-positive astroglia in a dose-dependent manner (Fig. 5, A–C), demonstrating that the effect was not specific for microglia only.
FIGURE 5.

RNS60 up-regulates IκBα in mouse astroglia. A, primary mouse astroglia were treated with different concentrations of RNS60 or NS for 1 h followed by monitoring the level of IκBα protein by Western blot. Rel., relative. B, bands were scanned, and results are presented as protein expression relative to actin. a, p < 0.001 versus control. C, astroglia were treated with RNS60 or NS for 1 h followed by monitoring IκBα protein by double-label immunofluorescence using GFAP as a marker for astroglia. Results represent three independent experiments.
RNS60 Activates PI 3-Kinase in Microglia
Next, we investigated mechanisms by which RNS60 may transduce signals for the up-regulation of IκBα. As activation of PI 3-kinase, a dual protein, and lipid kinase occurs in close association with the cell membrane, we investigated the effect of RNS60 on the activation of PI 3-kinase. Class IA PI 3-kinase, which is regulated by receptor tyrosine kinases, consists of a heterodimer of a regulatory 85-kDa subunit and a catalytic 110-kDa subunit (p85:p110α/β/δ). Class IB PI 3-kinase, however, consists of a dimer of a 101-kDa regulatory subunit and a p110γ catalytic subunit (p101/p110γ). In addition to p101, p84 has also been identified as a regulatory subunit of p110γ (41). Therefore, we first examined the presence of different subunits of PI 3-kinase in BV-2 microglial cells. As evident from Fig. 6A, p110α, p110β, p110δ, p110γ, and the new regulatory subunit p84 are present in BV-2 cells. Earlier, we also demonstrated the presence of p85α in BV-2 cells (21). When we monitored lipid kinase activity, we observed that RNS60, but not NS, markedly induced the activation of p85α-associated PI 3-kinase within 15 min of treatment (Fig. 6, B and C), suggesting that RNS60 induces the activation of class IA PI 3-kinase. Phosphatidylinositol (3,4,5)-triphosphate (PIP3) is the product of class I PI 3-kinase. Therefore, to confirm the activation of class I PI 3-kinase from another angle, we monitored PIP3 levels in RNS60-treated microglia by immunofluorescence. Although RNS60 was unable to increase the level of PIP3 within 5 min of stimulation, marked increase in PIP3 was observed at 15 min of stimulation (Fig. 6D). However, consistent with that observed in lipid kinase activity, NS remained unable to up-regulate the level of PIP3 (Fig. 6D). RNS60 did not, however, activate PI 3-kinase activity associated with either p101 (Fig. 6E) or p84 (Fig. 6F), suggesting that it is unable to activate class IB PI 3-kinase. Next, we tried to identify the catalytic subunit of class IA PI 3-kinase that is involved in this lipid kinase activity. By immuno-complex lipid kinase assay, we observed that RNS60-induced activation of PI 3-kinase was due to p110α (Fig. 6G) and p110β (Fig. 6H) but not p110δ (Fig. 6I).
FIGURE 6.

Activation of PI 3-kinase by RNS60 in mouse microglial cells. A, presence of different subunits of PI 3-kinase (p110α, p110β, p110δ, p110γ, and p84) was examined in BV-2 cells by Western blot. BV-2 microglial cells were treated with either 10% (v/v) RNS60 or NS in serum-free DMEM/F-12. After 15 min of treatment, cells were lysed, immunoprecipitated with antibodies against either p85α (B and C), p101 (E), or p84 (F), and the lipid kinase activity of immunoprecipitated PI 3-kinase was assayed. Lipids were detected by exposure to film at −70 °C (B, E, and F) and quantitated by densitometry (C). Results represent mean ± S.D. of three separate experiments. a, p < 0.001 versus control. D, cells were treated with RNS60 or NS and at different time points, and the level of PIP3 was monitored by immunofluorescence. DAPI was used to visualize nucleus. Cells were treated with RNS60 for 15 min, and cell lysates were immunoprecipitated with antibodies against p110α (G), p110β (H), or p110δ (I) followed by assaying PI 3-kinase activity in immunoprecipitates. Results represent three independent experiments.
RNS60 Activates Akt in Microglia
PI 3-kinases have been linked to a diverse group of cellular functions, and many of these functions are related to its ability to activate protein kinase B (PKB, also known as Akt). Therefore, we examined whether RNS60 could activate Akt in microglial cells. RNS60, but not NS, markedly induced the activation of Akt in microglia as evident from immunoblot (Fig. 7, A and B) and immunofluorescence analysis (Fig. 7C) of phospho-Akt, without increasing the level of total Akt (Fig. 7D). Although Akt is downstream of PI 3-kinase, recently it has been shown that the serine/threonine kinase IKBKE activates Akt through a PI 3-kinase-independent pathway (42). Therefore, we examined whether RNS60 required PI 3-kinase for the activation of Akt. Because RNS60 induced the activation of p110α and p110β, we used chemical inhibitors that are specific for these catalytic subunits. Although GDC-0941 blocks the activity of p110α, TGX-221 specifically inhibits p110β (43, 44). Strong inhibition of RNS60-induced phosphorylation of Akt by both GDC-0941 and TGX-221 suggests that RNS60 employs class IA PI 3-kinase p110α/β for the activation of Akt in microglial cells.
FIGURE 7.

RNS60 induces the activation of Akt in mouse microglial cells via PI 3-kinase. A, cells were stimulated with RNS60 or NS for different time periods followed by monitoring the levels of phospho-Akt and total Akt by Western blot. B, bands were scanned, and results are presented as phospho-Akt/total Akt. Results represent means ± S.D. of three separate experiments. a, p < 0.001 versus control. Levels of phospho-Akt (C) and total Akt (D) were also monitored by double-label immunofluorescence (C and D). E, cells preincubated with TGX-221 and GDC-0941 for 30 min were stimulated with RNS60 followed by monitoring the levels of phospho-Akt and total Akt by Western blot. F, bands were scanned and results presented as phospho-Akt/total Akt. Rel., relative. a, p < 0.001 versus control.
RNS60 Requires the PI 3-Kinase-Akt Signaling Pathway to Suppress the Expression of iNOS, Attenuate the Activation of NF-κB, and Up-regulate IκBα
Because RNS60 induced the activation of PI 3-kinase-Akt signaling pathway, we investigated the possibility that RNS60 was utilizing this pathway to exhibit its anti-inflammatory activity. We examined the effect of LY294002, an inhibitor of PI 3-kinase, and AktI, an inhibitor of Akt, on the RNS60-mediated anti-inflammatory effect. Both LY294002 and AktI abrogated the inhibitory effect of RNS60 on the induction of NO production (Fig. 8A) and the expression of iNOS protein (Fig. 8, B and C) in LPS-stimulated microglial cells. Similarly, LY294002 and AktI also abolished the RNS60-mediated up-regulation of IκBα protein and mRNA (Fig. 8, D and E). To identify the catalytic subunit of PI 3-kinase involved in RNS60-mediated up-regulation of IκBα, we used GDC-0941 (an inhibitor of p110α) (43), TGX-221 (an inhibitor of p110β) (44), IC-87114 (an inhibitor of p110δ) (45), and AS-605240 (an inhibitor of p110γ) (46). Suppression of RNS60-mediated up-regulation of IκBα by GDC-0941 (supplemental Fig. 2, A and B) and TGX-221 (supplemental Fig. 2, C and D), but not IC-87114 (supplemental Fig. 2, E and F) and AS-605240 (supplemental Fig. 2, G and H), suggests that p110α and p110β, but not p110δ and p110γ, are involved in RNS60-mediated increase in RNS60. This has been supported by siRNA knockdown studies as well. As evident from supplemental Fig. 3A, p110α siRNA decreased the level of p110α, but not p110δ. Similarly, p110δ siRNA reduced the level of p110δ but not p110α (supplemental Fig. 3A). However, siRNA against p110α, but not p110δ, suppressed the RNS60-induced up-regulation of IκBα in BV-2 microglial cells (supplemental Fig. 3, B and C).
FIGURE 8.

Inhibitors of PI 3-kinase-Akt pathway abrogate the anti-inflammatory effect of RNS60. Mouse BV-2 microglial cells preincubated with LY294002 (LY) (10 μm) or AktI (10 μm) for 30 min were treated with RNS60 (10% v/v) under serum-free conditions. After 1 h of treatment, cells were stimulated with 1 μg/ml LPS and 24 h after stimulation, and nitrite levels were assayed in supernatants by Griess reagent (A), and the level of iNOS protein was monitored by Western blot (B). Bands were scanned, and results are presented as protein expression relative to actin (C). Results are mean ± S.D. of three different experiments. a, p < 0.001 versus control; b, p < 0.001 versus LPS; c, p < 0.001 versus (LPS + RNS60). Cells preincubated with LY294002 or AktI for 30 min were treated with RNS60 (10% v/v) under serum-free conditions. Rel., relative. After 1 h of treatment, the level of IκBα was monitored by Western blot (D) and real time PCR (E). Results are mean ± S.D. of three different experiments. a, p < 0.001 versus control; b, p < 0.001 versus RNS60. F, cells were co-transfected with 0.25 μg of PBIIX-Luc and 12.5 ng of pRL-TK. Twenty four hours after transfection, cells were incubated with LY294002 or AktI for 30 min followed by treatment with RNS60 (10% v/v) under serum-free conditions. After 2 h of treatment, cells were stimulated with LPS for 4 h followed by assay of firefly and Renilla luciferase activities. Results are mean ± S.D. of three different experiments. a, p < 0.001 versus control; b, p < 0.01 versus LPS; c, p < 0.01 versus (LPS + RNS60).
Next we investigated whether PI 3-kinase-Akt pathway was also playing a role in RNS60-mediated inhibition of NF-κB activation. Abrogation of the inhibitory effect of RNS60 on NF-κB activation in LPS-stimulated microglial cells by LY and AktI (Fig. 8F) suggests that RNS60 affects NF-κB activity via PI 3-kinase-Akt pathway.
To confirm this observation further, we transfected BV-2 microglial cells with a dominant-negative mutant of p85α. As evidenced by Western blot analysis (Fig. 9A) and real time PCR (Fig. 9B), RNS60 induced the expression of IκBα protein and mRNA in empty vector-transfected, but not p85α-transfected, microglial cells. These results suggest that RNS60 up-regulates IκBα and exerts its anti-inflammatory effect in microglial cells via the PI 3-kinase-Akt signaling pathway.
FIGURE 9.
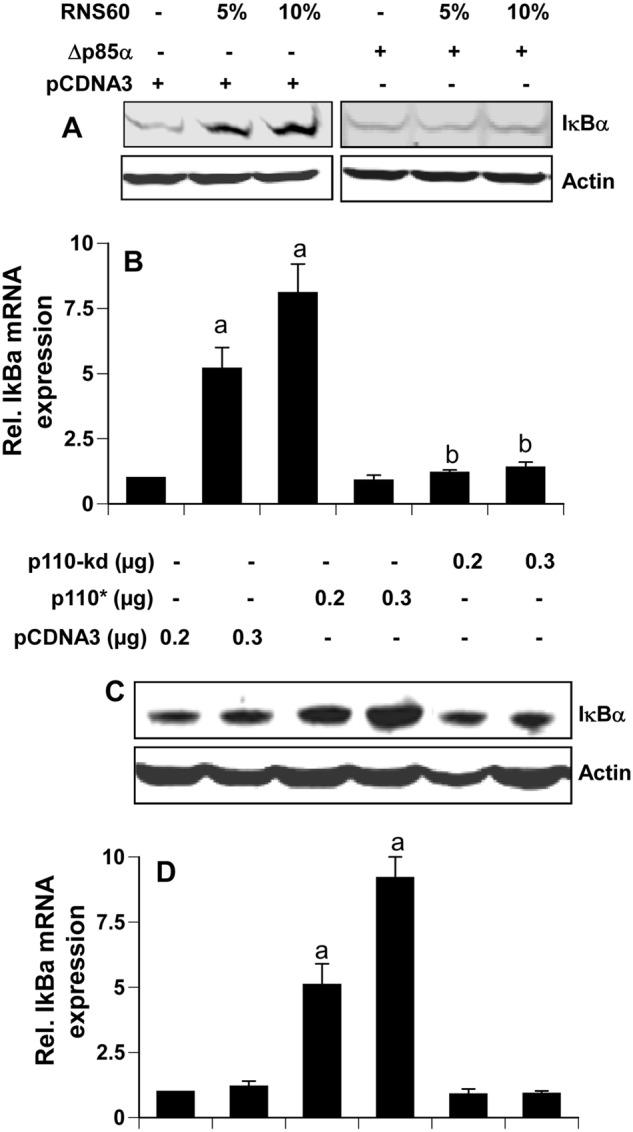
Expression of Δp85α (a dominant-negative mutant of p85α) and p110* (a catalytically active mutant of p110α/β) modulates the expression of IκBα. Mouse BV-2 microglial cells were transfected with 0.25 μg of either Δp85α or an empty vector. Twenty four hours after transfection, cells were incubated with RNS60 for 2 h followed by monitoring the expression of IκBα by Western blot (A) and real time PCR (B). Rel., relative. Cells were transfected with different amounts of p110*, p110-kd (a kinase-dead mutant) or an empty vector. Twenty four hours after transfection, cells were incubated in serum-free media for 2 h followed by monitoring the expression of IκBα by Western blot (C) and real time PCR (D). Results represent mean ± S.D. of three separate experiments. a, p < 0.001 versus empty vector; b, p < 0.001 versus empty vector-RNS60.
Activation of PI 3-Kinase Is Sufficient for the Up-regulation of IκBα in Microglia
In addition to activating PI 3-kinase, RNS60 may activate other signaling pathways, which may cooperate with the PI 3-kinase-Akt pathway for the up-regulation of IκBα. Therefore, we next investigated if the activation of PI 3-kinase alone by a constitutively active mutant of p110α/β (p110*) was sufficient to induce the expression of IκBα in microglia. It is evident from Fig. 9, C and D, that overexpression of p110* was capable of increasing mRNA and protein expression of IκBα. However, p110-kd (a kinase-dead mutant) had no effect on the expression of IκBα (Fig. 9, C and D). These results clearly suggest that activation of PI 3-kinase alone is sufficient to induce microglial expression of IκBα.
How Does the PI 3-Kinase-Akt Signaling Pathway Couple to the Up-regulation of IκBα in Microglia?
Upon analysis of the IκBα promoter using MatInspector, we have observed binding sites for many transcription factors, including NF-κB and CREB. Accordingly, several studies have delineated a role of NF-κB activation in the transcription of IκBα (14, 40). However, in this case, RNS60 inhibits the activation of NF-κB (Fig. 2, A and B), and so it is not likely to utilize this transcription factor for inducing IκBα up-regulation. Therefore, we were prompted to investigate if RNS60 was using CREB for the up-regulation of IκBα. RNS60, but not NS, induced the phosphorylation of CREB (Fig. 10, A and B). Consistently, RNS60 also induced the DNA binding activity (Fig. 10C) and transcriptional activity (Fig. 10D) of CREB in microglial cells, suggesting that RNS60 alone is sufficient for the activation of CREB. Furthermore, both LY294002 and AktI markedly suppressed RNS60-induced phosphorylation of CREB (Fig. 10E) and the transcriptional activation of CREB (Fig. 10F), suggesting the involvement of the PI 3-kinase-Akt pathway for the activation of CREB. Next, to examine if RNS60 required CREB for the up-regulation of IκBα in microglia, we utilized siRNA to silence CREB. CREB siRNA, but not control siRNA, decreased the expression of CREB protein (Fig. 11A) and abrogated RNS60-mediated up-regulation of IκBα mRNA (Fig. 11B) and protein (Fig. 11C). Taken together, these results clearly demonstrate that RNS60 induces the activation of CREB via PI 3-kinase-Akt signaling pathway and that CREB is required for increased transcription of IκBα.
FIGURE 10.
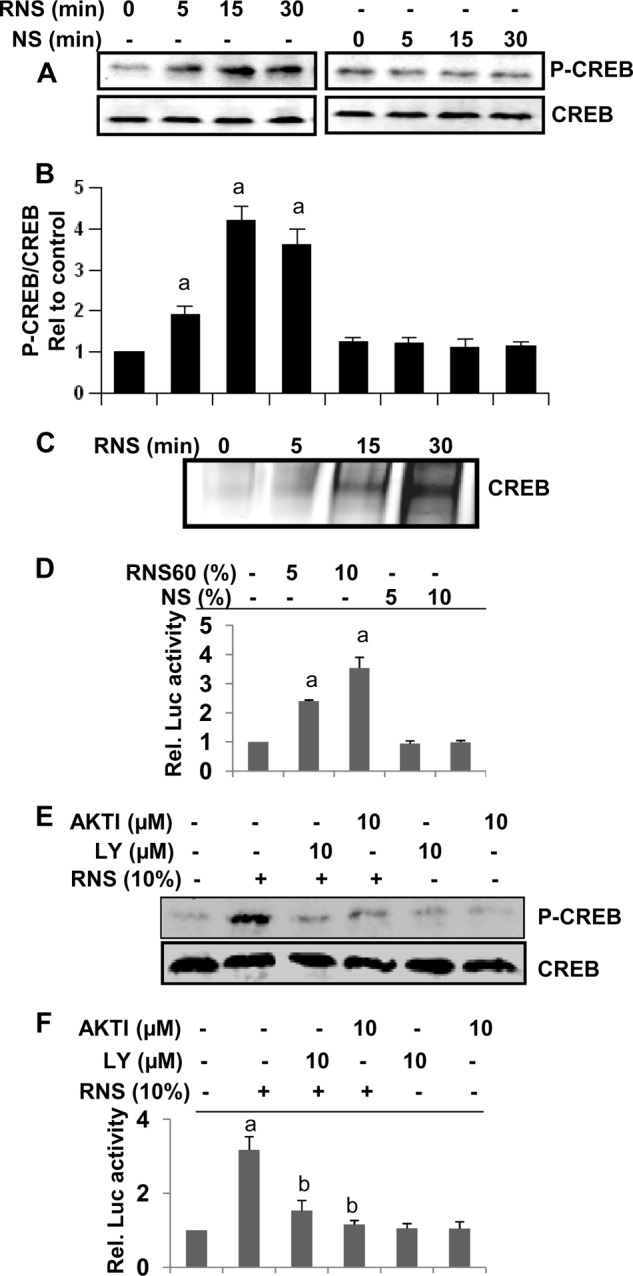
RNS60 induces the activation of CREB in mouse microglial cells via PI 3-kinase-Akt pathway. A, BV-2 microglial cells were stimulated with RNS60 and NS for different time periods followed by monitoring the levels of phospho-CREB and total CREB by Western blot. B, bands were scanned and results presented as phospho-CREB/total CREB. Results are mean ± S.D. of three different experiments. a, p < 0.001 versus control. C, cells were stimulated with RNS60 for different time periods followed by monitoring the DNA binding activity of CREB by EMSA. D, cells plated in 12-well plates were co-transfected with 0.25 μg of pCRE-Luc (an CREB-dependent reporter construct) and 12.5 ng of pRL-TK. Twenty four hours after transfection, cells received different concentrations of RNS60 and NS. After 2 h of incubation, firefly and Renilla luciferase activities were obtained by analyzing the total cell extract. a, p < 0.001 versus control. Rel., relative. E, cells preincubated with LY294002 (LY) (10 μm) or AktI (10 μm) for 30 min were treated with RNS60 (10% v/v) under serum-free conditions. After 30 min of treatment, the levels of phospho-CREB and total CREB were monitored by Western blot. F, effect of LY294002 and AktI was tested on the transcriptional activity of CREB using pCRE-Luc as described above. Results are mean ± S.D. of three different experiments. a, p < 0.001 versus control; b, p < 0.001 versus RNS60.
FIGURE 11.

RNS60 up-regulates the expression of IκBα in mouse microglial cells via CREB. A, BV-2 microglial cells plated in 12-well plates were transfected with 0.25 μg of either CREB siRNA or control (Cont) siRNA, and after 24 h of transfection, the level of CREB protein was monitored by Western blot. Actin was run as a control. Bands were scanned, and results are presented as protein expression relative to actin (B). Rel., relative. Results are mean ± S.D. of three different experiments. a, p < 0.001 versus control siRNA. Cells were transfected with 0.25 μg of either CREB siRNA or control siRNA, and after 24 h of transfection, cells were treated with different concentrations of RNS60. After 1 h of treatment, the expression of IκBα was monitored by real time PCR (C) and Western blot (D). Bands were scanned, and results are presented as protein expression relative to actin (E). Results represent mean ± S.D. of three separate experiments. a, p < 0.001 versus no RNS60-control siRNA; b, p < 0.001 versus RNS60–30-min control siRNA; c, p < 0.001 versus RNS60–60-min control siRNA.
DISCUSSION
Activation of glial cells (microglia and astroglia) has been implicated in the pathogenesis of a variety of neurodegenerative diseases, including Alzheimer disease, Parkinson disease, Creutzfeldt-Jacob disease, HIV-associated dementia, stroke, and multiple sclerosis (2, 9, 35). It has been found that activated microglia and astroglia accumulate at sites of injury or plaques in neurodegenerative CNS (35, 47). Although activated microglia scavenge dead cells from the CNS and secrete different neurotrophic factors for neuronal survival and activated astrocytes may have important beneficial effects in the recovery of injured CNS by actively monitoring and controlling the extracellular water, pH, and ion homeostasis, it is believed that severe activation of these brain cells causes various autoimmune responses leading to neuronal death and brain injury (2, 35, 47). During activation, microglia and astroglia express various genes related to inflammation, such as proinflammatory cytokines, proinflammatory enzymes, and proinflammatory adhesion molecules. Therefore, characterization of signaling pathways required for the activation of glial cells is an active area of investigation because compounds capable of antagonizing such signaling steps may have a therapeutic effect in neurodegenerative disorders.
Although other drugs and approaches exist to combat glial inflammation, here we introduce an elegantly simple, yet uniquely innovative, saline-based agent to suppress glial activation. Nanobubbles have been suggested for potential use in biomedical imaging and drug delivery (48, 49), but they have not demonstrated direct biological effects. Nanoparticles are known to interact with cells based upon their charge and size, and we propose that nanobubble-based structures within RNS60 act in a similar way. These stable and charged entities may possess the necessary energetics to affect the membrane and its voltage-sensitive elements. Because RNS60 does not contain any active pharmaceutical ingredient, these proposed “charge stabilized nanostructures” would be the effector of activity at the cell membrane level.
Several lines of evidence presented in this study clearly support the conclusion that RNS60 attenuates the activation of glial cells. Our conclusion is based on the following observations. First, LPS, a prototype inducer of inflammation in many cell types including microglia, and other proinflammatory molecules induced the expression of iNOS in mouse microglia. However, RNS60 attenuated the expression of iNOS in activated microglia without altering cell survival, suggesting that this attenuation is not due to any cell death. This inhibition was also specific as other saline preparations like NS (normal saline of the same batch), RNS10.3 (TCP-modified saline without excess oxygen), and PNS60 (saline containing oxygen in the absence of TCP modification) had no effect. Second, we extended the study beyond LPS and examined whether RNS60 was capable of suppressing microglial expression of iNOS induced by other neurotoxins. It is important that RNS60, but not NS, attenuated microglial production of NO induced by various neurotoxins such as H2O2, poly(IC), and IL-1β. Third, IL-1β is critical for inducing iNOS and activating astrocytes (34). RNS60, but not NS, also suppressed the expression of iNOS in primary mouse astrocytes. Because excess NO has been implicated in the pathogenesis of demyelinating and neurodegenerative diseases, our results provide a potentially important mechanism whereby RNS60 may ameliorate neural injury.
The signaling events required for the transcription of iNOS and proinflammatory cytokines are becoming clear. Although many transcription factors such as NF-κB, C/EBPβ, AP-1, STAT, IRF-1, etc. play a role, activation of NF-κB seems essential for the transcription of most of the proinflammatory molecules (17, 36–39). Therefore, for a drug to exhibit anti-inflammatory effects, it is almost mandatory to attenuate the activation of NF-κB. Treatment of microglia with RNS60 led to the inhibition of NF-κB activation at both DNA binding and transcriptional levels. Accordingly, we have seen that RNS60 suppresses the production of NO in response to LPS, IL-1β, H2O2, and poly(IC) because these stimuli require NF-κB for the expression of iNOS (21). In contrast, RNS60 remained unable to inhibit IFN-γ-induced NO production. This is consistent with a finding by Kleinert et al. (50) that delineates an important role of STAT1α in IFN-γ-induced expression of iNOS. Because RNS60 inhibits the activation of NF-κB, this TCP-modified saline is capable of attenuating the expression of only those proinflammatory molecules whose expression depends on the activation of NF-κB. However, IFN-γ induces the expression of iNOS independent of NF-κB (50); therefore, RNS60 is unable to inhibit IFN-γ-induced microglial production of NO. In resting cells, the classical p65:p50 heterodimer is arrested in the cytoplasm as an inactive complex by IκBα (15). Therefore, IκBα is an important endogenous anti-inflammatory molecule that selectively attenuates the activation of classical NF-κB. These data clearly demonstrate that RNS60 is a rapid inducer of IκBα mRNA and protein in glial cells.
How RNS60 up-regulates the expression of IκBα is an interesting question. Because of the facts that RNS60 may interact at the cell membrane, that activation of PI 3-kinase occurs in close association with cell membrane, and that PI 3-kinase activation induces the expression of IκBα mRNA in microglia (21), we were prompted to investigate the role of PI 3-kinase in RNS60-mediated up-regulation of IκBα. Here, we have discovered that RNS60 specifically induces the activation of class IA, but not class IB, PI 3-kinase in microglia. PI 3-kinase activation by RNS60 was due to p85α-associated p110α and p110β. Accordingly, chemical inhibitors of p110α and p110β, and Δp85α, a dominant-negative mutant of p85α, blocked RNS60-mediated up-regulation of IκBα. Alternatively, overexpression of p110*, a catalytically active mutant of p110α/p110β, alone increased the expression of IκBα. In contrast, overexpression of p110-kd, a kinase-dead mutant of p110α/p110β, had no such effect. Because in most cases, activation of PI 3-kinase leads to Akt activation, we also examined the role of Akt in the up-regulation of IκBα. RNS60 alone induced the phosphorylation of Akt, and this was not due to increased expression of Akt.
We also went further downstream to describe how RNS60 coupled type IA PI 3-kinase-Akt pathway to the transcription of IκBα. Interestingly, we have delineated that RNS60 activates CREB via PI 3-kinase-Akt pathway and then CREB drives the transcription of IκBα in response to RNS60. Rapid activation of the PI3K-Akt pathway suggests that RNS60 treatment may be beneficial for other important cellular pathologies involving this pathway. The exact mechanism by which RNS60 may interact with the cell membrane to activate PI3K is a challenging question that is currently under active investigation.
In summary, we have demonstrated a saline-based agent devoid of an active pharmaceutical ingredient to exhibit anti-inflammatory effects via class IA PI-3K-Akt-CREB-mediated up-regulation of IκBα and inhibition of NF-κB activation. Our results highlight novel biological properties of a physically modified saline and indicate that such approaches may be developed for therapeutic intervention in neuroinflammatory or neurodegenerative disorders as primary or adjunct therapy.
Supplementary Material
Acknowledgments
We thank Andreas Kalmes and Tony Mega of Revalesio Corporation for manuscript editing.
This work was supported by Revalesio Corporation.

This article contains supplemental Figs. S1–S3.
- iNOS
- inducible NO synthase
- TCP
- Taylor-Couette-Poiseuille
- NS
- normal saline
- PI
- phosphatidylinositol
- CREB
- cAMP-response element-binding protein
- AFM
- atomic force microscopy
- PIP3
- phosphatidylinositol (3,4,5)-triphosphate
- GFAP
- glial fibrillary acidic protein
- kd
- kinase-dead
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
REFERENCES
- 1. Giovannini M. G., Scali C., Prosperi C., Bellucci A., Vannucchi M. G., Rosi S., Pepeu G., Casamenti F. (2002) β-Amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo. Involvement of the p38MAPK pathway. Neurobiol. Dis. 11, 257–274 [DOI] [PubMed] [Google Scholar]
- 2. Dauer W., Przedborski S. (2003) Parkinson disease. Mechanisms and models. Neuron 39, 889–909 [DOI] [PubMed] [Google Scholar]
- 3. Gao H. M., Liu B., Zhang W., Hong J. S. (2003) Novel anti-inflammatory therapy for Parkinson disease. Trends Pharmacol. Sci. 24, 395–401 [DOI] [PubMed] [Google Scholar]
- 4. Barnum C. J., Tansey M. G. (2010) Modeling neuroinflammatory pathogenesis of Parkinson disease. Prog. Brain Res. 184, 113–132 [DOI] [PubMed] [Google Scholar]
- 5. Jantaratnotai N., Ryu J. K., Kim S. U., McLarnon J. G. (2003) Amyloid β peptide-induced corpus callosum damage and glial activation in vivo. Neuroreport 14, 1429–1433 [DOI] [PubMed] [Google Scholar]
- 6. Qureshi G. A., Baig S., Bednar I., Södersten P., Forsberg G., Siden A. (1995) Increased cerebrospinal fluid concentration of nitrite in Parkinson disease. Neuroreport 6, 1642–1644 [DOI] [PubMed] [Google Scholar]
- 7. Dehmer T., Lindenau J., Haid S., Dichgans J., Schulz J. B. (2000) Deficiency of inducible nitric-oxide synthase protects against MPTP toxicity in vivo. J. Neurochem. 74, 2213–2216 [DOI] [PubMed] [Google Scholar]
- 8. Nagatsu T., Mogi M., Ichinose H., Togari A. (2000) Changes in cytokines and neurotrophins in Parkinson disease. J. Neural Transm. Suppl. 60, 277–290 [DOI] [PubMed] [Google Scholar]
- 9. Ghosh A., Roy A., Liu X., Kordower J. H., Mufson E. J., Hartley D. M., Ghosh S., Mosley R. L., Gendelman H. E., Pahan K. (2007) Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 104, 18754–18759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mondal S., Roy A., Jana A., Ghosh S., Kordower J. H., Pahan K. (2012) Testing NF-κB-based therapy in hemiparkinsonian monkeys. J. Neuroimmune Pharmacol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dluska E. (2004) Interfacial area in gas-liquid Couette-Taylor flow reactor. Exp. Thermal Fluid Sci. 28, 467–472 [Google Scholar]
- 12. Forney L. J., Ye Z., Giorges A. (2005) Fast competitive reactions in Taylor-Couette flow. Ind. Eng. Chem. Res. 44, 7306–7312 [Google Scholar]
- 13. Hamakawa H., Yamasaki M., Iino M., Hori M., Mori H., Setoguchi T. (2008) J. Therm. Sci. 17, 175–180 [Google Scholar]
- 14. Hayden M. S., Ghosh S. (2008) Shared principles in NF-κB signaling. Cell 132, 344–362 [DOI] [PubMed] [Google Scholar]
- 15. Baeuerle P. A., Baltimore D. (1988) IκB. A specific inhibitor of the NF-κB transcription factor. Science 242, 540–546 [DOI] [PubMed] [Google Scholar]
- 16. Giulian D., Baker T. J. (1986) Characterization of ameboid microglia isolated from developing mammalian brain. J. Neurosci. 6, 2163–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roy A., Fung Y. K., Liu X., Pahan K. (2006) Up-regulation of microglial CD11b expression by nitric oxide. J. Biol. Chem. 281, 14971–14980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wood A., Archambeau G., Watson R. (2008) U. S. Patent 7,832,920
- 19. Brahmachari S., Fung Y. K., Pahan K. (2006) Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J. Neurosci. 26, 4930–4939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brahmachari S., Jana A., Pahan K. (2009) Sodium benzoate, a metabolite of cinnamon and a food additive, reduces microglial and astroglial inflammatory responses. J. Immunol. 183, 5917–5927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jana M., Jana A., Liu X., Ghosh S., Pahan K. (2007) Involvement of phosphatidylinositol 3-kinase-mediated up-regulation of IκBα in anti- inflammatory effect of gemfibrozil in microglia. J. Immunol. 179, 4142–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hara K., Yonezawa K., Sakaue H., Ando A., Kotani K., Kitamura T., Kitamura Y., Ueda H., Stephens L., Jackson T. R., et al. (1994) 1-Phosphatidylinositol 3-kinase activity is required for insulin-stimulated glucose transport but not for RAS activation in CHO cells. Proc. Natl. Acad. Sci. U.S.A. 91, 7415–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanti J. F., Grémeaux T., Grillo S., Calleja V., Klippel A., Williams L. T., Van Obberghen E., Le Marchand-Brustel Y. (1996) Overexpression of a constitutively active form of phosphatidylinositol 3-kinase is sufficient to promote Glut 4 translocation in adipocytes. J. Biol. Chem. 271, 25227–25232 [DOI] [PubMed] [Google Scholar]
- 24. Saha R. N., Jana M., Pahan K. (2007) MAPK p38 regulates transcriptional activity of NF-κB in primary human astrocytes via acetylation of p65. J. Immunol. 179, 7101–7109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jana A., Pahan K. (2010) Fibrillar amyloid-β-activated human astroglia kill primary human neurons via neutral sphingomyelinase. Implications for Alzheimer disease. J. Neurosci. 30, 12676–12689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saha R. N., Liu X., Pahan K. (2006) Up-regulation of BDNF in astrocytes by TNF-α. A case for the neuroprotective role of cytokine. J. Neuroimmune Pharmacol. 1, 212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ghosh A., Roy A., Matras J., Brahmachari S., Gendelman H. E., Pahan K. (2009) Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson disease. J. Neurosci. 29, 13543–13556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou Z., Xu Z., Finch J., Masliyah J., Chow R. (2009) On the role of cavitation in particle collection in flotation. A critical review. II. Minerals Engineering 22, 419–433 [Google Scholar]
- 29. Fan M., Tao D., Honaker R., Luo Z. (2010) Nanobubble generation and its effects on properties of microbubble and millimeter scale bubble solutions. Mining Sci. Technol. 20, 0001–0019 [Google Scholar]
- 30. Häbich A., Ducker W., Dunstan D. E., Zhang X. (2010) Do stable nanobubbles exist in mixtures of organic solvents and water? J. Phys. Chem. B 114, 6962–6967 [DOI] [PubMed] [Google Scholar]
- 31. Weijs J. H., Seddon J. R., Lohse D. (2012) Diffusive shielding stabilizes bulk nanobubble clusters. ChemPhysChem 13, 2197–2204 [DOI] [PubMed] [Google Scholar]
- 32. Craig V. S. (2011) Very small bubbles at surfaces. The nanobubble puzzle. Soft Matter 7, 40–48 [Google Scholar]
- 33. Mazumder M., Bhushan B. (2011) Propensity and geometrical distribution of surface nanobubbles: Effect of electrolyte, roughness, and substrate bias. Soft Matter 7, 9184–9196 [Google Scholar]
- 34. Jana M., Anderson J. A., Saha R. N., Liu X., Pahan K. (2005) Regulation of inducible nitric-oxide synthase in proinflammatory cytokine-stimulated human primary astrocytes. Free Radic. Biol. Med. 38, 655–664 [DOI] [PubMed] [Google Scholar]
- 35. González-Scarano F., Baltuch G. (1999) Microglia as mediators of inflammatory and degenerative diseases. Annu. Rev. Neurosci. 22, 219–240 [DOI] [PubMed] [Google Scholar]
- 36. Saha R. N., Pahan K. (2006) Signals for the induction of nitric-oxide synthase in astrocytes. Neurochem. Int. 49, 154–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jana M., Liu X., Koka S., Ghosh S., Petro T. M., Pahan K. (2001) Ligation of CD40 stimulates the induction of nitric-oxide synthase in microglial cells. J. Biol. Chem. 276, 44527–44533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pahan K., Sheikh F. G., Namboodiri A. M., Singh I. (1997) Lovastatin and phenylacetate inhibit the induction of nitric-oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J. Clin. Invest. 100, 2671–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xie Q. W., Kashiwabara Y., Nathan C. (1994) Role of transcription factor NF-κB/Rel in induction of nitric-oxide synthase. J. Biol. Chem. 269, 4705–4708 [PubMed] [Google Scholar]
- 40. Arenzana-Seisdedos F., Thompson J., Rodriguez M. S., Bachelerie F., Thomas D., Hay R. T. (1995) Inducible nuclear expression of newly synthesized IκBα negatively regulates DNA-binding and transcriptional activities of NF-κB. Mol. Cell. Biol. 15, 2689–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suire S., Coadwell J., Ferguson G. J., Davidson K., Hawkins P., Stephens L. (2005) p84, a new Gβγ-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110γ. Curr. Biol. 15, 566–570 [DOI] [PubMed] [Google Scholar]
- 42. Guo J. P., Coppola D., Cheng J. Q. (2011) IKBKE protein activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain malignant transformation. J. Biol. Chem. 286, 37389–37398 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43. Folkes A. J., Ahmadi K., Alderton W. K., Alix S., Baker S. J., Box G., Chuckowree I. S., Clarke P. A., Depledge P., Eccles S. A., Friedman L. S., Hayes A., Hancox T. C., Kugendradas A., Lensun L., Moore P., Olivero A. G., Pang J., Patel S., Pergl-Wilson G. H., Raynaud F. I., Robson A., Saghir N., Salphati L., Sohal S., Ultsch M. H., Valenti M., Wallweber H. J., Wan N. C., Wiesmann C., Workman P., Zhyvoloup A., Zvelebil M. J., Shuttleworth S. J. (2008) The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI 3-kinase for the treatment of cancer. J. Med. Chem. 51, 5522–5532 [DOI] [PubMed] [Google Scholar]
- 44. Jackson S. P., Schoenwaelder S. M., Goncalves I., Nesbitt W. S., Yap C. L., Wright C. E., Kenche V., Anderson K. E., Dopheide S. M., Yuan Y., Sturgeon S. A., Prabaharan H., Thompson P. E., Smith G. D., Shepherd P. R., Daniele N., Kulkarni S., Abbott B., Saylik D., Jones C., Lu L., Giuliano S., Hughan S. C., Angus J. A., Robertson A. D., Salem H. H. (2005) PI 3-kinase p110β. A new target for antithrombotic therapy. Nat. Med. 11, 507–514 [DOI] [PubMed] [Google Scholar]
- 45. Lee K. S., Lee H. K., Hayflick J. S., Lee Y. C., Puri K. D. (2006) Inhibition of phosphoinositide 3-kinase δ attenuates allergic airway inflammation and hyper-responsiveness in murine asthma model. FASEB J. 20, 455–465 [DOI] [PubMed] [Google Scholar]
- 46. Marone R., Cmiljanovic V., Giese B., Wymann M. P. (2008) Targeting phosphoinositide 3-kinase. Moving toward therapy. Biochim. Biophys. Acta 1784, 159–185 [DOI] [PubMed] [Google Scholar]
- 47. Carson M. J. (2002) Microglia as liaisons between the immune and central nervous systems. Functional implications for multiple sclerosis. Glia 40, 218–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bhaskar S., Tian F., Stoeger T., Kreyling W., de la Fuente J. M., Grazú V., Borm P., Estrada G., Ntziachristos V., Razansky D. (2010) Multifunctional nanocarriers for diagnostics, drug delivery, and targeted treatment across blood-brain barrier. Perspectives on tracking and neuroimaging. Part Fibre. Toxicol. 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rapoport N., Gao Z., Kennedy A. (2007) Multifunctional nanoparticles for combining ultrasonic tumor imaging and targeted chemotherapy. J. Natl. Cancer Inst. 99, 1095–1106 [DOI] [PubMed] [Google Scholar]
- 50. Kleinert H., Wallerath T., Fritz G., Ihrig-Biedert I., Rodriguez-Pascual F., Geller D. A., Förstermann U. (1998) Cytokine induction of NO synthase II in human DLD-1 cells. Roles of the JAK-STAT, AP-1, and NF-κB signaling pathways. Br. J. Pharmacol. 125, 193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.