

Background: Glucocorticoid receptor (GR) activation can inhibit apoptosis through mechanisms not fully clarified.
Results: GR activation induced PKCϵ expression and protected MCF10A-myc cells from p53-dependent apoptosis. PKCϵ silencing reversed the protective effect of GR.
Conclusion: Activated GR can inhibit p53-dependent apoptosis through induction of PKCϵ.
Significance: The results demonstrate PKCϵ plays an important role in the signaling pathway activated during GR-induced inhibition of apoptosis.
Keywords: Apoptosis, Breast Cancer, Cancer Biology, p53, Protein Kinase C (PKC)
Abstract
Glucocorticoid receptor (GR) is a ligand-dependent transcription factor that can promote apoptosis or survival in a cell-specific manner. Activated GR has been reported to inhibit apoptosis in mammary epithelial cells and breast cancer cells by increasing pro-survival gene expression. In this study, activated GR inhibited p53-dependent apoptosis in MCF10A cells and human mammary epithelial cells that overexpress the MYC oncogene. Specifically, GR agonists hydrocortisone or dexamethasone inhibited p53-dependent apoptosis induced by cisplatin, ionizing radiation, or the MDM2 antagonist Nutlin-3. In contrast, the GR antagonist RU486 sensitized the cells to apoptosis by these agents. Apoptosis inhibition was associated with maintenance of mitochondrial membrane potential, diminished caspase-3 and -7 activation, and increased expression at both the mRNA and protein level of the anti-apoptotic PKC family member PKCϵ. Knockdown of PKCϵ via siRNA targeting reversed the protective effect of dexamethasone and restored apoptosis sensitivity. These data provide evidence that activated GR can inhibit p53-dependent apoptosis through induction of the anti-apoptotic factor PKCϵ.
Introduction
The tumor suppressor protein p53 is a key regulator of multiple cellular processes, and evidence suggests that apoptosis is critical for its tumor suppressor function (1). The main function of p53 is to maintain genetic stability in response to various oncogenic challenges, such as DNA damage or inappropriate oncogene signaling. p53 carries out this function by inducing cell cycle arrest, apoptosis, or senescence. As such, p53 is often referred to as the “guardian of the genome.” p53 is negatively regulated by MDM2 through different mechanisms in coordination with MDMX (MDM4). Upon overexpression, MDM2 binds the transcription domain of p53 and blocks its ability to activate gene transcription (2–4). MDM2 also functions as an E3 ubiquitin ligase, mediating the ubiquitination and proteasome degradation of p53 (4–6). The type of response that follows p53 activation depends on a number of factors. Importantly, oncogenic transformation can cause a switch in the cell's response to p53 activation from growth arrest to programmed cell death (7). As a result, transformed tumor cells may be more prone to undergo apoptosis following p53 activation than corresponding normal cells. Inactivation of p53 not only promotes tumorigenesis and cancer progression but also confers cancer cells with an ability to evade death induced by therapeutic agents (8).
Breast cancer is the leading cause of cancer-related death among women worldwide. In 2011, according to the American Cancer Society, more than 230,000 new cases of breast cancer and ∼40,000 breast cancer-related deaths in women have been registered in the United States alone (American Cancer Society Breast Cancer Facts and Figures). A major factor contributing to the development of breast cancer is inactivation of p53 (9). Thus, inactivating point mutations in the P53 gene are observed in 20–30% of breast cancers (10, 11). Furthermore, wild-type p53 is often inactivated through alternative mechanisms in cancers where the P53 gene is not mutated. Mechanisms for wild-type p53 inactivation in breast cancer include overexpression of MDM2, silencing of p14Arf (which causes hyper-activation of MDM2), and cytoplasmic sequestration (12, 13). Many cytotoxic drugs (e.g. cisplatin) and irradiation can damage DNA and can activate wild-type p53 (14, 15). In several reports, p53 wild-type cancer cells respond better to DNA-damaging therapeutics than p53-mutated or p53-null cancer cells due to activation of wild-type p53 growth-inhibitory pathways (16, 17).
Conzen and co-workers (18–21) have reported that activation of the glucocorticoid receptor (GR)2 inhibits apoptosis and promotes survival of breast cancer and breast epithelial cells by increasing expression of pro-survival genes. Recent studies indicate that GR activation is associated with poor prognosis in estrogen receptor-negative breast cancer (22). Glucocorticoid synthesis is enhanced following stressful conditions leading and acting mostly through GR to regulate inflammatory and immune responses, as well as cellular proliferation and apoptosis. Most glucocorticoid-mediated effects result from the ability of activated GR to act as a transcription factor, either through a DNA binding-dependent mechanism or through cross-talk and/or interference with other transcription factors such as activator protein-1 (AP-1) (23), signal transducers and activators of transcription-5 (Stat-5) (24), and nuclear factor-κB (NF-κB) (25). The accumulated evidence shows that GR activation has a dual and cell type-specific role in cell death regulation. GR is able to induce apoptosis in lymphocytes, leukemia, lymphoma, and multiple myeloma cells (26, 27). However, in other cell types such as hepatocytes, vascular endothelial cells, osteoclasts, and particularly in mammary epithelial cells, GR can inhibit apoptosis induced by a variety of signaling events (18). Several groups have observed that glucocorticoids can inhibit chemotherapy-induced apoptosis in vitro (18, 19) and in vivo (28).
We wished to gain insight into the mechanisms by which GR activation inhibits apoptosis and promotes survival in breast epithelial cells. To this end, we addressed the involvement of protein kinase Cϵ (PKCϵ) as a potential mediator, because overexpression of PKCϵ is found in various cancers, including breast cancer, and is considered an important marker of negative disease outcome (29). PKC is a family of serine/threonine kinases involved in several processes, including proliferation, differentiation, survival, apoptosis, and migration (30–32). Based on the structure of the regulatory domain, PKC isoforms are divided into three subgroups as follows: classical (PKCα, -βI, -βII, and -γ), novel (PKCδ, -ϵ, -η, and -θ), and atypical (PKCζ and -ι/λ). Classical and novel PKCs contain a diacylglycerol (DAG)-binding C1 domain and are therefore regulated by activation of pathways that lead to DAG generation. Atypical PKCs are DAG-insensitive and regulated in a different manner (31). Previous studies have implicated the DAG-sensitive classical and novel PKC isoforms in promoting malignant features of breast cancer cells. In addition, earlier studies reported that the synthetic GR agonist dexamethasone (Dex) could increase PKCϵ expression (33, 34). However, the potential involvement of PKCϵ in GR-regulated inhibition of apoptosis has not been explored.
Here, we demonstrate that the GR agonists Dex and hydrocortisone can protect MCF10Amyc and HMEC+myc mammary epithelial cells from p53-induced apoptosis. Activation of GR by hydrocortisone/Dex and attenuation of p53-induced apoptosis is associated with increased expression of PKCϵ mRNA and protein, maintenance of mitochondrial membrane potential, and diminished caspase-3/7 activation. In contrast, the GR antagonist RU486 suppressed the anti-apoptotic effect of GR and enhanced apoptosis in MCF10Amyc cells. Finally, siRNA-mediated knockdown of PKCϵ reversed the protective effect of Dex, rendering the cells susceptible to apoptosis. These data suggest that PKCϵ plays an important role in the signaling pathway activated by Dex during GR-induced inhibition of apoptosis.
MATERIALS AND METHODS
Cell Lines and Culture Conditions
The c-Myc-transformed human breast epithelial cell line MCF10Amyc was a gift from Dr. Suzanne Conzen, Department of Medicine, University of Chicago. Ras-transformed human breast epithelial cell line MCF10ANeoT was a gift from Dr. Jitesh Pratap, Department of Anatomy and Cell Biology, Rush University Medical Center, Chicago. Normal HMECs and HMECs made to overexpress c-Myc were gifts from Dr. Steven Elledge (35). Nontransformed MCF10A cells were purchased from American Type Culture Collection. All these cells were grown in DMEM/F-12 supplemented with 5 μg/ml insulin, 10 ng/ml EGF, 0.5 μg/ml hydrocortisone, 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin. These cells were plated 24 h before being treated with Nutlin-3 (5 μm, Sigma), irradiation (4, 7, or 10 Gy), cisplatin (1, 5, or 15 μm; Bedford Laboratory), or as indicated. Dexamethasone and mifepristone (alternative name, RU486) were purchased from Sigma. Tetramethylrhodamine ethyl ester perchlorate was purchased from Invitrogen.
Flow Cytometry
For cell cycle and apoptosis analysis, cells were harvested and fixed in 25% ethanol overnight. The cells were then stained with propidium iodide (10 μg/ml, Calbiochem). Flow cytometry analysis was performed on FACS Canto (BD Biosciences) and analyzed with CellQuest (BD Biosciences) and FlowJo (Treestar Inc.). For each sample, 10,000 events were collected. For annexin-V staining, cells were stained with annexin V-PE and 7-aminoactinomycin D (Pharmingen) according to manufacturer's instructions. For mitochondrial potential (ΔΨm) analysis, cells were harvested and stained with tetramethylrhodamine ethyl ester (0.1 μm).
Apoptosis Assay/Caspase-Glo® 3/7 Assay
Apoptosis was analyzed in triplicate assays with the Caspase-Glo-3/7 Assay (Promega Biotech, Madison, WI). Briefly, 10,000 cells per well were seeded in 96-well plates, and the cells were incubated for 24 h with 2.5, 5.0, or 10 μm Nutlin in 100 μl of medium. Subsequently, 100 μl of the caspase-3/7 reconstituted reagent was added to the cells and incubated for 30 min in the incubator. After incubation, luminescence of each sample was measured in a plate-reading luminometer (Wallac Victor 2/PerkinElmer Life Sciences) at 485/527 nm. Results were presented as the mean (signal-to-noise ratios) ± S.E. of the triplicate assays.
Cell Fractionation
For preparation of soluble and particulate fractions, the cells were homogenized in buffer A consisting of 10 mm Tris-HCl, pH 7.5, 1.5 mm MgCl2, 10 mm KCl, mini protease inhibitor mixture tablets from Roche Diagnostics, and 10.6 mm 2-mercaptoethanol. Cells were allowed to swell in buffer A for at least 15 min. Triton X-100 was then added to a final concentration of 0.2%, and cells were incubated on ice for 15 min with light vortexing every 5 min, followed by centrifugation (7,000 rpm for 5 min). The supernatant (Triton X-100-soluble (0.2%) fraction) was removed, and the pellet was resuspended in buffer A with 400 mm NaCl and 0.4% Triton X-100 for 30 min on ice with light vortexing every 10 min. The insoluble material was removed by centrifugation at 14,000 rpm for 15 min, and the supernatant was collected (Triton X-100-soluble (0.4%) fraction). The remaining pellet (Triton X-100-insoluble) was resuspended in RIPA (radioimmunoprecipitation) lysis buffer.
Immunoblotting
Whole cell extracts were prepared by resuspending cell pellets in lysis buffer, resolved by SDS-PAGE, and transferred to polyvinylidene difluoride membranes (PerkinElmer Life Sciences). Antibodies to PKCϵ, GR, p53, MDM2, p21, and poly(ADP-ribose) polymerase were purchased from Santa Cruz Biotechnology; antibodies to Noxa and Bax were purchased from Calbiochem and Upstate, respectively. Primary antibodies were detected with goat anti-mouse or goat anti-rabbit antibodies conjugated to horseradish peroxidase (Jackson ImmunoResearch) using enhanced chemiluminescence (PerkinElmer Life Sciences).
Reverse Transcription and Quantitative Real Time PCR
To investigate the levels of PUMA, Bax, p21, Noxa, and PKCϵ mRNA, quantitative real time PCR was performed. After treatment with 10 μm Nutlin for 24 h, the complementary DNA (cDNA) was prepared by reverse transcription of 1 μg of RNA in a final volume of 20 μl using SuperScript III First-strand Synthesis SuperMix system (Invitrogen) following the manufacturer's instructions. The quantitative real time PCR was run in a 7300 Real Time PCR System (Applied Biosystems, Foster, CA) using SYBR Green PCR master mix (Applied Biosystems, Foster City, CA) following the manufacturer's instructions. Thermocycling was done in a final volume of 20 μl containing 2 μl of cDNA and 400 nmol/liter of primers as follows: PKCϵ forward, 5′-CAT GTT GGC AGA ACT CAA GGG CAA-3′, and reverse, 5′-TGC AGT CCA CGT CAT CAT CCT GAA-3′; p21 forward, 5′-GGA CCT GGA GACTCT CA-3′, and reverse 5′-CCT CTT GGA GAA GAT CAG-3′; PUMA forward, 5′-GAC CTC AAC GCA CAGTA-3′, and reverse 5′-CTA ATT GGG CTC CAT CT-3′; Bax forward. 5′-TCA GCA CAG ATT AGT TTC TG-3′, and reverse, 5′-GGG ATT ACA GGC ATG AGC TA-3′; Noxa forward, 5′-TGGAAGTCGAGTGTGCTACTCAACT-3′, and reverse, 5′-AGATTCAGAAGTTTCTGCCGGAA-3′; GAPDH forward, 5′-GTC GGA GTC AAC GGA TTT GGT-3′, and reverse 5′-TTA AAA GCA GCC CTG GTG ACC-3′. All samples were amplified in triplicate using the following cycle scheme: after initial denaturation of the samples at 95 °C for 2 min, 40 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s were done, fluorescence was measured in every cycle, and mRNA levels were normalized by the GAPDH values in all samples. A melting curve was run after PCR by increasing the temperature from 50 to 96 °C (0.5 °C increments). A single peak was obtained for targets, supporting the specificity of the reaction.
siRNA-mediated Transient Knockdown
PKCϵ and p53 RNAi and control RNAi (On-target plus siControl nontargeting pool) were purchased from Dharmacon and transfected according to the manufacturer's guidelines using DharmaFECT I reagent. Treatments were applied 24 h after transfection.
RESULTS
GR Activation Suppresses Nutlin-induced Apoptosis in MCF10Amyc Cells
When cultured in defined (serum-free) medium, mammary epithelial cells require glucocorticoids, epidermal growth factor (EGF), and insulin for survival and growth (36). EGF and insulin can promote survival, in part, through activation of the AKT pathway (37, 38). Glucocorticoids (e.g. hydrocortisone) promote survival through activation of GR. At least two mechanisms have been described through which activated GR can promote survival as follows: 1) through direct interaction with and inhibition of p53 (39, 40), and 2) through its ability to function as a transcription factor and increase expression of anti-apoptotic genes such as SGK-1, MPK1, and BCL-XL (19, 21, 41). MCF10Amyc cells are immortalized mammary epithelial cells that overexpress the MYC oncogene. In the first experiments, MCF10Amyc cells were cultured in serum-free medium in the presence (+ALL) or absence (−ALL) of hydrocortisone, EGF, and insulin. The cells were then untreated or treated with the small molecule MDM2 antagonist and p53 activator Nutlin (42) and visually examined 24 h later. Cells in growth factor-enriched medium were largely unaffected by Nutlin treatment (Fig. 1A). In contrast, cells cultured in the absence of growth factors (−ALL) were rounded up and apparently killed by Nutlin treatment. We used annexin-V staining and sub-G1 DNA content to quantify the percent cells undergoing apoptosis. Nutlin caused cell cycle arrest with relatively low apoptosis in growth factor-enriched medium (+ALL), but it caused abundant apoptosis (70–80% of cells) in the absence of growth factors (−ALL) (Fig. 1, B and C; see DNA histograms in supplemental Fig. 1). Each growth factor was then removed individually from the medium, and similar experiments were conducted. As shown in Fig. 1, B and C, removal of hydrocortisone alone rendered MCF10Amyc cells highly susceptible to apoptosis by Nutlin, whereas removal of either EGF or insulin had a relatively small effect.
FIGURE 1.
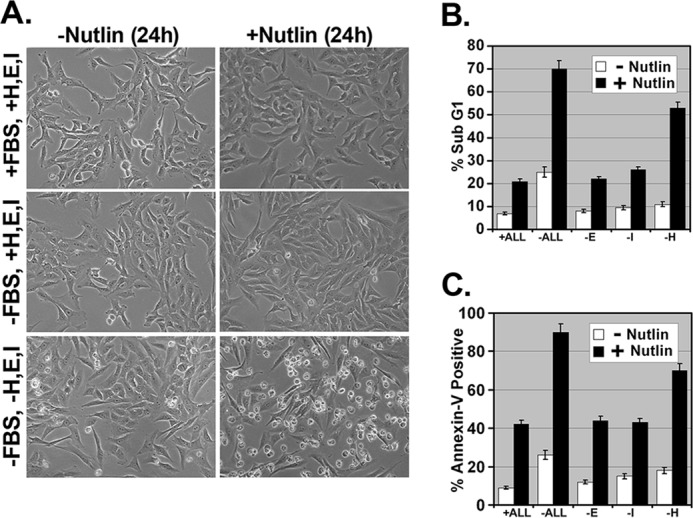
Nutlin induces apoptosis in MCF10Amyc cell line in the absence of hydrocortisone. A, MCF10Amyc cells were starved of FBS (−FBS, +H, E, I) or FBS along with hydrocortisone (H), EGF (E), insulin (I) (−FBS, −H, E, I) or grown in normal media (+FBS, +H, E, I) for 30 min and treated with or without Nutlin (5 μm) for 24 h in the same condition. The representative images were taken under the magnification ×200. B and C, MCF10Amyc cells were starved of either FBS along hydrocortisone, EGF, insulin (−ALL), or EGF alone (−E) or insulin alone (−I), or hydrocortisone alone (−H) for 30 min, and these cells were further treated with or without Nutlin (5 μm) in the same condition for 24 h. Cells were harvested after 24 h and stained with propidium iodide to measure DNA content (B) (the percentage of sub-G1 cells), or stained for the percentage annexin V-positive cells (C). Samples in B and C were plated in three independent experiments.
We reasoned that if hydrocortisone inhibits apoptosis by activating GR, then other GR agonists would inhibit Nutlin-induced apoptosis, whereas GR antagonists would sensitize cells to Nutlin. To test this, MCF10Amyc cells were cultured in serum-free medium in the absence of growth factors (−ALL) and treated with Nutlin. In some cases, the cells were pretreated 30 min with the synthetic GR agonist Dex prior to Nutlin treatment. Apoptosis was determined 48 h later by sub-G1 DNA content, decreased mitochondrial membrane potential, and caspase-3 and -7 activation. As shown in Fig. 2, Nutlin-induced abundant apoptosis in these experiments using all three apoptosis criteria. Importantly, Dex pretreatment provided potent protection against Nutlin-induced apoptosis. Next, we asked whether the GR antagonist RU486 could overcome the protective effects of Dex. Although Dex protected cells from Nutlin-induced apoptosis, cells pretreated with a combination of Dex and RU486 underwent apoptosis when exposed to Nutlin (Fig. 2, A and B). This demonstrated that RU486 could abrogate the protective effects of Dex. Finally, we asked whether RU486 could sensitize cells to Nutlin under growth factor-enriched conditions (+ALL). As shown in Fig. 2, A and B, when cells in growth factor-enriched medium (+ALL) were treated with Nutlin alone they underwent relatively little apoptosis, but these same cells underwent abundant apoptosis when treated with a combination of Nutlin and RU486. In sum, GR agonists (hydrocortisone and Dex) inhibited Nutlin-induced apoptosis, whereas GR antagonists (RU486) sensitized cells to Nutlin. We conclude activated GR inhibits Nutlin-induced apoptosis in MCF10Amyc cells.
FIGURE 2.

GR activation protects against Nutlin-induced apoptosis and mitochondrial depolarization in MCF10Amyc cells. MCF10Amyc cells were starved of FBS/hydrocortisone/EGF/insulin (−ALL) or grown in hydrocortisone, EGF, and insulin without serum (+ALL), and these cells were treated with either vehicle, Dex (1 μm), Dex (1 μm)/RU486 (0.1 μm), or Dex (1 μm)/RU486 (1.0 μm) for 30 min. After 30 min of treatment, MCF10Amyc cells were further treated with or without Nutlin (Nut) (5 μm) in the same condition. A, percentage of sub-G1 cells (propidium iodide staining) was analyzed 72 h post-treatment. B, percentage of cells with low ΔΨm (tetramethylrhodamine ethyl ester staining) was analyzed 72 h post treatment. C, caspase-Glo-3/7 assay was performed with (+Dex) or without (−Dex) Dex for 30 min, and these cells were further treated with or without indicated Nutlin doses (2.5, 5.0, or 10.0 μm) for 24 h. The analysis was done in triplicate assays, and data are presented as mean ± S.E. (n = 3).
Dexamethasone Inhibits Nutlin-induced Apoptosis without Altering p53 Levels or Transcriptional Activity
There are multiple ways GR activation could inhibit Nutlin-induced apoptosis. First, Wasylyk and co-workers (39, 40) reported that GR binds p53 in a ligand (Dex)-dependent manner in neuroblastoma and umbilical vein endothelial cells and inhibits p53 by causing its MDM2-dependent degradation. Thus, GR could inhibit Nutlin-induced apoptosis by promoting p53 degradation. Second, GR may bind p53 on DNA and inhibit p53 from activating/repressing expression of pro- or anti-apoptotic genes. This model has recently been described for estrogen receptor inhibition of p53 (43, 44). Third, GR is a ligand-dependent transcription factor that can directly activate expression of pro-survival genes, such as SGK-1 and BCL-XL (21, 41). Thus, GR could inhibit Nutlin-induced apoptosis by activating one or more pro-survival genes that can overcome and prevent the apoptotic signals arising from Nutlin-induced p53. To examine these possibilities, MCF10Amyc cells were treated with Nutlin in the presence or absence of glucocorticoid (Dex). Levels of p53 and downstream p53 targets were determined at various time points after treatment (Fig. 3). If GR inhibits Nutlin-induced apoptosis by targeting p53 for degradation, then p53 would be increased to a lesser extent by Nutlin in the presence of Dex than in the absence of Dex. However, as shown in Fig. 3A, p53 increased to comparable levels and with similar kinetics in the absence or presence of Dex, suggesting that GR is not inhibiting Nutlin-induced apoptosis by targeting p53 for degradation. p53 downstream targets MDM2, p21, PUMA, and Bax were also induced by Nutlin in the presence or absence of Dex to comparable levels and with similar kinetics (Fig. 3, A and B). This demonstrates p53 transcriptional activity is not inhibited by Dex, at least for these p53 targets. Notably, expression of the p53 target gene NOXA was only minimally increased by Nutlin treatment, suggesting it may not play a role in Nutlin-induced apoptosis, although Noxa mRNA was decreased by Dex treatment. Although Dex did not inhibit p53 levels or activity in Nutlin-treated cells, it did diminish poly(ADP-ribose) polymerase cleavage at the 12- and 24-h time points and the level of activated (cleaved) caspase-3 (Fig. 3A). In total, the results suggest activated GR inhibits Nutlin-induced apoptosis by maintaining mitochondrial membrane potential (Fig. 2B) and diminishing caspase-3 activation (Fig. 3A), but not by inhibiting p53 transcriptional activity or targeting p53 for degradation.
FIGURE 3.
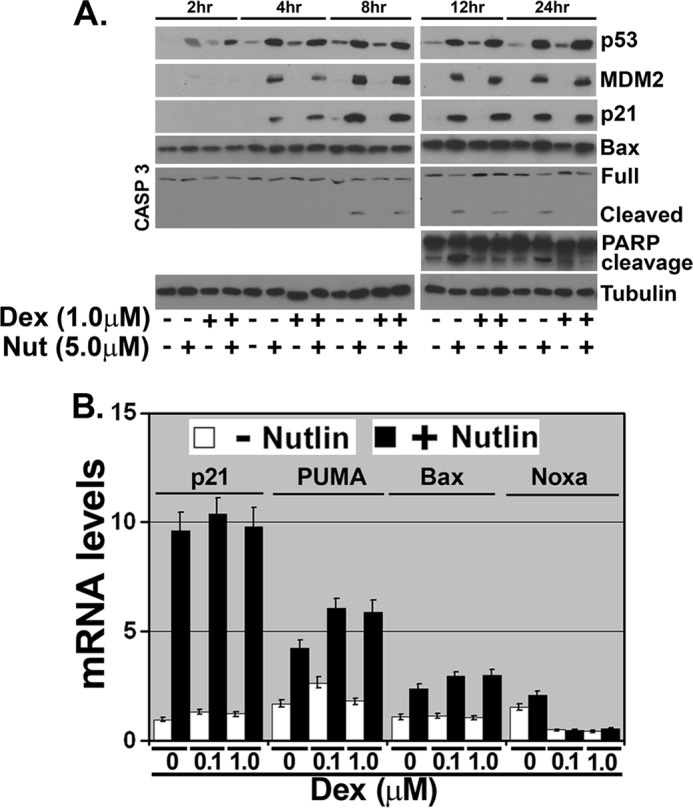
Dexamethasone protects against cell death but without altering p53 induction or transcriptional activity. MCF10Amyc cells starved of FBS/hydrocortisone/EGF/insulin (−ALL) were exposed to Dex (1 μm) for 30 min. The cells were then treated with Nutlin (Nut) (5 μm) or untreated for 2, 4, 8, 12, and 24 h in the continued presence of Dex. A, cell lysates were collected and analyzed by immunoblot analysis with indicated antibodies. B, total RNA was extracted from each treated group after 24 h and quantitative real time PCR (qRT-PCR) was performed using p21, PUMA, Bax, and Noxa-specific primer pairs.
GR Activation Increases PKCϵ mRNA and Protein Levels
Next, we sought out genes that are potentially activated by GR and that could inhibit apoptosis. In preliminary studies, mRNA levels for SGK-1 and BCL-XL were not increased in MCF10Amyc cells treated with Dex for 24 h (not shown), and we therefore sought out other potential GR targets. Earlier studies reported that Dex treatment was associated with increased PKCϵ expression in the rat brain and in coronary arteries (33, 34), suggesting PKCϵ could be a direct or indirect transcription target of GR. We analyzed PKCϵ expression in Dex- and Nutlin-treated MCF10Amyc cells. A strong induction of PKCϵ protein expression was detected after 2 h and increased up to 24 h following Dex treatment in MCF10Amyc (Fig. 4A). Nutlin appeared to decrease PKCϵ expression slightly at early time points (2 and 4 h), but this change was not detected at later time points. We further investigated the dose-dependent effect of Dex treatment after 24 h at both the protein (Fig. 4B) and mRNA (Fig. 4C) levels. GR is targeted for proteasomal degradation upon ligand binding/activation as part of an autoregulatory feedback loop that limits GR activity (45, 46). Consistent with this, GR levels were decreased in Dex-treated cells (Fig. 4B). We found a significant induction of PKCϵ mRNA and protein at Dex doses as low as 0.5 μm, and the expression of PKCϵ was largely unchanged at higher doses (1.0 and 2.0 μm) (Fig. 4, B and C). To validate that increased expression of PKCϵ resulted from GR activation, we analyzed PKCϵ expression in the presence of different growth factors or the absence/presence of the GR antagonist RU486. As shown in Fig. 4D, PKCϵ protein levels were higher in cells grown in medium with GR activators (Dex or hydrocortisone) as compared with cells grown in medium without GR activators. Moreover, when cells were treated with either RU486 in +ALL medium or RU486 with Dex in −ALL medium, we found the GR antagonist down-regulated PKCϵ expression (Fig. 4E). These results support GR activation increasing expression of PKCϵ.
FIGURE 4.
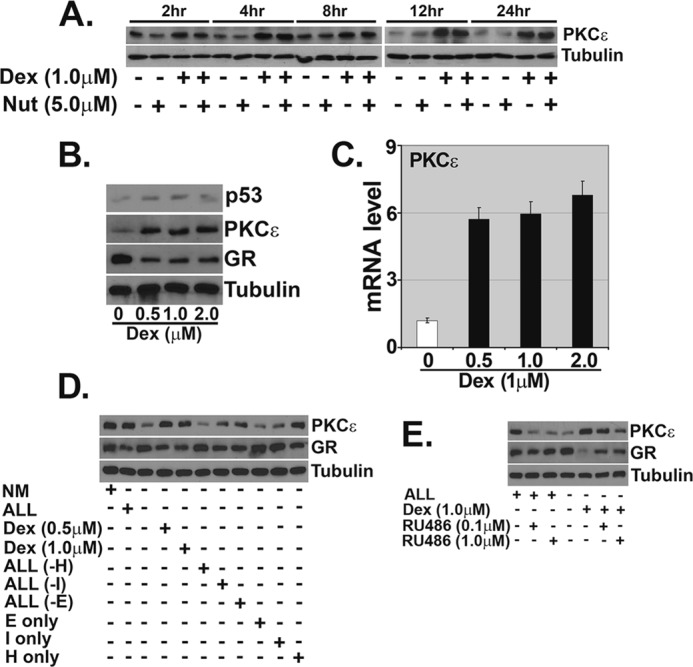
GR activation increases PKCϵ mRNA and protein levels. MCF10Amyc cells were treated as described in the legend to Fig. 3A. A, cell lysates were collected and analyzed by immunoblot analysis with PKCϵ antibody. Tubulin was probed as a loading control. B, MCF10Amyc cells were incubated with Dex (0–2.0 μm) for 24 h. At the end of the incubation, expression of p53, PKCϵ, and GR was measured by immunoblot analysis. C, qRT-PCR analysis of PKCϵ gene expression was performed after 24 h of incubation with Dex (0–2.0 μm). D, MCF10Amyc cells were serum-starved and grown in either normal media (NM) with serum, hydrocortisone (H)/EGF (E)/insulin (I) (+ALL), without hydrocortisone/EGF/insulin (−ALL), Dex (0.5 or 1.0 μm) only, EGF/insulin (ALL(−H)), hydrocortisone/EGF (ALL(−I)), hydrocortisone/insulin (ALL(−E)), EGF only (E only), insulin only (I only), and hydrocortisone only (H only) for 24 h. Cell lysates were collected and analyzed by immunoblot analysis with PKCϵ and GR antibodies. Tubulin was probed as a loading control. E, MCF10Amyc were serum-starved and treated with +ALL, +ALL+RU486 (0.1 μm), +ALL+RU486 (1.0 μm), −ALL, −ALL+Dex, −ALL+RU486 (0.1 μm), and −ALL+RU486 (1.0 μm). After 24 h, cell lysates were collected, and PKCϵ and GR expression levels were examined by immunoblot analysis. Nut, Nutlin.
Knockdown of PKCϵ by siRNA Abrogates Dexamethasone Protection from Nutlin-induced Apoptosis
Next, we wished to determine whether PKCϵ was required for the inhibition of apoptosis in Dex-treated cells. To this end, we employed siRNA to knock down PKCϵ in MCF10Amyc cells before treatment with Dex or Dex plus Nutlin. Immunoblotting showed PKCϵ effectively knocked down in cells transfected with PKCϵ siRNA (Fig. 5C). Visual examination and sub-G1 analysis showed that Dex could inhibit Nutlin-induced apoptosis in untransfected and cells transfected with control siRNA (siCon) but not in cells transfected with PKCϵ siRNA (Fig. 5, A and B; DNA histograms in supplemental Fig. 2). These results demonstrate PKCϵ is required for apoptosis inhibition by Dex. Immunoblotting again showed p53 and its downstream targets (p21, MDM2, and Bax) were induced comparably by Nutlin in either the presence or absence of Dex and were also induced comparably in cells transfected with control siRNA (siCon) or siRNA against PKCϵ (Fig. 5C). Consistent with results from Fig. 3B, Noxa expression increased very slightly in Nutlin-treated cells at the 48-h time point, but it was largely inhibited by Dex. In total, the results suggest activated GR inhibits Nutlin-induced apoptosis, at least in part, by increasing expression of PKCϵ and that this occurs without altering p53 levels or activity.
FIGURE 5.

Inhibition of PKCϵ abrogates dexamethasone protection from Nutlin-induced apoptosis in MCF10Amyc cells. MCF10Amyc cells were transfected with nontargeting siRNA (siCon) or PKCϵ specific siRNA (siPKCϵ). After 24 h, Dex (1 μm) was added for 30 min before adding Nutlin (5 μm) in −ALL condition. A, after 48 h post-Nutlin (Nut) treatment, the representative images were taken under the magnification ×100. B, cells were harvested at 48 h post-Nutlin treatment and stained with propidium iodide to measure DNA content. C, cell lysates were collected 24 and 48 h post-Nutlin treatment and analyzed by immunoblot analysis with indicated antibodies.
Phorbol Ester Inhibits Nutlin-induced Apoptosis in a PKCϵ-dependent Manner
Next, we asked whether activation of PKCϵ using a stimuli alternative to GR agonists could also inhibit Nutlin-induced apoptosis. Phorbol ester (PE) activates PKCϵ, and this is associated with a movement of PKCϵ from a soluble to an insoluble cell fraction (47, 48). MCF10Amyc cells were pretreated with PE and then tested for Nutlin-induced apoptosis. As expected, PE induced translocation of PKCϵ from Triton-soluble fractions to a more insoluble pellet fraction (Fig. 6C). In contrast, PKCδ localization was unaffected by PE treatment. Importantly, PE-treated cells were largely protected from Nutlin-induced apoptosis (Fig. 6A; DNA histograms in supplemental Fig. 3). We used siRNA knockdown to ask whether apoptosis protection in PE-treated cells required PKCϵ. Immunoblotting showed PKCϵ expression was lost in the siPKCϵ-transfected cells (Fig. 6B). As shown in Fig. 6A, PE pretreatment protected si-control transfected cells from Nutlin-induced apoptosis but failed to protect PKCϵ knockdown cells (DNA histograms in supplemental Fig. 3). These results suggest PE can inhibit Nutlin-induced apoptosis in a manner dependent on PKCϵ.
FIGURE 6.

Activation of PKCϵ by phorbol ester inhibits Nutlin-induced apoptosis in MCF10Amyc. A, MCF10Amyc cells were transfected with nontargeting siRNA (siCon) or PKCϵ specific siRNA (siPKCϵ). After 24 h, phorbol ester (PE) (100 nm) was added for 6 h before Nutlin (Nut) (5 μm) in −ALL condition. Cells were harvested at 48 h post-Nutlin treatment and stained with propidium iodide to measure DNA content. B, cell lysates were collected 24 h post-Nutlin treatment and analyzed by immunoblot analysis with indicated antibodies. C, MCF10Amyc cells were treated with either −ALL or −ALL+PE (100 nm) for 6 h and whole, Triton X-100 (0.2%)-soluble, Triton X-100 (0.4%)-soluble cell lysate and final pellet in RIPA buffer were collected to analyze PKCϵ and PKCδ expression levels by immunoblot analysis.
GR-activated PKCϵ and Nutlin-induced Apoptosis Is MCF10Amyc-specific
To confirm and extend our findings, we used two additional cell lines, nontransformed MCF10A cells and MCF10ANeoT (MCF10A cells transformed by the H-RAS oncogene). As shown in Fig. 7A, immunoblot analysis showed the expression of c-Myc, PKCϵ, GR, p53, and MDM2 after 24 h of Dex treatment in all three cell lines. Interestingly, PKCϵ protein expression was increased after Dex treatment only in MCF10Amyc but not in MCF10A and MCF10ANeoT cell lines. Similarly, PKCϵ mRNA was also more highly induced following Dex treatment in MCF10Amyc cells than in MCF10A or MCF10ANeoT (Fig. 7B). Next, we monitored apoptosis induction by Nutlin in all three cell lines and the potential for Dex to inhibit this apoptosis. p53 was induced comparably by Nutlin in all three cell lines, and p53 downstream targets p21 and MDM2 were induced comparably in MCF10Amyc and MCF10ANeoT (supplemental Fig. 4A). p21 and MDM2 were less induced by Nutlin in MCF10A cells (supplemental Fig. 4D). As shown in Fig. 7C, Nutlin induced abundant apoptosis in MCF10Amyc cells at 24–72-h time points, and this apoptosis was inhibited by Dex. In contrast, MCF10A and MCF10ANeoT cells were largely resistant to Nutlin-induced apoptosis and not protected by Dex (Fig. 7C: DNA histograms in supplemental Fig. 4, B–D). Similar results were obtained when apoptosis was monitored by annexin V staining and caspase3/7 activation (supplemental Fig. 4, E and F). These results indicate MCF10Amyc cells are more sensitive to apoptosis by Nutlin than the other MCF10A variants and are significantly protected by Dex and increased expression of PKCϵ.
FIGURE 7.
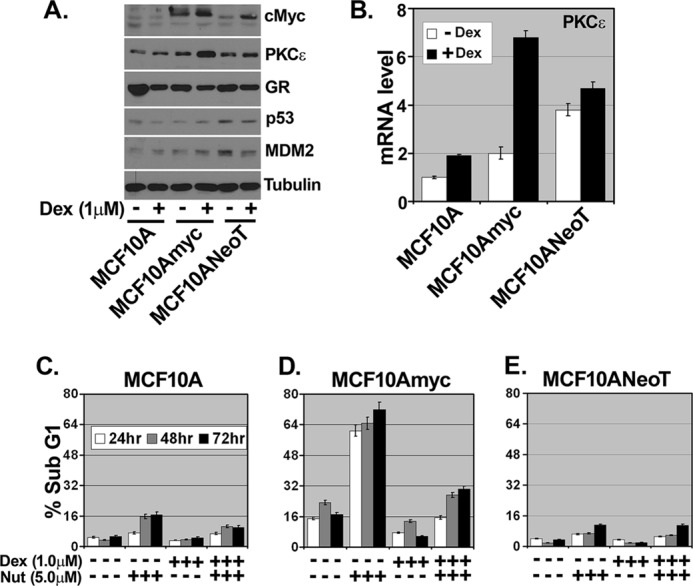
GR-activated PKCϵ and Nutlin-induced apoptosis are MCF10Amyc-specific. MCF10A, MCF10Amyc, and the ras-transformed human breast epithelial cell line MCF10ANeoT were serum-starved and then treated with Dex (1 μm) or untreated for 24 h. A, cell lysates were collected and analyzed by immunoblot analysis with indicated antibodies. B, total RNA was collected and qRT-PCR was performed using PKCϵ-specific primer pair. MCF10A (C), MCF10Amyc (D), and MCF10A NeoT (E) were starved of FBS/hydrocortisone/EGF/insulin (−ALL) and treated with Dex (1 μm) for 30 min. The cells were then treated with or without Nutlin (Nut) (5 μm) in the same condition for 24, 48, and 72 h. Cells were harvested and stained with propidium iodide to measure DNA content. The analysis was done in triplicate assays, and data are presented as mean ± S.E. (n = 3).
c-Myc Overexpression Can Render Cells Sensitive to Nutlin-induced Apoptosis
The results in Fig. 7 suggest mammary epithelial cells expressing high levels of c-Myc may be especially sensitive to Nutlin-induced apoptosis. Consistent with this possibility, c-Myc knockdown in MCF10Amyc cells via siRNA rendered the cells largely resistant to Nutlin-induced apoptosis (Fig. 8, A and B; DNA histograms in supplemental Fig. 5A). To examine this further, we tested Nutlin sensitivity in HMECs that do or do not overexpress c-Myc. As shown in Fig. 8C, the HMEC+myc cells were sensitive to Nutlin-induced apoptosis compared with control HMECs, which were largely resistant (DNA histograms in supplemental Fig. 5B). Next, we asked whether HMEC+myc cells could be protected from apoptosis by Dex and whether this required PKCϵ. Pretreatment with Dex protected HMEC+myc cells from Nutlin-induced apoptosis in two separate experiments (Fig. 9, A and B; DNA histograms in supplemental Fig. 6), coincident with increased PKCϵ expression (Fig. 9, C and D). Importantly, PKCϵ knockdown via siRNA abrogated to a large extent the protective effects of Dex in these cells, confirming apoptosis protection was largely PKCϵ-dependent.
FIGURE 8.

c-Myc overexpression can render cells sensitive to Nutlin-induced apoptosis. A, MCF10Amyc cells were transfected with nontargeting siRNA (siCon) or c-Myc-specific siRNA (sic-Myc). After 24 h, Nutlin (Nut) (5 μm) was added in −ALL condition. Cells were harvested at 48 h post-Nutlin treatment and stained with propidium iodide to measure DNA content. B, cell lysates were collected 24 h post-Nutlin treatment and analyzed by immunoblot analysis with indicated antibodies. C, HMEC and HMECmyc cells were treated with Nutlin (5 μm) for 72 h. Cells were harvested and stained with propidium iodide to measure DNA content. D, cell lysates were collected 24 h post-Nutlin treatment and analyzed by immunoblot analysis with indicated antibodies.
FIGURE 9.

Inhibition of PKCϵ abrogates dexamethasone protection from Nutlin-induced apoptosis in HMECmyc cells. A and B, HMECmyc cells were transfected with nontargeting siRNA (siCon) or PKCϵ-specific siRNA (siPKCϵ). After 24 h, Dex (1 μm) was added for 30 min before Nutlin (Nut) (5 μm) treatment in −ALL condition. Cells were harvested at 72 h post-Nutlin treatment and stained with propidium iodide to measure DNA content. C and D, cell lysates were collected 24 h post-Nutlin treatment and analyzed by immunoblot analysis with indicated antibodies. The analysis was done in duplicate assays, and both are presented separately.
GR-activated PKCϵ Also Protects against Cisplatin- and Irradiation-induced Apoptosis in MCF10Amyc
Next, we wished to test whether GR activation could inhibit apoptosis induced by conventional therapy agents, Cis and IR, and, if yes, whether this requires PKCϵ. To this end, MCF10Amyc cells were treated with Cis (0, 5, and 15 μm) or IR (0, 4, and 10 Gy) for 72 h, and percent apoptosis was determined by sub-G1 DNA content. Results in Fig 10, A and B, show that both Cis and IR could induce abundant apoptosis. Importantly, Dex treatment diminished Cis- and IR-induced apoptosis in both untransfected cells and cells transfected with control siRNA (siCon) but did not inhibit apoptosis in cells transfected with PKCϵ siRNA (Fig 10, A and B; DNA histograms in supplemental Fig. 7, A and B). Immunoblotting showed PKCϵ was effectively knocked down in siRNA-transfected cells (Fig 10, C and D). p53 was induced comparably by Cis or IR, regardless of whether PKCϵ was knocked down (Fig 10, C and D), consistent with the notion that PKCϵ inhibits Cis- and IR-induced apoptosis without altering p53 levels.
FIGURE 10.

GR activated PKCϵ also protects against Cisplatin/Irradiation induced apoptosis in MCF10Amyc. MCF10Amyc cells were transfected with nontargeting siRNA (siCon) or PKCϵ-specific siRNA (siPKCϵ). After 24 h, Dex (1 μm) was added for 30 min before treating. A, Cis (Cis; 5, 15 μm); B, irradiation (IR; 4, 10 Gy) in −ALL condition. After 48 h post-treatment, cells were harvested and stained with propidium iodide to measure DNA content. C and D, cell lysates were collected and analyzed by immunoblot analysis with indicated antibodies. The analysis was done in duplicate assays, and data are presented in C as means ± S.E. (n = 2).
Apoptosis in Response to Nutlin/Cisplatin/Irradiation Is at Least in Part p53-dependent
Finally, we addressed the extent to which the apoptosis induced by Nutlin, Cis, and IR in our experiments is p53-dependent. MCF10Amyc cells were transfected with control siRNA (siCon) or siRNA against p53 and then treated with Nutlin, Cis, or irradiation. Apoptosis was monitored by sub-G1 DNA content and p53 protein expression monitored by immunoblotting. In Nutlin-treated cells, an approximate 75% decrease in p53 expression was achieved in sip53 transfectants (Fig. 11B). This coincided with an ∼45% decrease in apoptosis (Fig 11A), indicating the vast majority of apoptosis in Nutlin-treated cells is p53-dependent. In Cis- and IR-treated cells, an ∼75% decrease in p53 expression was achieved in sip53 transfectants (Fig 11, E and F). This coincided with an ∼35–45% decrease in apoptosis (Fig 11, C and D), demonstrating the Cis- and IR-induced apoptosis is dependent in large part on p53 (representative DNA histograms for Fig 11 are provided in supplemental Fig. 8). Results in Figs. 5, 9, and 10 show that Dex and GR activation inhibits Nutlin/Cis/IR-induced apoptosis by increasing PKCϵ expression. Insofar as this apoptosis is p53-dependent, we conclude PKCϵ induced by activated GR can inhibit p53-dependent apoptosis in Nutlin-, Cis-, and IR-treated MCF10Amyc cells.
FIGURE 11.
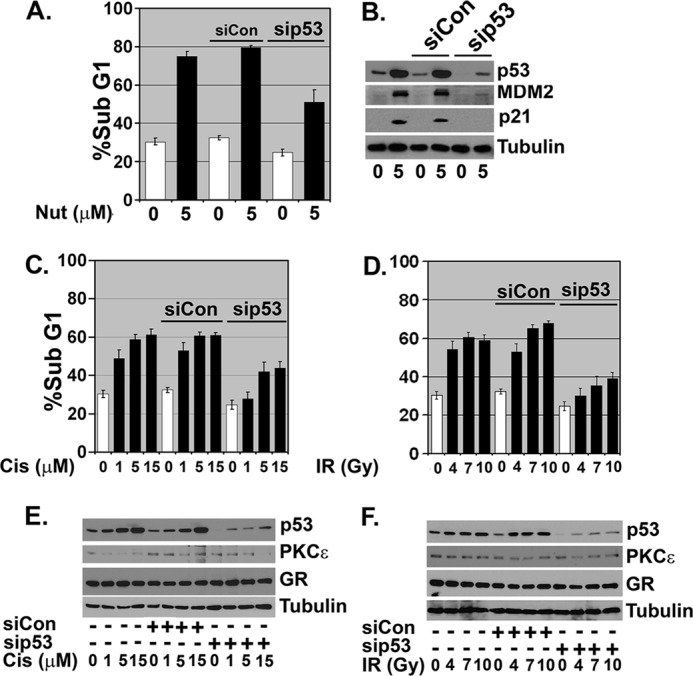
Nutlin/cisplatin/irradiation-induced apoptosis is partially p53-dependent in MCF10Amyc. MCF10Amyc cells were transfected with nontargeting siRNA (siCon) or p53-specific siRNA (sip53). 24 h post-siRNA treatment, these cells were further treated for 48 h with the following: A, Nutlin (Nut) (5 mm); C, Cis (1, 5 or 15 μm); D, IR (4, 7, or 10 Gy), or untreated. Cells were harvested and stained with propidium iodide to measure DNA content. The analysis was done in duplicate assays, and data are presented as mean ± S.E. (n = 2). B, C, and D, cell lysates were collected and analyzed by immunoblot analysis with indicated antibodies.
DISCUSSION
Wild-type p53 and GR have opposing effects on the response of breast cancer cells to chemotherapeutic drugs. P53 promotes growth arrest or death in breast cancer cells exposed to chemotherapeutic drugs (9), whereas GR promotes survival (18, 20). PKCϵ is a novel PKC isoform with anti-apoptotic activity that is expressed, through an unknown mechanism, at high levels in multiple cancer types (49). In breast cancer, increased PKCϵ is linked with high histologic grade and poor disease-free survival (29). In this study, the GR agonists Dex and hydrocortisone promoted PKCϵ expression in MCF10A and HMECs that overexpress the MYC oncogene. Importantly, this effect was reversed by the GR antagonist RU486, confirming increased PKCϵ expression resulted from activation of GR. Moreover, Dex protected the cells from apoptosis that was at least partly p53-dependent and induced by Cis, IR, or the p53 activator Nutlin. Knockdown of PKCϵ abrogated the protective effect of Dex and sensitized the cells to Cis, IR, and Nutlin. Together, these data demonstrate that GR activation can inhibit therapy- and p53-dependent apoptosis and that this occurs through increased expression of PKCϵ.
Wasylyk and co-workers (39, 40) have demonstrated negative cross-talk between GR and p53. In their studies, activation of endogenous GR by Dex treatment inhibited p53-induced cell cycle arrest in stressed cells, whereas activation of endogenous p53 by DNA damage inhibited GR transcriptional activity. Mechanistically, they found that GR could form a ligand-dependent (Dex-dependent) complex with p53. MDM2 was recruited to this GR-p53 complex via p53 and promoted the ubiquitin-dependent degradation of both p53 and GR, thus inhibiting both proteins. Their model suggested that activated GR binds and inhibits p53 by promoting its MDM2-mediated degradation. This model is unlikely to explain GR protection against p53-dependent apoptosis in our system for two reasons. First, p53 was induced comparably by Nutlin, Cis, and IR in the presence or absence of Dex (Figs. 3 and 7). Thus, degradation was not limiting the accumulation of p53 when GR was activated. Second, Dex inhibited the apoptosis induced by Nutlin, even though Nutlin disrupts binding between p53 and MDM2. This suggests activated GR can inhibit p53-induced apoptosis in a manner that does not involve MDM2 interaction with p53. Rather, our results demonstrate Dex treatment increases levels of the anti-apoptotic PKC family member PKCϵ. This increase is at both the mRNA and protein level. The GR antagonist RU486 blocks the increase in PKCϵ expression. Our results are consistent with those of Dwivedi and Pandey (33) and Maddali et al. (34), who reported that administration of Dex coincided with increased PKCϵ expression in coronary arteries and in the rat brain. We speculate PKCϵ is either a direct or indirect transcription target of GR that is required for apoptosis inhibition.
How does PKCϵ induced by Dex treatment inhibit Nutlin-, Cis-, and IR-induced apoptosis? siRNA knockdown studies showed apoptosis in response to all three agents is, in part, p53-dependent in MCF10Amyc cells (Fig. 11). Interestingly, p53 transcriptional activity was apparently unaffected by Dex under conditions in which Dex induced PKCϵ expression and inhibited apoptosis. This is based on the fact that multiple p53 target genes (P21, MDM2, PUMA, and Bax) were induced comparably by Nutlin treatment in the presence or absence of Dex (Fig. 3). Thus, it is unlikely PKCϵ blocks apoptosis by inhibiting p53 transcriptional activity. p53-responsive factors PUMA and Bax promote the formation of pores in the mitochondrial membrane that causes loss of membrane potential and release of factors that activate caspases and drive apoptosis. We found that mitochondrial membrane potential was maintained by Dex treatment in cells exposed to Nutlin, and caspase-3/7 activation was diminished (Fig. 2, B and C). Based on this, we speculate PKCϵ blocks apoptosis by somehow maintaining mitochondrial membrane potential and blocking caspase activation. In this regard, it is worth pointing out that PKCϵ has been reported to localize in cardiac mitochondria, associate with mitochondrial ATPase and MAPKs, and promote phosphorylation and inhibition of the pro-apoptotic factor Bad (50, 51). These activities of PKCϵ are believed to confer protection from ischemic/reperfusion injury in the heart (52, 53). However, it is unknown whether mitochondrial PKCϵ would maintain mitochondrial membrane integrity and block apoptosis in cardiac cells exposed to chemotherapeutic drugs or Nutlin. Likewise, it is unknown whether or the extent to which PKCϵ may localize in mitochondria in our experiments. Interestingly, pro-apoptotic Noxa expression was decreased in Dex-treated cells, although it was not significantly up-regulated by Nutlin/p53 in our studies (Fig. 3B). It is unclear whether decreased Noxa expression may contribute to apoptosis protection by Dex.
An interesting finding from our studies is that Nutlin-induced apoptosis in MCF10Amyc cells but not MCF10A parental cells or MCF10NeoT cells. Furthermore, siRNA-mediated Myc knockdown diminished Nutlin-induced apoptosis in MCF10Amyc cells, and HMEC+myc cells were susceptible to apoptosis by Nutlin although control HMECs were not (Fig. 8). These data suggest MYC expression can render MCF10A cells susceptible to p53-dependent apoptosis. Previous studies reported MYC is directly recruited to the p21 gene promoter by the DNA-binding protein Miz-1 and inhibits p21 expression, favoring the initiation of apoptosis (54). We do not believe MYC sensitizes MCF10A cells through this mechanism in our studies, because p21 was highly expressed in MCF10Amyc cells that were treated with Nutlin and targeted for apoptosis (Fig. 3). It is also interesting that MCF10ANeoT cells are transformed, but remain resistant to apoptosis (Fig. 7). This suggests sensitivity to Nutlin is a consequence of increased MYC expression and does not result from cellular transformation alone. Finally, it is interesting that Dex treatment caused a more pronounced induction of PKCϵ expression in MCF10-Amyc cells compared with either MCF10A or MCF10ANeoT (Fig. 7B). How MYC may affect PKCϵ expression and sensitivity to apoptosis remains to be determined.
Conzen and co-workers (18, 19, 21) previously identified GR target genes associated with survival and apoptosis inhibition in MCF10Amyc cells and different breast cancer cell lines. Their studies included gene expression analysis to identify GR targets induced at early time points after Dex treatment (30 min after Dex). They reported two GR target genes, SGK1 and MKP1, conferred survival in MCF10Amyc cells, and inhibited apoptosis following either serum deprivation or paclitaxel treatment (18, 19, 21). Our results identify PKCϵ as another factor whose increased expression after Dex treatment confers protection against apoptosis. PKCϵ expression increases in response to GR agonists (Dex and hydrocortisone) and decreases in response to GR antagonists (RU486), suggesting PKCϵ is a direct or indirect target of GR. The results suggest GR agonists can promote expression of multiple factors (SGK1, MKP1, and PKCϵ) that confer apoptosis protection.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant 1RO1CA137598-01A1 from NCI (to C. G. M.).

This article contains supplemental Figs. 1–8.
- GR
- glucocorticoid receptor
- HMEC
- human mammary epithelial cell
- Dex
- dexamethasone
- Cis
- cisplatin
- IR
- ionizing radiation
- Gy
- gray
- DAG
- diacylglycerol
- PE
- phorbol ester.
REFERENCES
- 1. Zilfou J. T., Lowe S. W. (2009) Tumor-suppressive functions of p53. Cold Spring Harbor Perspect. Biol. 1, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen J., Marechal V., Levine A. J. (1993) Mapping of the p53 and mdm-2 interaction domains. Mol. Cell Biol. 13, 4107–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oliner J. D., Pietenpol J. A., Thiagalingam S., Gyuris J., Kinzler K. W., Vogelstein B. (1993) Oncoprotein MDM2 conceals the activation domain of tumor suppressor p53. Nature 362, 857–860 [DOI] [PubMed] [Google Scholar]
- 4. Huang L., Yan Z., Liao X., Li Y., Yang J., Wang Z. G., Zuo Y., Kawai H., Shadfan M., Ganapathy S., Yuan Z. M. (2011) The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 12001–12006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haupt Y., Maya R., Kazaz A., Oren M. (1997) Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 [DOI] [PubMed] [Google Scholar]
- 6. Kubbutat M. H., Jones S. N., Vousden K. H. (1997) Regulation of p53 stability by Mdm2. Nature 387, 299–303 [DOI] [PubMed] [Google Scholar]
- 7. Lowe S. W., Ruley H. E., Jacks T., Housman D. E. (1993) p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 74, 957–967 [DOI] [PubMed] [Google Scholar]
- 8. O'Connor P. M., Jackman J., Bae I., Myers T. G., Fan S., Mutoh M., Scudiero D. A., Monks A., Sausville E. A., Weinstein J. N., Friend S., Fornace A. J., Jr., Kohn K. W. (1997) Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 57, 4285–4300 [PubMed] [Google Scholar]
- 9. Gasco M., Shami S., Crook T. (2002) The p53 pathway in breast cancer. Breast Cancer Res. 4, 70–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Soussi T. (2000) The p53 tumor suppressor gene. From molecular biology to clinical investigation. Ann. N.Y. Acad. Sci. 910, 121–137 [DOI] [PubMed] [Google Scholar]
- 11. Tennis M., Krishnan S., Bonner M., Ambrosone C. B., Vena J. E., Moysich K., Swede H., McCann S., Hall P., Shields P. G., Freudenheim J. L. (2006) p53 mutation analysis in breast tumors by a DNA microarray method. Cancer Epidemiol. Biomark. Prev. 15, 80–85 [DOI] [PubMed] [Google Scholar]
- 12. Vogelstein B., Lane D., Levine A. J. (2000) Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 13. Moll U. M., Riou G., Levine A. J. (1992) Two distinct mechanisms alter p53 in breast cancer. Mutation and nuclear exclusion. Proc. Natl. Acad. Sci. U.S.A. 89, 7262–7266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bragado P., Armesilla A., Silva A., Porras A. (2007) Apoptosis by cisplatin requires p53-mediated p38α MAPK activation through ROS generation. Apoptosis 12, 1733–1742 [DOI] [PubMed] [Google Scholar]
- 15. Impicciatore G., Sancilio S., Miscia S., Di Pietro R. (2010) Nutlins and ionizing radiation in cancer therapy. Curr. Pharm. Des. 16, 1427–1442 [DOI] [PubMed] [Google Scholar]
- 16. Wu G. S., El-Deiry W. S. (1996) Apoptotic death of tumor cells correlates with chemosensitivity, independent of p53 or bcl-2. Clin. Cancer Res. 2, 623–633 [PubMed] [Google Scholar]
- 17. Wu G. S., Ding Z. (2002) Caspase 9 is required for p53-dependent apoptosis and chemosensitivity in a human ovarian cancer cell line. Oncogene 21, 1–8 [DOI] [PubMed] [Google Scholar]
- 18. Wu W., Chaudhuri S., Brickley D. R., Pang D., Karrison T., Conzen S. D. (2004) Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 64, 1757–1764 [DOI] [PubMed] [Google Scholar]
- 19. Wu W., Pew T., Zou M., Pang D., Conzen S. D. (2005) Glucocorticoid receptor-induced MAPK phosphatase-1 (MPK-1) expression inhibits paclitaxel-associated MAPK activation and contributes to breast cancer cell survival. J. Biol. Chem. 280, 4117–4124 [DOI] [PubMed] [Google Scholar]
- 20. Moran T. J., Gray S., Mikosz C. A., Conzen S. D. (2000) The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res. 60, 867–872 [PubMed] [Google Scholar]
- 21. Mikosz C. A., Brickley D. R., Sharkey M. S., Moran T. W., Conzen S. D. (2001) Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J. Biol. Chem. 276, 16649–16654 [DOI] [PubMed] [Google Scholar]
- 22. Pan D., Kocherginsky M., Conzen S. D. (2011) Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 71, 6360–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jonat C., Rahmsdorf H. J., Park K. K., Cato A. C., Gebel S., Ponta H., Herrlich P. (1990) Antitumor promotion and anti-inflammation. Down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell 62, 1189–1204 [DOI] [PubMed] [Google Scholar]
- 24. Wyszomierski S. L., Yeh J., Rosen J. M. (1999) Glucocorticoid receptor/signal transducer and activator of transcription 5 (STAT5) interactions enhance STAT5 activation by prolonging STAT5 DNA binding and tyrosine phosphorylation. Mol. Endocrinol. 13, 330–343 [DOI] [PubMed] [Google Scholar]
- 25. Brostjan C., Anrather J., Csizmadia V., Stroka D., Soares M., Bach F. H., Winkler H. (1996) Glucocorticoid-mediated repression of NFκB activity in endothelial cells does not involve induction of IκBα synthesis. J. Biol. Chem. 271, 19612–19616 [DOI] [PubMed] [Google Scholar]
- 26. Distelhorst C. W. (2002) Recent insights into the mechanism of glucocorticosteroid-induced apoptosis. Cell Death Differ. 9, 6–19 [DOI] [PubMed] [Google Scholar]
- 27. Greenstein S., Ghias K., Krett N. L., Rosen S. T. (2002) Mechanisms of glucocorticoid-mediated apoptosis in hematological malignancies. Clin. Cancer Res. 8, 1681–1694 [PubMed] [Google Scholar]
- 28. Herr I., Ucur E., Herzer K., Okouoyo S., Ridder R., Krammer P. H., von Knebel Doeberitz M., Debatin K. M. (2003) Glucocorticoid cotreatment induces apoptosis resistance toward cancer therapy in carcinomas. Cancer Res. 63, 3112–3120 [PubMed] [Google Scholar]
- 29. Pan Q., Bao L. W., Kleer C. G., Sabel M. S., Griffith K. A., Teknos T. N., Merajver S. D. (2005) Protein kinase Cϵ is a predictive biomarker of aggressive breast cancer and a validated target for RNA interference anticancer therapy. Cancer Res. 65, 8366–8371 [DOI] [PubMed] [Google Scholar]
- 30. Aziz M. H., Sundling K. E., Dreckschmidt N. E., Verma A. K. (2009) Protein kinase Cϵ inhibits UVR-induced expression of FADD, an adaptor protein, linked to both Fas- and TNFR1-mediated apoptosis. J. Invest. Dermatol. 129, 2011–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aziz M. H., Hafeez B. B., Sand J. M., Pierce D. B., Aziz S. W., Dreckschmidt N. E., Verma A. K. (2010) Protein kinase Cvarϵ mediates Stat3Ser-727 phosphorylation, Stat3-regulated gene expression, and cell invasion in various human cancer cell lines through integration with MAPK cascade (RAF-1, MEK1/2, and ERK1/2). Oncogene 29, 3100–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bourguignon L. Y., Spevak C. C., Wong G., Xia W., Gilad E. (2009) Hyaluronan-CD44 interaction with protein kinase Cϵ promotes oncogenic signaling by the stem cell marker Nanog and the production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance in breast tumor cells. J. Biol. Chem. 284, 26533–26546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dwivedi Y., Pandey G. N. (1999) Administration of dexamethasone up-regulates protein kinase C activity and the expression of γ and κ protein kinase C isozymes in the rat brain. J. Neurochem. 72, 380–387 [DOI] [PubMed] [Google Scholar]
- 34. Maddali K. K., Korzick D. H., Turk J. R., Bowles D. K. (2005) Isoform-specific modulation of coronary artery PKC by glucocorticoids. Vascul. Pharmacol. 42, 153–162 [DOI] [PubMed] [Google Scholar]
- 35. Kessler J. D., Kahle K. T., Sun T., Meerbrey K. L., Schlabach M. R., Schmitt E. M., Skinner S. O., Xu Q., Li M. Z., Hartman Z. C., Rao M., Yu P., Dominguez-Vidana R., Liang A. C., Solimini N. L., Bernardi R. J., Yu B., Hsu T., Golding I., Luo J., Osborne C. K., Creighton C. J., Hilsenbeck S. G., Schiff R., Shaw C. A., Elledge S. J., Westbrook T. F. (2012) A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hammond S. L., Ham R. G., Stampfer M. R. (1984) Serum-free growth of human mammary epithelial cells. Rapid clonal growth in defined medium and extended serial passage with pituitary extract. Proc. Natl. Acad. Sci., U.S.A. 81, 5435–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou B. P., Liao Y., Xia W., Zou Y., Spohn B., Hung M. C. (2001) HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 3, 973–982 [DOI] [PubMed] [Google Scholar]
- 38. Ozes O. N., Akca H., Mayo L. D., Gustin J. A., Maehama T., Dixon J. E., Donner D. B. (2001) A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc. Natl. Acad. Sci. U.S.A. 98, 4640–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sengupta S., Vonesch J. L., Waltzinger C., Zheng H., Wasylyk B. (2000) Negative cross-talk between p53 and the glucocorticoid receptor and its role in neuroblastoma cells. EMBO J. 19, 6051–6064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sengupta S., Wasylyk B. (2001) Ligand-dependent interaction of the glucocorticoid receptor with p53 enhances their degradation by Hdm2. Genes Dev. 15, 2367–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ní Chonghaile T., Concannon C. G., Szegezdi E., Gorman A. M., Samali A. (2006) Dexamethasone inhibits apoptosis in C6 glioma cells through increased expression of Bcl-XL. Apoptosis 11, 1247–1255 [DOI] [PubMed] [Google Scholar]
- 42. Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., Liu E. A. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 43. Liu W., Konduri S. D., Bansal S., Nayak B. K., Rajasekaran S. A., Karuppayil S. M., Rajasekaran A. K., Das G. M. (2006) Estrogen receptor-α binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 281, 9837–9840 [DOI] [PubMed] [Google Scholar]
- 44. Sayeed A., Konduri S. D., Liu W., Bansal S., Li F., Das G. M. (2007) Estrogen receptor α inhibits p53-mediated transcriptional repression: implications for the regulation of apoptosis. Cancer Res. 67, 7746–7755 [DOI] [PubMed] [Google Scholar]
- 45. Wallace A. D., Cidlowski J. A. (2001) Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J. Biol. Chem. 276, 42714–42721 [DOI] [PubMed] [Google Scholar]
- 46. Webster J. C., Jewell C. M., Bodwell J. E., Munck A., Sar M., Cidlowski J. A. (1997) Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J. Biol. Chem. 272, 9287–9293 [DOI] [PubMed] [Google Scholar]
- 47. Petiti J. P., De Paul A. L., Gutiérrez S., Palmeri C. M., Mukdsi J. H., Torres A. I. (2008) Activation of PKCϵ induces lactotroph proliferation through ERK1/2 in response to phorbol ester. Mol. Cell. Endocrinol. 289, 77–84 [DOI] [PubMed] [Google Scholar]
- 48. Holden N. S., Squires P. E., Kaur M., Bland R., Jones C. E., Newton R. (2008) Phorbol ester-stimulated NF-κB-dependent transcription. Roles for isoforms of novel protein kinase C. Cell. Signal. 20, 1338–1348 [DOI] [PubMed] [Google Scholar]
- 49. Gorin M. A., Pan Q. (2009) Protein kinase Cϵ. An oncogene and emerging tumor biomarker. Mol. Cancer 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baines C. P., Zhang J., Wang G. W., Zheng Y. T., Xiu J. X., Cardwell E. M., Bolli R., Ping P. (2002) Mitochondrial PKCϵ and MAPK form signaling modules in the murine heart. Enhanced mitochondrial PKCϵ-MAPK interactions and differential MAPK activation in PKCϵ-induced cardioprotection. Circ. Res. 90, 390–397 [DOI] [PubMed] [Google Scholar]
- 51. Jabůrek M., Costa A. D., Burton J. R., Costa C. L., Garlid K. D. (2006) Mitochondrial PKC epsilon and mitochondrial ATP-sensitive K+ channel copurify and coreconstitute to form a functioning signaling module in proteoliposomes. Circ. Res. 99, 878–883 [DOI] [PubMed] [Google Scholar]
- 52. Cohen M. V., Baines C. P., Downey J. M. (2000) Ischemic preconditioning. From adenosine receptor to KATP channel. Annu. Rev. Physiol. 62, 79–109 [DOI] [PubMed] [Google Scholar]
- 53. Baines C. P., Pass J. M., Ping P. (2001) Protein kinases and kinase-modulated effectors in the late phase of ischemic preconditioning. Basic Res. Cardiol. 96, 207–218 [DOI] [PubMed] [Google Scholar]
- 54. Seoane J., Le H. V., Massagué J. (2002) Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 419, 729–734 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.