

Background: Role of SPDEF in tumor biology remains hotly debated.
Results: SPDEF suppressed tumor metastasis in part by modulating MMP9 and MMP13.
Conclusion: SPDEF may be a modifiable therapeutic target in prostate tumors.
Significance: This is the first study directly implicating SPDEF as a tumor metastasis suppressor, in any system, in vivo.
Keywords: Cancer, Cancer Biology, ETS Family Transcription Factor, Matrix Metalloproteinase (MMP), Prostate Cancer, Cancer Metastasis Suppressor
Abstract
Emerging evidence suggests that the SAM pointed domain containing ETS transcription factor (SPDEF) plays a significant role in tumorigenesis in prostate, breast, colon, and ovarian cancer. However, there are no in vivo studies with respect to the role of SPDEF in tumor metastasis. The present study examined the effects of SPDEF on tumor cell metastasis using prostate tumor cells as a model. Utilizing two experimental metastasis models, we demonstrate that SPDEF inhibits cell migration and invasion in vitro and acts a tumor metastasis suppressor in vivo. Using stable expression of SPDEF in PC3-Luc cells and shRNA-mediated knockdown of SPDEF in LNCaP-Luc cells, we demonstrate for the first time that SPDEF diminished the ability of disseminated tumors cells to survive at secondary sites and establish micrometastases. These effects on tumor metastasis were not a result of the effect of SPDEF on cell growth as SPDEF expression had no effect on cell growth in vitro or subcutaneous tumor xenograft-growth in vivo. Transcriptional analysis of several genes associated with tumor metastasis, invasion, and the epithelial-mesenchymal transition demonstrated that SPDEF expression selectively down-regulated MMP9 and MMP13 in prostate cancer cells. Further analysis indicated that forced MMP9 or MMP13 expression rescued the invasive phenotype in SPDEF expressing PC3 cells in vitro, suggesting that the effects of SPDEF on tumor invasion are mediated, in part, through the suppression of MMP9 and MMP13 expression. These results demonstrate for the first time, in any system, that SPDEF functions as a tumor metastasis suppressor in vivo.
Introduction
The development of a metastatic tumor continues to represent the most lethal and least treatable hallmark of cancer (1). Metastases can occur years or decades after successful treatment of the primary tumor, which is due in part to tumor dormancy, the persistence of solitary cells at secondary sites, which can persist for extended periods of time in a secondary organ (2–4). As the temporal and spatial order of biological events in metastasis continue to be elucidated in the laboratory setting; clinically, there remains no curative treatment for metastatic disease; therefore, continued experimental manipulation of the metastatic process to understand its biological mechanisms is warranted.
ETS (E-twenty-six transformation-specific) transcription factors are involved in a multitude of normal and pathological cellular processes. Many ETS factors are deregulated and are thought to be key players in the generation of several types of cancer (5). In prostate cancer, chromosomal rearrangements such as the ETS family member ERG-TMPRSS2 fusion represent an early event driving the development of prostate neoplasia progression. SPDEF (SAM pointed domain containing ETS transcription factor; also known as prostate-derived ETS factor) is the latest ETS family member discovered whose expression is limited to epithelial cells of the prostate, breast, lung, ovary, and colon. However, the role(s) SPDEF plays in tumorigenesis remains a subject of continued debate (reviewed in Refs. 6 and 7). Although several reports link SPDEF expression to tumor promotion, others demonstrate a role of tumor suppression in various models. In particular, Turner et al. (8) demonstrated reduced invasive capacity of breast cancer cell lines when SPDEF was introduced and increased migration when SPDEF was knocked down via siRNA, whereas Gunawardane et al. (9) demonstrated increased breast cell migration when SPDEF was overexpressed. Moreover, the single in vivo study published to date, using transformed mouse breast epithelial cells, found that SPDEF overexpression retarded both tumor incidence and growth in vivo (10); a mechanism dependent upon increased p21 expression. However, the precise role(s) of SPDEF in tumor growth and metastasis is not completely understood.
Prostate cancer is the most frequently diagnosed, non-skin cancer in the United States and is the second leading cause of death in men.2 Although conventional therapies produce a high cure rate for patients presenting with localized disease, there remains no curative treatment once the tumor has metastasized. Prostate tumor metastasis is the primary cause of mortality in prostate cancer patients. In addition, there are few, if any, reliable biomarker(s) that distinguish an indolent tumor, one which will respond well to conventional therapy from aggressive tumors, which require more aggressive therapy. In our previously published studies, we observed that SPDEF expression is reduced or lost in advanced stages of prostate cancer (12). These studies also suggested that SPDEF expression suppressed an aggressive phenotype in prostate cancer. Our findings are in contrast to a report by Sood et al. (13) but have been confirmed and even extended by two additional groups (14, 15). In fact, Ghadersohi et al. (15) associated SPDEF expression with a favorable prognosis in localized prostate cancer. Taken together, based on these limited studies, the loss of SPDEF appears to be an indicator of aggressive prostate cancer.
Experimental metastasis models do not recapitulate all steps of the metastatic cascade; however, the use of these models allows the study of the survival of disseminated tumor cells in the circulation, extravasation, and growth at secondary sites (16, 17). MMP9,3 also known as gelatinase B, is an extracellular matrix-degrading enzyme that is regarded as one of the classic metastasis-promoting genes (18, 19) through either direct effects on substrate degradation, thereby promoting migration and invasion, or indirectly through prometastatic microenvironmental remodeling of the metastatic niche (19–21). MMP13, also known as collagenase-3, is known to degrade collagen and has been shown to be predominantly expressed at the invading front of tumor cells and is also produced by tumor-associated stromal fibroblasts (22). SPDEF and MMP9 levels have been found to inversely correlate in tissue samples, demonstrating that decreased SPDEF levels are associated with increased MMP9 expression in advanced disease (12). Numerous studies have shown a role for SPDEF in cell migration and invasion in vitro using cell lines of multiple tissue origin (6, 12, 23, 24); however, to date, no in vivo studies have been performed investigating the effect of SPDEF on tumor metastasis. In the present study, we evaluated the functions of SPDEF using luciferase-expressing prostate cancer cell lines (PC3-Luc and LNCaP-Luc). Results presented herein demonstrate for the first time that stable expression of SPDEF in PC3-Luc cells decreased, whereas stable knockdown of SPDEF in LNCaP-Luc prostate tumor cells increased, the ability of these cells to survive at metastatic sites. Finally, we found that SPDEF-mediated down-regulation of MMP9 and MMP13 represents a significant mechanism of action in that MMP9 or MMP13 overexpression is sufficient to overcome the inhibitory effects of SPDEF expression on tumor cell invasion. To the best of our knowledge, this is the first study, in any model, demonstrating a tumor metastasis suppressor function of SPDEF in vivo.
EXPERIMENTAL PROCEDURES
Cell Culture
Cells were cultured, and the cloning of SPDEF and creation of stable SPDEF expressing and vector control PC3 cell lines were described previously (12). To engineer luciferase expression, 6 × 106 cells were seeded in six-well plates. The following day, cells were infected with a luciferase delivering lentivirus virus with the addition of 8 μg/ml polybrene. Clonal selection was performed, and clones were screened for luciferase expression.
Creation of shRNA Containing Lentiviral Particles
shRNA constructs were obtained from the University of Colorado Functional Genomics core facility in the pLKO1 backbone. The plasmid along with a packaging and envelop plasmid (Addgene) was transfected into 8 × 105 293T cells using Effectene (Qiagen) according to the manufacturer's instructions. After 24 h, the medium was changed (20 mm HEPES, 30% FBS/DMEM, 2 mm sodium butyrate). After 24 h, the medium was collected, filtered through 0.45-μm filters, and flash frozen.
Lentiviral Delivery of shRNA
Cells were infected with lentiviral particles at a multiplicity of infection of 1. Two shRNA sequences were used for SPDEF. Clones were picked from cell pools selected via puromycin and were tested for knockdown efficiency via Western blot. The scrambled (Scr) shRNA vector contains four base pair mismatches within the short hairpin sequence to any known human gene, serving as a negative control. Each SPDEF-specific shRNA construct had the same phenotypic effect (data not shown), thus eliminating off-target effects of the shRNA and between cell clones. Multiple cell clones from both shRNAs demonstrated similar results.
Reagents
The following antibodies were used: p21 (1:1000) mouse monoclonal, p27 (1:1000) rabbit polyclonal, and CDK2 (1:1000) rabbit monoclonal were purchased from Cell Signaling; PDEF N-14 (1:250) goat polyclonal from Santa Cruz Biotechnology, and actin (1:1000) rabbit polyclonal and anti-FLAG were purchased from Sigma.
Protein Isolation and Western Blotting
These were performed as described previously (25).
Cell Proliferation Assay
MTT assays were performed as described previously (26).
Cell Migration Assays
Wound healing assays were performed as described previously (12).
Transient Transfection
A TrueClone MMP9 or MMP13 expression plasmid was purchased from OriGene, and 0.4 μg of DNA was transfected using Effectene (Qiagen) according to the manufacturer's instructions. Cells were transfected in suspension and seeded into Matrigel-coated Boyden chambers as described below.
RT-PCR
RNA was isolated per manufacturer's instructions (Qiagen; RNeasy). cDNA was synthesized using cDNA iScript (Bio-Rad) per the manufacturer's instructions. PCR was then performed using standard Taq polymerase (Fermentas) with the following cycles: 95° C for 4 min; (95° C for 30 s, annealing temperature of 1 min, 72° C for 1 min) ×38 cycles; 72° C for 10 min. Annealing temperatures were 2° C lower than the primer melting temperatures found in Table 1. Primer sequences can also be found in Table 1.
TABLE 1.
RT-PCR primer sequences
| Gene | RT-PCR primer sequences |
|||
|---|---|---|---|---|
| Forward | Temperature (°C) | Reverse | Temperature (°C) | |
| Cyclin B | 5′-TTGATACTGCCTCTCCAAGCCCAA-3′ | 60.3 | 5′-TTGGTCTGACTGCTTGCTCTTCCT-3′ | 60.3 |
| Cyclin D | 5′-CCTTTGGTGCCAACTGGTGTTTGA-3′ | 60.3 | 5′-TCAGATGACTCTGGGAAACGCCAA-3′ | 60.3 |
| Cyclin E | 5′-TGCAGAGCTGTTGGATCTCTGTGT-3′ | 60.3 | 5′-ACAACATGGCTTTCTTTGCGCGGG-3′ | 60.3 |
| CDK2 | 5′-AGCCAGAAACAAGTTGACGGGAGA-3′ | 60.5 | 5′-AAGAGGAATGCCAGTGAGAGCAGA-3′ | 60.0 |
| p27 | 5′-AGTCCATTTGATCAGCGGAGACTCG-3′ | 60.4 | 5′-TCGCACGTTTGACATCTTTCTCCC-3′ | 59.5 |
| p21 | 5′-TTCGACTTTGTCACCGAGACACCA-3′ | 60.2 | 5′-AGGCACAAGGGTACAAGACAGTGA-3′ | 60.1 |
| VASP | 5′-GTAAGAGTAACACTGTAGCCGCCA-3′ | 58.5 | 5′-ATCATAAAGCATCACAGTGGCCCG-3′ | 59.6 |
| Survivin | 5′-GAGGCTGGCTTCATCCACTG-3′ | 58.2 | 5′-CAGCTGCTCGATGGCACGGC-3′ | 64.1 |
| Snail | 5′-TGCCAATGCTCATCTGGGACTCT-3′ | 60.5 | 5′-GCCTCCAAGGAAGAGACTGAAGTA-3′ | 57.9 |
| MMP9 | 5′-TACCACCTCGAACTTTGACAGCGA-3′ | 60.1 | 5′-AAAGGCACAGTAGTGGCCGTAGAA-3′ | 60.3 |
| Slug | 5′-ACTACAGTCCAAGCTTTCAGACCC-3′ | 58.6 | 5′-CCGCAGATCTTGCAAACACAAGGT-3′ | 60.2 |
| MMP13 | 5′-AACATCCAAAAACGCCAGAC-3′ | 53.9 | 5′-GGAAGTTCTGGCCAAAATGA-3′ | 53.3 |
| SPDEF | 5′-CAGGTGAAGTCCGCTCTTTC-3′ | 55.7 | 5′-AATGTGCAGAAGTGGCTCCT-3′ | 56.9 |
| IL-6 | 5′-TACCCCCAGGAGAAGATTCC-3′ | 55.7 | 5′-AAAGCTGCGCAGAATGAGAT-3′ | 55.0 |
| CXCR4 | 5′-TGGCCTTATCCTGCCTGGTATTGT-3′ | 60.2 | 5′-TGGCTCCAAGGAAAGCATAGAGGA-3′ | 59.9 |
| GAPDH | 5′-AAGGTCGGAGTCAACGGATTTGGT-3′ | 60.5 | 5′-AGTGATGGCATGGACTGTGGTCAT-3′ | 60.5 |
Densitometry
Densitometry was performed using ImageJ software. All mRNA bands were compared with GAPDH bands.
Immunohistochemistry
Immunohistochemistry was performed as described previously (12).
Invasion Assay
The invasion assay was performed as described previously (27), with the exception of 10% serum containing medium in the bottom chamber. 1% serum conditions were used in the top chamber in cells that were transfected with MMP9 or MMP13 constructs.
Three-dimensional Colony Assay
Cells were grown in eight-well chamber slides coated with Matrigel. After cell seeding, 4% Matrigel was added to the top of the cells. Medium was changed every other day for 10–15 days. See Ref. 12 for more details.
Subcutaneous in Vivo Experiments
Six- to 8-week-old male nude mice (Taconic NCRNU) were injected subcutaneous with 1.5 × 106 cells in 100 μl PBS/Matrigel (50:50) subcutaneously (n = 5 VC; n = 5 SPDEF OE). Tumors became measurable by day 22 post-implantation and were measured with calipers twice per week. Tumor volumes were calculated by the following equation: volume = π/6 × length × width2. Mice were euthanized at 42 days post-implantation, and tumors were surgically removed and fixed in 10% formaldehyde or flash frozen. All animal experiments were performed in accordance with guidelines set by the University of Colorado Institutional Animal Care and Use Committee.
In Vivo Experimental Metastasis Models
Six- to 8-week-old male nude mice (Taconic-NCR-nu) were injected via either intracardiac or tail vein with 1 × 106 cells in 100 μl of PBS. An insulin needle was used for the tail vein injections, whereas a 26-gauge needle was used for the intracardiac injection. Mice were imaged for luciferase signal by an intraperitoneal injection of luciferin (Caliper Life Sciences) (15 μg in 100 μl of PBS) every other week for 12 weeks using a Xenogen system.
Statistical Analysis
GraphPad Software (Prism, version 3.0) was utilized to perform all statistics. Mann-Whitney T-tests were performed to indicate statistical significance. All error bars display the S.E.
RESULTS
Stable Expression of SPEDF in PC3-Luc Cells and Knockdown of SPDEF in LNCaP-Luc Cells
The expression levels of SPDEF varies among prostate tumor cell lines (12). PC3 prostate tumor cells express low to undetectable levels of SPDEF protein and are known to have high metastatic potential in nude mouse models, whereas LNCaP tumor cells express relatively high levels of SPDEF and lack metastatic potential in vivo. Stable expression of FLAG-tagged SPDEF in PC3 cells was achieved as described previously (12). SPDEF-targeted shRNA in a lentiviral vector was used to create stable knockdowns of SPDEF in LNCaP cells, whereas vector control LNCaP cells received Scr-shRNA via a lentiviral vector. All four cell lines VC-PC3 cells, FLAG-tagged SPDEF expressing (SPDEF-PC3) cells, Scr shRNA LNCaP cells and SPDEF knockdown (SPDEF-KD) LNCaP cells were stably transfected with luciferase using a lentiviral vector to generate VC-PC3-Luc, SPDEF-PC3-Luc, Scr-LNCaP-Luc, and SPDEF-KD-LNCaP-Luc cells, respectively. Luciferase was visualized via bioluminescence imaging, and no significant difference in luciferase expression was detected between the cells lines (Fig. 1A). The expression and knockdown of SPDEF was confirmed in luciferase-expressing cells as evidenced via an anti-SPDEF and/or anti-FLAG Western blot (Fig. 1B).
FIGURE 1.

SPDEF expression reduces in vitro tumor cell migration, invasion, and aggressive phenotype. A, representative bioluminescent image quantitation of VC, SPDEF-expressing (SPDEF OE-Luc) PC3 cells, Scr shRNA, and SPDEF KD-Luc LNCaP cells, confirming luciferase expression. B, Western blot analysis indicating the expression levels of SPDEF protein in the SPDEF OE-Luc cells (both anti-SPDEF and anti-FLAG antibodies) and knockdown levels in two different shRNA LNCaP-Luc clones. Tubulin was used as a protein loading control. C, SPDEF overexpression reduces whereas knockdown increases cell migration. Shown are images and quantitation of wound healing assays performed between the four cell lines. D, SPDEF overexpression reduces and knockdown increases tumor cell invasion. SPDEF OE-Luc PC3 cells are less invasive, whereas SPDEF KD-Luc LNCaP cells are more invasive as determined using Matrigel-coated Boyden chambers as described under “Experimental Procedures.” E, representative images of three-dimensional Matrigel culture of PC3-Luc and LNCaP-Luc cells with and without SPDEF overexpression or knockdown. VC-Luc, VC-PC3-Luc; SPDEF OE-Luc, SPDEF-PC3-Luc; Scr-Luc, Scr-LNCaP-Luc; SPDEF KD-Luc, SPDEF-KD-LNCaP-Luc). *, statistical significance (p < 0.05) compared with VC-Luc or Scr-Luc cells.
Stable Expression of SPDEF Decreases, whereas SPDEF Knockdown Increases Cell Migration and Invasion in Vitro
For these studies, we used Luc-tagged VC-PC3-Luc, SPDEF-PC3-Luc, Scr-LNCaP-Luc, and SPDEF-KD-LNCaP-Luc cells. We found that stable expression of SPDEF expression resulted in decreased cell motility as SPDEF-PC3-Luc cells demonstrated significantly decreased motility in a directional migration assay as compared with VC-PC3-Luc cells. In addition, SPDEF-KD-LNCaP-Luc cells were more motile compared with Scr-LNCaP-Luc control cells, suggesting that SPDEF expression results in decreased cell motility (Fig. 1C). Moreover, modulation of SPDEF expression affected cell invasion using a Matrigel-coated Boyden chamber assay. We observed that SPDEF-PC3-Luc cells had significantly decreased ability to invade as compared with VC-PC3-Luc cells, whereas SPDEF-KD-LNCaP-Luc cells were more invasive as compared with Scr-LNCaP-Luc control cells, suggesting that SPDEF expression was an impediment to cell invasion (Fig. 1D). Lastly, stable expression of SPDEF appeared to reverse the aggressive pattern of growth of these cells in three-dimensional cell culture (Fig. 1E), whereas SPDEF knockdown in LNCaP-Luc cells caused a more subtle increase in the aggressive appearance of LNCaP-Luc cells in a three-dimensional culture (Fig. 1E). These results are in agreement with numerous studies demonstrating the relationship between phenotypic three-dimensional spheroid formation with cell migration and invasion (28, 29). Thus, our results suggest that SPDEF modulation effects cell migration and invasion, namely when SPDEF expression is high, cells are less motile and invasive, and conversely, when SPDEF expression is low, cells demonstrate increased motility and invasion.
SPDEF Expression Decreases Prostate Tumor Metastasis
Because SPDEF expression decreased tumor cell migration and invasion in vitro, the hypothesis that SPDEF expression effects tumor metastasis was analyzed. Two experimental metastasis models were used. First, VC-PC3-Luc cells or SPDEF-PC3-Luc cells were injected into either the tail vein or into the arterial circulation (intracardiac injection) of nude mice. Mice were imaged 1 week following the injection for up to 12 weeks (Fig. 2, A and B). Both the VC-PC3-Luc cells and SPDEF-PC3-Luc cells were found to have circulated well, and no significant difference in the metastatic seeding ability was detected between these two cell lines at 1 week following injections. However, two mice in the intracardiac model (numbered two and five in Fig. 2B) had tumor cell growth near the heart. These two mice were eliminated from further imaging and all quantitation to quantitate only those cells that were successfully disseminated in to circulation. At the end of 12 weeks, in vivo bioluminescent imaging (Fig. 2, A and B) and quantitation (Fig. 2, C and D) revealed that the SPDEF-PC3-Luc cells significantly failed to survive and develop micrometastases as compared with the VC-PC3-Luc cells, suggesting that SPDEF expression reduces disseminated cell survival in both the tail vein and intracardiac metastasis models.
FIGURE 2.

SPDEF expression in PC3-Luc cells reduces tumor metastasis. A and B, representative images of bioluminescent images of mice injected with VC-Luc (n = 7) and SPDEF OE-Luc (n = 5) cells (n represents each model). Representative mice are shown. Shown are images at week 1 and week 12 post injection. C and D, quantitation of luciferase signal in the mice at the various time points shown. A significant decrease in disseminated tumor cells (luciferase activity) were detected in the SPDEF OE-Luc cells compared with the VC-Luc cells at 12 weeks. VC-Luc, VC-PC3-Luc; SPDEF OE-Luc, SPDEF-PC3-Luc. *, statistical significance (p < 0.05) compared with VC-Luc cells at 12 weeks.
In parallel experiments, we utilized the SPDEF-KD-LNCaP-Luc cells and Scr-LNCaP-Luc cells and injected them into either the tail vein or into the arterial circulation (intracardiac injection) of nude mice. Both the SPDEF-KD-LNCaP-Luc cells and Scr-LNCaP-Luc cells were found to have circulated well, and no significant difference in the metastatic seeding ability was detected between these two cell lines at week 1. At the end of 12 weeks, in vivo bioluminescent imaging (Fig. 3, A and B) and quantitation (Fig. 3, C and D) revealed that the SPDEF-KD-LNCaP-Luc cells survived significantly better and developed micrometastases compared with the Scr-LNCaP-Luc cells, suggesting that SPDEF knockdown increases survival of disseminated cell in both the tail vein and intracardiac metastasis models. Taken together, these results demonstrate the ability of SPDEF to inhibit the ability of circulating tumor cells to develop into successful metastasis.
FIGURE 3.

SPDEF knockdown in LNCaP-Luc cells increases tumor metastasis. A and B, representative images of bioluminescent images of mice injected with Scr-Luc (n = 7) and SPDEF KD-Luc (n = 5) cells (n represents each model), representative mice are shown. Shown are images at week 1 and week 12 post injection. C and D, quantitation of luciferase signal in the mice at the various time points shown. A significant increase in disseminated tumor cells (luciferase activity) were detected in the SPDEF KD-Luc cells compared with the Scr-Luc cells at 12 weeks. Scr-Luc, Scr-LNCaP-Luc; SPDEF KD-Luc, SPDEF-KD-LNCaP-Luc. *, statistical significance (p < 0.05) compared with Scr-Luc cells at 12 weeks.
SPDEF Does Not Affect Cell Growth in Vitro or Tumor Growth in Xenograft Models in Vivo
To test whether or not SPDEF modulated tumor metastasis in vivo by altering tumor cell growth, we evaluated the effects of differential SPDEF expression on cell growth in vitro. For these studies, we compared growth curves of VC-PC3-Luc cells and SPDEF-PC3-Luc cells as well as SPDEF-KD-LNCaP-Luc cells and Scr-LNCaP-Luc cells in culture. Results presented in Fig. 4A, show that modulation of SPDEF expression levels had no effect on cell growth rates in the PC3 or LNCaP prostate tumor cells grown in vitro. Moreover, SPDEF expression in PC3-Luc cell lines had no effect on mRNA levels of the following cell cycle regulatory genes: cyclins B, D, and E, CDK2, p27, and p21 (Fig. 4B).
FIGURE 4.

Modulation of SPDEF expression does not affect tumor cell growth rates. A, MTT assay shows that VC-Luc and SPDEF-Luc cells as well as Scr-Luc and SPDEF OE-Luc cells proliferate at the same rate in vitro. B, SPDEF does not modulate the cycle regulatory gene, RT-PCR (left) and densitometry (right). VC-Luc, VC-PC3-Luc; SPDEF OE-Luc, SPDEF-PC3-Luc; Scr-Luc, Scr-LNCaP-Luc; SPDEF KD-Luc, SPDEF-KD-LNCaP-Luc). Densitometry is the average of three independent experiments of the RT-PCR data compared with GAPDH control bands. *, statistical significance (p < 0.05) compared with VC-Luc cells.
Because tumor cell lines often behave differently when grown in vivo compared with in vitro, we investigated the role of SPDEF on tumor growth in vivo. For these studies, VC-PC3-Luc cells and SPDEF-PC3-Luc cells were subcutaneously injected into the hind flanks of 6- to 8-week-old male nude mice. Tumors were manually measured starting 15 days post injection for up to 42 days. Results demonstrate that there was no significant difference in tumor incidence or tumor growth of xenografts between VC-PC3-Luc cells and SPDEF-PC3-Luc cells over the course of the experiment (Fig. 5B). At the conclusion of this study (42 days post-implantation), the tumors were visualized by bioluminescent imaging (Fig. 5A), again indicating no difference in tumor growth. Moreover, there was no significant difference in tumor weight (Fig. 5C). These studies are in contrast to the findings of a previous study using transformed mouse breast epithelial cells, which demonstrated a p21-dependent reduction in tumor growth in tumors expressing SPDEF (10). We also performed Western blot analysis to determine whether SPDEF expression modulated p21, p27, or CDK2 levels in our PC3 xenograft model. We found no significant difference in the protein expression levels of p21, p27, or CDK2 either by Western blot analysis (Fig. 5D) or by immunohistochemical methods (p21 and p27) (Fig. 5E). Moreover, we found no discernible changes in the mRNA levels of cell cycle regulatory genes including cyclins B, D, and E, CDK2, and p21 between the two groups of xenograft tumors (Fig. 5F). Taken together, these results demonstrate that, in the context of prostate cancer, SPDEF expression has no effect on tumor cell growth in vitro or tumor xenograft growth in vivo. These results suggest that modulation of tumor cell metastasis by SPDEF is not a consequence of changes in cell growth.
FIGURE 5.

SPDEF expression has no effect on subcutaneous tumor xenograft growth. A and B, nude mice (n = 5 per group) were subcutaneously injected with either VC-Luc or SPDEF OE-Luc cells. Tumors growth was followed for 42 days, and tumor volume was measured. A, representative image of xenografts using in vivo bioluminescent imaging. B, quantitation of tumor volumes over the experimental period. C, at the end of the experimental period, tumors were dissected and weighed. D, Western blot analysis of cell cycle regulatory proteins. Note that SPDEF expression was increased and was used as an internal positive control. E, immunohistochemistry of the cell cycle inhibitors p21 and p27. F, RT-PCR analysis of expression of cell cycle regulatory genes. Graphs represent densitometry of three independent experiments of the RT-PCR data compared with GAPDH control. VC-Luc, VC-PC3-Luc; SPDEF OE-Luc, SPDEF-PC3-Luc. *, statistical significance (p < 0.05) compared with VC-Luc cells.
MMP9 and MMP13 mRNA Levels Are Differentially Regulated by SPDEF
Because SPDEF has been shown to regulate several genes in various cell lines, many of which are involved in epithelial-to-mesenchymal transition, and to begin to dissect a mechanism of SPDEF-mediated modulation of tumor metastasis, we evaluated the expression of genes involved in cell motility, invasion, and metastasis among those genes previously described to be regulated by SPDEF (6). The differential gene expression from two cell lines and the cell-derived subcutaneous tumors were analyzed. The genes analyzed are shown in Fig. 6 with representative gels on the left and densitometry on the right, averaged from three separate experiments. Of the 10 genes tested, only two genes, MMP9 and MMP13, were found to be differentially down-regulated in the context of SPDEF expression in the cell lines in vitro. It is interesting to point out that MMP9 was also down-regulated in the subcutaneous in vivo tumors; however, we did not detect the down-regulation of MMP13. This could be due to contamination by tumor-associated stromal fibroblasts as they are known to express MMP13 (22). Although MMP9 levels have previously been shown to be inversely corrected with SPDEF levels (12), this is the first study to suggest that MMP13 gene expression is regulated by SPDEF.
FIGURE 6.

MMP9 and MMP13 mRNA levels are down-regulated in response to SPDEF expression. RT-PCR was performed on a host of genes that have either previously been reported to be regulated by SPDEF or have a known role in prostate tumor progression and/or metastasis. MMP9 mRNA levels were significantly down-regulated in both the cell line and in the subcutaneous tumors overexpressing SPDEF. In addition, we found that MMP13 was also significantly down-regulated in the cell lines in vitro but not in the in vivo tumors. Expression measured was relative to housekeeping gene GAPDH. Graphs represent densitometry of three independent experiments of the RT-PCR data relative to the GAPDH. VC-Luc, VC-PC3-Luc; SPDEF OE-Luc, SPDEF-PC3-Luc. *, statistical significance (p < 0.05) compared with VC-Luc cells.
Forced MMP9 or MMP13 Expression Is Sufficient to Restore Cell Invasion in SPDEF Cells
SPDEF levels have been shown to be inversely related in human prostate tumor samples where high levels of SPDEF are associated with low MMP9 levels (12). MMP13 levels have been shown to correlate with the invasive and metastatic capacity of head and neck squamous cell carcinomas. However, the significance of MMP9 or MMP13 loss in response to SPDEF expression has not been reported. Therefore, we reintroduced MMP9 or MMP13 back in the SPDEF-PC3-Luc cells in transient transfection experiments. Results presented in Fig. 7A demonstrate that mRNA levels of MMP9 and MMP13 were up-regulated upon transfection. Furthermore, results of cell invasion in Matrigel-coated Boyden chamber assays demonstrated that MMP9 or MMP13 expression abrogates the effect of SPDEF expression on cell invasion, as the SPDEF-PC3-Luc/MMP9 as well as the SPDEF-PC3-Luc/MMP13 cells invaded to the same level as VC-PC3-Luc cells (Fig. 7B), suggesting that MMP9 or MMP13 expression alone is sufficient to overcome SPDEF-induced inhibition of cell invasion.
FIGURE 7.
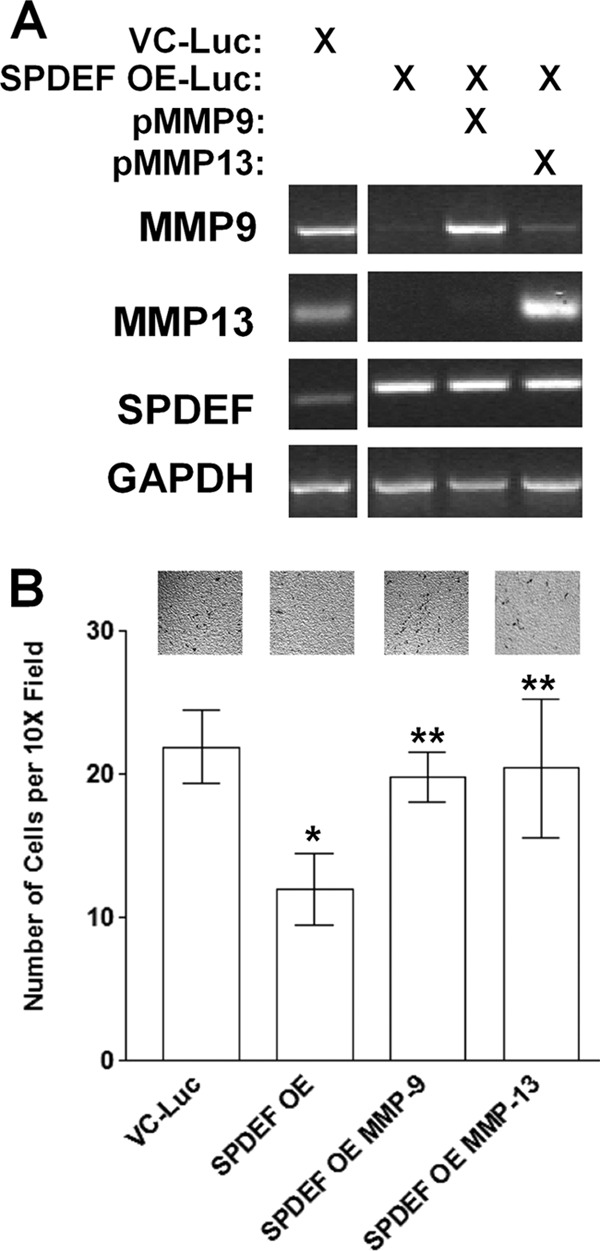
MMP9 and MMP13 expression rescues the cells from SPDEF-mediated inhibition of cell invasion. A, RT-PCR was performed in parallel to confirm levels of MMP9, MMP13, and SPDEF mRNA expression in these cells upon transfection with a MMP9 or a MMP13 plasmid. B, cell invasion was analyzed via Matrigel-coated Boyden chambers. Note that forced expression of MMP9 or MMP13 in SPDEF OE-Luc cells abrogates the inhibitory effect of SPDEF on cell invasion. VC-Luc, VC-PC3-Luc; SPDEF OE-Luc, SPDEF-PC3-Luc. *, statistical significance (p < 0.05) compared with VC-Luc cells; **, statistical significance (p < 0.05) compared with SPDEF OE-Luc cells.
DISCUSSION
Metastasis is the leading perpetrator of tumor-associated deaths, including that of prostate cancer. Although the knowledge of the metastatic process and the players involved continues to increase, no curative treatments for metastatic prostate cancer exist, which lends credence to the investigation of novel proteins that may be involved in the metastatic process. Discovered just more than a decade ago, SPDEF is one such protein, which appears to function as a tumor metastasis suppressor in this model system.
SPDEF has been shown to regulate several gene products and be involved in several physiological and pathophysicological processes, including epithelial-to-mesenchymal transition, since its first appearance in the literature in 2000 (30). In fact, reports on the role of SPDEF in prostate (7), breast (23, 31), ovarian (32, 33), and colon (34) tumors have yielded exciting results regarding the gene products regulated by SPDEF. As previously reviewed (6, 7), the role of SPDEF in different tumor cell lines remains debatable, and additional studies are required to determine the role of SPDEF in tumor cells from different tissue origin. While the debate regarding role of SPDEF in tumor metastasis continues, no studies to date have examined the role SPDEF plays in tumor metastasis in vivo in any system. Results presented here demonstrate that forced SPDEF expression in cells that have decreased or no detectible SPDEF expression (PC3-Luc) impairs the ability of these cells to establish successful metastasis in vivo, whereas SPDEF knockdown in cells that have abundant SPDEF expression (LNCaP-Luc) enhances the ability of these cells to establish successful metastasis by increasing tumor cell survival at metastatic sites. Moreover, these effects of SPDEF on tumor cell metastasis are not an indirect result of modulation of tumor cell growth characteristics in vitro or in vivo. Our findings, that SPDEF expression limits the ability of prostate tumor cells to survive in the circulation and thereby establish metastases, are concordant with studies showing that SPDEF protein expression is decreased or undetectable in a majority of advanced/metastatic human prostate tumors (12, 14), suggesting that the loss of SPDEF may in part contribute to prostate tumor metastasis, at least in subsets of patients. This would explain the positive correlation between the loss of SPDEF expression in clinical specimens of prostate cancer and cancer specific death in these patients, as observed recently (15). Our exciting results suggest that SPDEF may represent a modifiable therapeutic target to prevent or decrease tumor cell survival or growth at secondary sites in prostate cancer and perhaps other malignancies. Moreover, the fact that SPDEF knockdown in LNCaP-Luc cells allows these cells to become metastatic is in itself exciting, as LNCaP tumor cells are known to be a non-aggressive, non-metastatic cell line. Thus, it appears that the loss of SPDEF may play a central role in conferring the metastatic capacity to tumor cells.
These affects of SPDEF expression modulation on tumor metastasis were independent of effects on cell growth. We did not detect any differences in growth patterns of prostate cancer cells following modulation of SPDEF levels. In addition, we did not detect any difference in p21 levels following modulation of SPDEF levels in our cells. These results are in contrast to the results of Schaefer et al. (10), who observed that SPDEF overexpression decreased proliferation in transformed mouse breast epithelial cells due to increased p21 expression. These discrepancies could be attributed to either context specific functions of SPDEF as SPDEF may function at different levels in epithelial cells from breast and prostate, or the discrepancy in the observations could have resulted from expression of human SPDEF in mouse breast epithelial cells, as Schaefer et al. (10) expressed human cell-derived SPDEF in mouse breast cells, whereas this study expressed human SPDEF in the context of human prostate cancer cells. Additional studies are needed to confirm or rule out the underlying causes of these discrepancies.
Of the genes that have been suggested to be modulated by SPDEF in the context of other cells, we report that SPDEF expression in cells that have decreased or no detectible SPDEF expression (PC3-Luc) consistently suppressed expression of MMP9 and MMP13. The observations about regulation of MMP9 are in line with our previous findings (12), whereas discovery of regulation of MMP13 by SPDEF is a new and novel finding. These findings, coupled with our observations that forced expression of MMP9 or MMP13 in SPDEF-expressing cells abrogates the effects of SPDEF on cell invasion, suggest that both MMP9 and MMP13 are not innocent bystanders but are significant downstream players of SPDEF-mediated effects on tumor invasion. Moreover, we have previously shown that there exists an inverse correlation between MMP9 and SPDEF expression levels in human prostate cancer tissue (12). Taken together, these findings are exciting given that MMP9 is a well known extracellular matrix degrading enzyme, which plays a significant role in remodeling of the metastatic niche (18, 19) and is correlated with increased invasive and metastatic tumors, including nasopharyngeal carcinoma (35), colorectal cancer (36), pancreatic cancer (37), gastric carcinoma (38), breast cancer (39), and prostate cancer (40, 41). Moreover, MMPs are known to have a considerable effect on the capability of tumor cells to grow at secondary sites (42). Of particular interest, MMP9 was reported to be required for murine prostate carcinoma metastasis but not primary tumor growth (43), and a separate study reported reduced lung metastasis in MMP9-deficient hosts (21). Moreover, MMP9 expression has a profound effect on the development of angiogenic vasculature where MMP9 has been shown to break down the basement membrane to release VEGF, thereby causing an increase in angiogenesis (44). This release of VEGF has also been shown to promote MMP9 and MMP13 expression, further perpetuating tumor growth and metastases (11). Thus, SPDEF may indirectly modulate several additional pathways required for successful metastases.
Given the implications of our important findings, additional studies are warranted to further characterize the role and mechanism of SPDEF-mediated effects on tumor metastasis. Of immediate interest is the potential use of SPDEF, MMP9, and/or MMP13 as a diagnostic and/or prognostic biomarkers to distinguish indolent from aggressive disease as previously proposed (12). Moreover, the use of MMP9 and/or MMP13 as downstream readouts of SPDEF expression may offer an excellent secreted biomarker that could be used in place of or in addition to prostate specific antigen testing. In addition, the mechanism(s) by which SPDEF is regulated is a significant question that must be answered. The ability to induce the expression of SPDEF through a gene therapy approach, small molecule inducer, or other regulatory mechanisms are important questions for the prevention or management of tumor metastasis.
Acknowledgments
We thank Douglas Lim for assistance measuring the subcutaneous tumors and Thomas Johnson for generating the luciferase expression plasmid and lentiviral constructs.
This work was supported in part by chair commitment (to H. K. K.) and Department of Surgery (School of Medicine Academic Enrichment Funds) (to H. K. K.).
American Cancer Society (2012) Cancer Facts and Figures 2012, Atlanta, GA.
- MMP9
- matrix metalloproteinase 9
- Luc
- luciferase
- VC
- vector control
- SPDEF-KD
- SPDEF knockdown
- OE
- overexpressed.
REFERENCES
- 1. Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 2. Chambers A. F., Groom A. C., MacDonald I. C. (2002) Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2, 563–572 [DOI] [PubMed] [Google Scholar]
- 3. Naumov G. N., MacDonald I. C., Chambers A. F., Groom A. C. (2001) Solitary cancer cells as a possible source of tumour dormancy? Semin. Cancer Biol. 11, 271–276 [DOI] [PubMed] [Google Scholar]
- 4. Naumov G. N., MacDonald I. C., Weinmeister P. M., Kerkvliet N., Nadkarni K. V., Wilson S. M., Morris V. L., Groom A. C., Chambers A. F. (2002) Persistence of solitary mammary carcinoma cells in a secondary site. Cancer Res. 62, 2162–2168 [PubMed] [Google Scholar]
- 5. Sharrocks A. D. (2001) The ETS-domain transcription factor family. Nat. Rev. Mol. Cell Biol. 2, 827–837 [DOI] [PubMed] [Google Scholar]
- 6. Steffan J. J., Koul H. K. (2011) Prostate-derived ETS factor (PDEF): A putative tumor metastasis suppressor. Cancer Lett. 310, 109–117 [DOI] [PubMed] [Google Scholar]
- 7. Sood A. K., Kim H., Geradts J. (2012) PDEF in prostate cancer. Prostate 72, 592–596 [DOI] [PubMed] [Google Scholar]
- 8. Turner D. P., Moussa O., Sauane M., Fisher P. B., Watson D. K. (2007) Prostate-derived ETS factor is a mediator of metastatic potential through the inhibition of migration and invasion in breast cancer. Cancer Res. 67, 1618–1625 [DOI] [PubMed] [Google Scholar]
- 9. Gunawardane R. N., Sgroi D. C., Wrobel C. N., Koh E., Daley G. Q., Brugge J. S. (2005) Novel role for PDEF in epithelial cell migration and invasion. Cancer Res. 65, 11572–11580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schaefer J. S., Sabherwal Y., Shi H. Y., Sriraman V., Richards J., Minella A., Turner D. P., Watson D. K., Zhang M. (2010) Transcriptional regulation of p21/CIP1 cell cycle inhibitor by PDEF controls cell proliferation and mammary tumor progression. J. Biol. Chem. 285, 11258–11269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghosh S., Basu M., Roy S. S. (2012) ETS-1 protein regulates vascular endothelial growth factor-induced matrix metalloproteinase-9 and matrix metalloproteinase-13 expression in human ovarian carcinoma cell line SKOV-3. J. Biol. Chem. 287, 15001–15015 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Johnson T. R., Koul S., Kumar B., Khandrika L., Venezia S., Maroni P. D., Meacham R. B., Koul H. K. (2010) Loss of PDEF, a prostate-derived Ets factor is associated with aggressive phenotype of prostate cancer: Regulation of MMP 9 by PDEF. Mol Cancer 9, 148. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13. Sood A. K., Saxena R., Groth J., Desouki M. M., Cheewakriangkrai C., Rodabaugh K. J., Kasyapa C. S., Geradts J. (2007) Expression characteristics of prostate-derived Ets factor support a role in breast and prostate cancer progression. Hum. Pathol. 38, 1628–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsujimoto Y., Nonomura N., Takayama H., Yomogida K., Nozawa M., Nishimura K., Okuyama A., Nozaki M., Aozasa K. (2002) Utility of immunohistochemical detection of prostate-specific Ets for the diagnosis of benign and malignant prostatic epithelial lesions. Int. J. Urol. 9, 167–172 [DOI] [PubMed] [Google Scholar]
- 15. Ghadersohi A., Sharma S., Zhang S., Azrak R. G., Wilding G. E., Manjili M. H., Li F. (2011) Prostate-derived Ets transcription factor (PDEF) is a potential prognostic marker in patients with prostate cancer. Prostate 71, 1178–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bodenstine T. M., Welch D. R. (2008) Metastasis suppressors and the tumor microenvironment. Cancer Microenviron. 1, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alix-Panabières C., Riethdorf S., Pantel K. (2008) Circulating tumor cells and bone marrow micrometastasis. Clin. Cancer Res. 14, 5013–5021 [DOI] [PubMed] [Google Scholar]
- 18. van Kempen L. C., Coussens L. M. (2002) MMP9 potentiates pulmonary metastasis formation. Cancer Cell 2, 251–252 [DOI] [PubMed] [Google Scholar]
- 19. Deryugina E. I., Quigley J. P. (2006) Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 25, 9–34 [DOI] [PubMed] [Google Scholar]
- 20. Itoh T., Tanioka M., Matsuda H., Nishimoto H., Yoshioka T., Suzuki R., Uehira M. (1999) Experimental metastasis is suppressed in MMP-9-deficient mice. Clin. Exp. Metastasis 17, 177–181 [DOI] [PubMed] [Google Scholar]
- 21. Hiratsuka S., Nakamura K., Iwai S., Murakami M., Itoh T., Kijima H., Shipley J. M., Senior R. M., Shibuya M. (2002) MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2, 289–300 [DOI] [PubMed] [Google Scholar]
- 22. Cazorla M., Hernández L., Nadal A., Balbín M., López J. M., Vizoso F., Fernández P. L., Iwata K., Cardesa A., López-Otín C., Campo E. (1998) Collagenase-3 expression is associated with advanced local invasion in human squamous cell carcinomas of the larynx. J. Pathol. 186, 144–150 [DOI] [PubMed] [Google Scholar]
- 23. Feldman R. J., Sementchenko V. I., Gayed M., Fraig M. M., Watson D. K. (2003) Pdef expression in human breast cancer is correlated with invasive potential and altered gene expression. Cancer Res. 63, 4626–4631 [PubMed] [Google Scholar]
- 24. Turner D. P., Findlay V. J., Moussa O., Semenchenko V. I., Watson P. M., LaRue A. C., Desouki M. M., Fraig M., Watson D. K. (2011) Mechanisms and functional consequences of PDEF protein expression loss during prostate cancer progression. Prostate 71, 1723–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Steffan J. J., Snider J. L., Skalli O., Welbourne T., Cardelli J. A. (2009) Na+/H+ Exchangers and RhoA regulate acidic extracellular pH-induced lysosome trafficking in prostate cancer cells. Traffic 10, 737–753 [DOI] [PubMed] [Google Scholar]
- 26. Kumar B., Koul S., Khandrika L., Meacham R. B., Koul H. K. (2008) Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 68, 1777–1785 [DOI] [PubMed] [Google Scholar]
- 27. Steffan J. J., Cardelli J. A. (2010) Thiazolidinediones induce Rab7-RILP-MAPK-dependent juxtanuclear lysosome aggregation and reduce tumor cell invasion. Traffic 11, 274–286 [DOI] [PubMed] [Google Scholar]
- 28. Findlay V. J., Turner D. P., Yordy J. S., McCarragher B., Shriver M. R., Szalai G., Watson P. M., Larue A. C., Moussa O., Watson D. K. (2011) Prostate-derived ETS factor regulates epithelial-to-mesenchymal transition through both SLUG-dependent and independent mechanisms. Genes Cancer 2, 120–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moss N. M., Liu Y., Johnson J. J., Debiase P., Jones J., Hudson L. G., Munshi H. G., Stack M. S. (2009) Epidermal growth factor receptor-mediated membrane type 1 matrix metalloproteinase endocytosis regulates the transition between invasive versus expansive growth of ovarian carcinoma cells in three-dimensional collagen. Mol. Cancer Res. 7, 809–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oettgen P., Finger E., Sun Z., Akbarali Y., Thamrongsak U., Boltax J., Grall F., Dube A., Weiss A., Brown L., Quinn G., Kas K., Endress G., Kunsch C., Libermann T. A. (2000) PDEF, a novel prostate epithelium-specific Ets transcription factor, interacts with the androgen receptor and activates prostate-specific antigen gene expression. J. Biol. Chem. 275, 1216–1225 [DOI] [PubMed] [Google Scholar]
- 31. Ghadersohi A., Sood A. K. (2001) Prostate epithelium-derived Ets transcription factor mRNA is overexpressed in human breast tumors and is a candidate breast tumor marker and a breast tumor antigen. Clin. Cancer Res. 7, 2731–2738 [PubMed] [Google Scholar]
- 32. Ghadersohi A., Odunsi K., Zhang S., Azrak R. G., Bundy B. N., Manjili M. H., Li F. (2008) Prostate-derived Ets transcription factor as a favorable prognostic marker in ovarian cancer patients. Int. J. Cancer 123, 1376–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodabaugh K. J., Mhawech-Fauceglia P., Groth J., Lele S., Sood A. (2007) Prostate-derived Ets factor is overexpressed in serous epithelial ovarian tumors. Int. J. Gynecol. Pathol. 26, 10–15 [DOI] [PubMed] [Google Scholar]
- 34. Moussa O., Turner D. P., Feldman R. J., Sementchenko V. I., McCarragher B. D., Desouki M. M., Fraig M., Watson D. K. (2009) PDEF is a negative regulator of colon cancer cell growth and migration. J. Cell Biochem. 108, 1389–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Z., Li L., Yang Z., Luo W., Li X., Yang H., Yao K., Wu B., Fang W. (2010) Increased expression of MMP9 is correlated with poor prognosis of nasopharyngeal carcinoma. BMC Cancer 10, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bendardaf R., Buhmeida A., Hilska M., Laato M., Syrjänen S., Syrjänen K., Collan Y., Pyrhönen S. (2010) MMP-9 (gelatinase B) expression is associated with disease-free survival and disease-specific survival in colorectal cancer patients. Cancer Invest. 28, 38–43 [DOI] [PubMed] [Google Scholar]
- 37. Tian M., Cui Y. Z., Song G. H., Zong M. J., Zhou X. Y., Chen Y., Han J. X. (2008) Proteomic analysis identifies MMP-9, DJ-1 and A1BG as overexpressed proteins in pancreatic juice from pancreatic ductal adenocarcinoma patients. BMC Cancer 8, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao Z. S., Wang Y. Y., Ye Z. Y., Tao H. Q. (2009) Prognostic value of tumor-related molecular expression in gastric carcinoma. Pathol. Oncol. Res. 15, 589–596 [DOI] [PubMed] [Google Scholar]
- 39. Provatopoulou X., Gounaris A., Kalogera E., Zagouri F., Flessas I., Goussetis E., Nonni A., Papassotiriou I., Zografos G. (2009) Circulating levels of matrix metalloproteinase-9 (MMP-9), neutrophil gelatinase-associated lipocalin (NGAL) and their complex MMP-9/NGAL in breast cancer disease. BMC Cancer 9, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Castellano G., Malaponte G., Mazzarino M. C., Figini M., Marchese F., Gangemi P., Travali S., Stivala F., Canevari S., Libra M. (2008) Activation of the osteopontin/matrix metalloproteinase-9 pathway correlates with prostate cancer progression. Clin. Cancer Res. 14, 7470–7480 [DOI] [PubMed] [Google Scholar]
- 41. Zhang L., Shi J., Feng J., Klocker H., Lee C., Zhang J. (2004) Type IV collagenase (matrix metalloproteinase-2 and -9) in prostate cancer. Prostate Cancer Prostatic Dis. 7, 327–332 [DOI] [PubMed] [Google Scholar]
- 42. Chambers A. F., Matrisian L. M. (1997) Changing views of the role of matrix metalloproteinases in metastasis. J. Natl. Cancer Inst. 89, 1260–1270 [DOI] [PubMed] [Google Scholar]
- 43. Sehgal G., Hua J., Bernhard E. J., Sehgal I., Thompson T. C., Muschel R. J. (1998) Requirement for matrix metalloproteinase-9 (gelatinase B) expression in metastasis by murine prostate carcinoma. Am. J. Pathol. 152, 591–596 [PMC free article] [PubMed] [Google Scholar]
- 44. Bergers G., Brekken R., McMahon G., Vu T. H., Itoh T., Tamaki K., Tanzawa K., Thorpe P., Itohara S., Werb Z., Hanahan D. (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2, 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]