

Background: [Ca2+] in ductal saliva decreases as it flows toward the oral cavity; the mechanism of Ca2+ reabsorption is unknown.
Results: Apical Ca2+-sensing receptor (CSR) in submandibular gland (SMG) responds to extracellular [Ca2+] and activates TRPC3 channel.
Conclusion: CSR-induced Ca2+ entry regulates Ca2+ reabsorption from saliva.
Significance: Ca2+ reabsorption in SMG duct can contribute to regulation of saliva [Ca2+], a critical factor in sialolithiasis.
Keywords: Amino Acid, Calcium Signaling, Calcium Transport, G Protein-coupled Receptor (GPCR), Salivary Gland
Abstract
Ca2+ is secreted from the salivary acinar cells as an ionic constituent of primary saliva. Ions such as Na+ and Cl− get reabsorbed whereas primary saliva flows through the salivary ductal system. Although earlier studies have shown that salivary [Ca2+] decreases as it flows down the ductal tree into the oral cavity, ductal reabsorption of Ca2+ remains enigmatic. Here we report a potential role for the G protein-coupled receptor, calcium-sensing receptor (CSR), in the regulation of Ca2+ reabsorption by salivary gland ducts. Our data show that CSR is present in the apical region of ductal cells where it is co-localized with transient receptor potential canonical 3 (TRPC3). CSR is activated in isolated salivary gland ducts as well as a ductal cell line (SMIE) by altering extracellular [Ca2+] or by aromatic amino acid, l-phenylalanine (l-Phe, endogenous component of saliva), as well as neomycin. CSR activation leads to Ca2+ influx that, in polarized cells grown on a filter support, is initiated in the luminal region. We show that TRPC3 contributes to Ca2+ entry triggered by CSR activation. Further, stimulation of CSR in SMIE cells enhances the CSR-TRPC3 association as well as surface expression of TRPC3. Together our findings suggest that CSR could serve as a Ca2+ sensor in the luminal membrane of salivary gland ducts and regulate reabsorption of [Ca2+] from the saliva via TRPC3, thus contributing to maintenance of salivary [Ca2+]. CSR could therefore be a potentially important protective mechanism against formation of salivary gland stones (sialolithiasis) and infection (sialoadenitis).
Introduction
Ca2+ reabsorption in epithelia is a crucial determinant of calcium balance (1). Salivary ducts have minimal fluid secretory function but are proposed to be involved in reabsorption of ions, including Na+ (2–4). The role of these cells in Ca2+ handling has not yet been examined. Primary saliva secreted from the acinar cell contains a high concentration of Ca2+ (5), which falls steadily as saliva travels down the ductal system (6). Although the mechanism involved in the decrease in salivary [Ca2+] in ducts is not known, maintenance of the [Ca2+] within a critical range is essential (7) because high [Ca2+] in saliva can lead to salivary gland stone formation (sialolithiasis) which obstructs the duct (8). This often results in bacterial infections as well, creating a condition referred to as sialoadenitis. Typically, epithelia that mediate Ca2+ reabsorption appear to have two major pathways: (i) the paracellular pathway which moves Ca2+ through tight junctions and (ii) the transcellular pathway that moves Ca2+ across the epithelium (9). The paracellular pathway is suggested to operate to efficiently transport bulk Ca2+ through the tight junctions, whereas the transcellular route fine-tunes the Ca2+ balance (10).
Ca2+-sensing receptor (CSR),2 a G-protein coupled receptor that senses the extracellular levels of Ca2+, is expressed in the plasma membranes of a wide variety of epithelial tissues including parathyroid, kidney, and gastrointestinal tract (11). CSR detects small changes in the extracellular Ca2+ concentration ([Ca2+]o) which itself together with other physiological agonists such as l-amino acids and peptides serves to activate the receptor signaling cascade which leads to PLC activation and Ca2+ entry (2, 12, 14, 15). Several recent studies have demonstrated that CSR might regulate Ca2+ influx via TRPC channels, suggesting a possible mechanism for Ca2+ reabsorption (16–18). The threshold for activation of CSR by Ca2+ is ∼1.2 mm, and the half-maximum effective concentration is ∼1.8 mm (12) in the absence of any allosteric modulators. The [Ca2+] in the intercalated ductal fluids (measured in rats) ranges from 1.5 to 3 mm (2). Therefore, [Ca2+] in primary (ductal) saliva would be sufficient to activate CSR. In addition, the presence of l-amino acids and peptides (14, 15) in saliva may sensitize CSR to a smaller change in extracellular [Ca2+] ([Ca2+]o) and modulate its function as in gastric epithelium (19). Based on this and our previous finding that TRPC3 is localized in the apical region of salivary gland ductal cells, we examined the presence and function of CSR in salivary gland ducts.
Here we report that functional CSRs are present in the apical region of submandibular gland (SMG) ductal cells. CSR agonists stimulate Ca2+ entry into these cells that is mediated by TRPC3. CSR co-localizes with TRPC3 in the apical region of polarized salivary gland ductal cells, and inhibition of CSR or TRPC3 function pharmacologically or by knockdown of the expression of either protein reduces CSR-stimulated Ca2+ entry. We suggest that CSR-regulated Ca2+ entry via the apical membrane of ductal cells represents a potential Ca2+ reabsorption mechanism that can account for the decrease in [Ca2+] in primary saliva as it flows down the ductal system.
EXPERIMENTAL PROCEDURES
Antibodies and Chemicals
All of the chemicals used were analytical grade. Anti-TRPC3 antibody was produced (rabbit polyclonal) and purified as described previously (20). All other specific chemicals and antibodies used are described in respective sections where they were used.
Cell Culture and Transfection
Submandibular immortalized epithelial (SMIE) cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen), supplemented with 10% fetal calf serum, 2 mm glutamine, 1% penicillin/streptomycin at 37 °C in 5% CO2. SMIE cells were transiently transfected with 1 μg of required plasmids (shTRPC3) using Lipofectamine reagent 2000 (Invitrogen). CSR-specific siRNA (siCSR) and a scrambled (control) siRNA (Santa Cruz Biotechnology, Santa Cruz, CA) were transiently transfected according to the supplier's instructions.
Immunocytochemistry of Submandibular Gland Sections
Adult male C57BL/6 strain of mice (weighing 25–35 g) were kept under environmentally controlled conditions (12:12-h light/dark cycle, 20–22 °C) with food and water available ad libitum until used. All animals were treated according to guidelines recommended by National Institutes of Health Animal Care and Use. SMGs were excised from the animals and fixed in 10% formalin solution for 24 h for immunocytochemistry, dehydrated in graded concentrations of ethanol, embedded in paraffin, and used to prepare 5–10-μm thick sections. Immunocytochemistry was performed on paraffin sections of mouse SMG as described previously (20). Briefly, sections were dewaxed, rehydrated, and permeabilized with 0.5% Triton X-100 non-ionic detergent, commonly used in phosphate-buffered saline (PBS), pH 7.5. For labeling of CSR, anti-CSR antibody (rabbit, 1:100 dilutions) was used, and in control sections, rabbit IgG was used instead of primary antibody. We used a labeling kit (Invitrogen) that utilizes diaminobenzidine, an alcohol-insoluble chromogen, and generates a brown precipitate.
Dispersed Cell Preparation from Mouse SMG and [Ca2+]i Measurements
SMGs were removed and placed in an ice-cold external solution with 0.02% soybean trypsin inhibitor and 0.1% bovine serum albumin. Each gland was cleaned and finely minced. The minced tissue was transferred to 8 ml of external solution containing 4 mg of collagenase P. The tube was gassed and capped. After a 15–20-min incubation at 37 °C, the digest was washed twice with the normal external solution followed by a 2–4-min centrifugation at 100 × g and resuspended in approximately 4 ml of physiological solution. For microfluorometry, dispersed cells were loaded with fura-2 for 45–60 min at 30 °C and placed in a poly-l-lysine-coated glass-bottom dish (Matek Corporation) and allowed to attach. Fluorescence measurements were made using a Till Photonics-Polychrome IV spectrofluorometer and MetaFluor Imaging System (Universal Imaging Corporation). Fura-2 fluorescence in freshly dispersed submandibular cells was measured as described earlier (20, 21) using an Olympus 50× microscope, with an ORCA-ER camera (Hamamatsu) attached to a Polychrome IV (Till Photonics LLC). Ducts and acinar cells from the same glands were morphologically identified by their distinctive appearance under microscopic examination (22, 23). We only imaged the ductal cells for measurement of Ca2+ signal. MetaFluor (Moleular Devices) was used to acquire images and processing the data. Analog plots of the fluorescence ratio (340/380) in single cells are shown. In some experiments we used Fluo-4/M (2–5 μm; Invitrogen) to load the cells for 20 min at 25 °C as described previously (24). Cells were washed with several volumes of bathing solution and left for 20 min before recording. We used standard bathing solution including 140 mm NaCl, 4 mm KCl, 10 mm HEPES, 10 mm glucose, 1–2 mm CaCl2, 1 mm MgCl2, pH 7.3, for the experiment. The Fluo-4/AM dye (Molecular Probes) was excited at 480 ± 15 nm. Emitted fluorescence was filtered with a 535 ± 25 bandpass filter captured by a SOPT RT digital camera (Diagnostic Instruments, Sterling Heights, MI) and read into a computer. Analysis was performed offline using Simple PCI software (Compix Inc., Sewickley, PA).
Polarized Monolayer Cell Culture and Measurement of Transepithelial Electrical Resistance (TER)
SMIE cells (as a gift from Dr. Bruce J. Baum, NIDCR) were plated in 12- or 24-mm Costar Transwell polycarbonate membrane plates at densities of 0.5 ± 105 or 2 ± 105 cells/well, respectively, in DMEM/10%FBS. Formation of tight junctions and integrity of the monolayers were determined by serial measurement of TER. Fully polarized monolayers were achieved at 72–96 h after cell plating. TER across cell monolayers was measured as previously described to ensure the polarity (25). TER measurements were made with a Millicell ERS epithelial volt-ohmmeter (Millipore) as described by the manufacturer. Experiments were performed in triplicate with three separate cell preparations. Data are presented as bar diagrams as mean ± S.E. A t test was used for statistical evaluation of the values; p < 0.05 was considered to be statistically significant.
Immunoprecipitation, Immunoblotting, and Cell Surface Biotinylation
SMIE cells were grown in a Transwell filter with serum-free DMEM and washed once by adding ice-cold PBS containing 1% (v/v) aprotinin (Sigma) before harvesting the cells. RIPA buffer containing protease inhibitors was added directly onto the filter for cell lysis and frozen at −80 °C for at least 2 h before use as described previously (20, 26). Protein concentration was determined by using the Bio-Rad protein assay (microassay procedure). Immunoprecipitation from the cell lysate was done as described previously (20, 27). Immunocomplexes were pulled down by adding anti-TRPC3 (1:100 dilution), anti-CSR (1:100 dilution) antibody followed by the addition of Sepharose beads. Protein-bound beads were separated by brief centrifugation. The supernatant was saved (unbound fraction). The beads were washed, and bound proteins (IP-fraction) were eluted by boiling in SDS-PAGE sample buffer for 5 min and were separated by SDS-PAGE. Proteins were detected by Western blotting as described previously (20) using anti-TRPC3 (1:500 dilution) and anti-CSR (1:400 dilution) primary antibody, the required secondary antibody, and treatment with ECL reagent. SMIE cells grown as described were incubated for 3–5 min with or without l-phenylalanine (l-Phe; 10 μm) at 37 °C in 1.2 mm Ca2+-containing SES medium (145 mm NaCl, 5 mm KCl, 1 mm MgCl2, 10 mm Na-HEPES, 10 mm glucose, pH 7.4), washed, and incubated for another 20 min with 0.5 mg/ml Sulfo-NHS-Biotin (Pierce) on ice. In some experiment cells were preincubated with CSR inhibitor (1 μm NPS-2143; Tocris Bioscience, Ellisville, MO) for 30 min before adding l-Phe. The cells were washed with buffer containing 0.1 m glycine and solubilized with of RIPA buffer. Biotinylated proteins were pulled down with NeutrAvidin-linked beads (Pierce). Bound fraction was washed and released with SDS-PAGE sample buffer and analyzed by Western blotting. Proteins were visualized with anti-TRPC3 (1:500 dilution) and anti-Na+,K+-ATPase (Sigma; 1:1000 dilution) antibody. Details of these methods were described previously (27). In some experiments freshly isolated dispersed cells from mouse SMG were used for co-immunoprecipitation. Cells were solubilized by RIPA buffer as described previously (27). Precleared lysates were incubated with the anti-CSR antibody (mouse; 1:100 dilution). Interacting proteins bound to Sepharose beads were separated, released with SDS-PAGE sample buffer, and detected by Western blotting as described (20) previously using anti-TRPC3 (1:500 dilution) antibody.
Immunofluorescence and Confocal Imaging
All steps were performed at room temperature unless otherwise mentioned. The cells were rinsed with PBS once and then fixed with 3% paraformaldehyde in PBS, pH 7.4, for 30 min. Next, we rinsed again with PBS, and treated with 100 mm glycine in PBS for 20 min. Cells were then washed and permeabilized with methanol at −20 °C for 5 min. Following incubation with a blocking solution containing 5% donkey serum and 0.5% BSA in PBS (PBS-BSA) for 20 min, the cells were incubated with primary antibodies for 1 h at room temperature, washed with PBS-BSA, and probed with the required FITC- or rhodamine-conjugated secondary antibody (Jackson Immuno Research, West Grove, PA). The filter containing the cells was excised from its support and mounted onto a slide with antifade reagent (Ted Pella). Fluorescence images were taken using a confocal laser scanning microscope Leica TCS-SP2 attached to an upright Leica DM-RE7 microscope. Details of the images are indicated in the figure legends.
Co-staining of TRPC3 and AQP5/CSR was performed on paraffin sections from mouse SMGs. Sections of glands were prepared as described above after fixation and permeabilization. The sections were then rehydrated followed by an antigen retrieval process. Afterward, samples were blocked and treated with anti-TRPC3 (rabbit, 1:200 dilution), and anti-CSR (mouse, 1:100 dilution) antibody for overnight at 4 °C. After washing, the sections were probed using secondary antibodies; Alexa Fluor 488 donkey anti-rabbit (for TRPC3) and Alexa Fluor 568 donkey anti-mouse (for CSR) for 30 min (1:100 dilutions). Anti-AQP5 primary antibody (1 μg) was prelabeled with Alexa Fluor 594 secondary antibody using a labeling kit (Invitrogen). Images were collected using Olympus FluoView confocal microscopes and further analyzed using MetaMorph software (Molecular Devices, Sunnyvale, CA). Control labeling for antibodies (for background subtraction) was performed using isotype (instead of primary antibody) plus the secondary antibody (data not shown). Additionally, co-localization analysis of SMG ducts labeled by anti-CSR and anti-TRPC3 antibody was performed using Volocity (version 6.0.1, PerkinElmer Life Sciences) software.
Measurements of Fluo-4 Fluorescence by Confocal Microscopy in Polarized SMIE Cells
SMIE cells were grown in Transwell filters, and TER was measured to confirm the establishment of polarization as described previously (20, 25). Thereafter, cells were incubated with 2–5 μm Fluo-4/AM ester at 37 °C for 1 h in SES medium. Samples were scanned on a TCS SP2 Leica confocal microscope system using a 40 × 0.8 NA dipping objective. Single Transwell inserts were washed, 2 ml of SES medium was added (apical chamber), and the inserts were placed in 35-mm chambers containing 2.5 ml of SES medium (basal chamber). All additions were introduced to the apical chamber of the Transwell filter. Samples were scanned in XZ-time mode for 5-min total time with 12-s increments between sampling. Calculations of the mean intensity in a defined region of interest were made using the histogram function in the Leica LCS Lite software and analyzing whole cell, basal, and apical regions throughout the time series.
RESULTS
Localization and Activation of CSR at SMG Duct
As reported in other epithelial cells (28), CSR is localized both at the luminal and basolateral region of the salivary duct (Fig. 1A, ii and iii). More specifically, antibody labeling demonstrates intense signal (Fig. 1Aiii) in striated ducts with some positive signal (Fig. 1Aii) in the granular convoluted ducts. We detected relatively less signal in the acini, which were not the focus of the present study (Fig. 1Aiii). Fig. 1Ai shows control labeling (rabbit IgG + secondary antibody). Dispersed mouse salivary gland ducts were used for [Ca2+]i measurements to provide functional evidence of CSR. Although the preparation contained both ductal fragments and acini, Ca2+ measurements were made only in ducts (selected by imaging, see inset in Fig. 1B) (20, 21). The function of CSR was examined by application of the potent agonist Gd3+ (200 μm) to cells in a Ca2+-free medium (Fig. 1B) that triggered a sharp rise in [Ca2+]i. Further, the activation of CSR by l-amino acids such as l-Phe has been shown to be modulated by extracellular Ca2+ in the kidney and gastrointestinal tracts (19). We found that addition of 10 mm l-Phe, but not l-Tyr, in the presence of >0.5 mm Ca2+ markedly increased [Ca2+]i (Fig. 1C) in the ductal cells (addition of l-Phe in a Ca2+-free medium did not induce any changes in [Ca2+]i; data not shown). In contrast, application of neomycin (500 μm), another CSR agonist (Fig. 1D), increased [Ca2+]i even in the absence of external Ca2+, i.e. representing intracellular release, followed by a second rise in [Ca2+]i due Ca2+ influx that occurred when Ca2+ was added to the medium. Similarly, when l-Phe was added together with 0.5 mm external Ca2+ a small increase in [Ca2+]i was seen. This was greatly enhanced when external Ca2+ was increased to 1.5 mm and subsequently decreased when Ca2+ was reduced to 0.5 mm (Fig. 1E). The modulation of CSR by l-Phe could have a physiological relevance in salivary duct because l-Phe has been shown to be present in saliva (14, 29). To confirm that CSR is involved, a specific antagonist, NPS-2143 (30) was used which completely blocked the l-Phe and Ca2+-induced responses (Fig. 1E).
FIGURE 1.

Localization and function of CSR in salivary duct. A, immunocytochemical localization of CSR in SMG tissue section showing labeling (diaminobenzidine; brown precipitate) with control (rabbit IgG) (i) and anti-CSR antibody (ii) to granular convoluted (GCT) and striated ducts (SD). Yellow arrows in iii (enlargement of middle panel) indicate dense signal at the apical region in striated ducts. B, inset, representative of freshly dispersed SMG cells isolated, showing highly distinguishable ducts and acini. These cells are loaded with fura-2 and used for [Ca2+]i measurement. Ductal cells were selected under the microscope for imaging. B–E, mean fluorescence traces (obtained from 3–4 separate experiments, 30–40 cells) of fura-2-loaded SMG ducts. Gd3+ (200 μm) was added (B) to cells in a Ca2+-free medium. C, application of l-Phe/l-Tryptophan (l-Try) (10 mm) in medium containing 1 mm Ca2+. D and E, activation of CSR and Ca2+ entry by neomycin (500 μm) (D) and l-Phe (5 mm) (E) at different external [Ca2+]. A complete block of CSR by NPS-2143 is shown in cells incubated with l-Phe (E, red traces). E, inset, peak [Ca2+]i significantly decreased (p < 0.01) in response to control (l-Phe) and NPS-2143- (1 μm) treated cells. Changes in external [Ca2+] and other conditions are otherwise indicated in the figure.
Function of CSR and TRPC3 in SMG Ducts
Immunofluorescence measurements revealed a strong TRPC3 signal in the apical region of SMG ductal cells, with relatively weak signal in the acini under these conditions (anti-AQP5 was used as a marker for acinar cells) (Fig. 2A). Furthermore, CSR and TRPC3 were co-localized (Table 1) in luminal region of the ducts (Fig. 2B, marked by white arrows) and co-immunoprecipitated from cell lysates (Fig. 2C). Interestingly, treatment of l-Phe with 1.2 mm Ca2+ enhanced the CSR-TRPC3 complex formation (Fig. 2D). The function of TRPC3 was assessed in these ducts using the activator, diacylglycerol analog, OAG, as well as the relatively specific TRPC3 inhibitor, Pyr3 (31). Whereas OAG increased Ca2+ entry into ducts, inclusion of Pyr3 in the medium suppressed the activity indicating the presence of functional TRPC3 channels in SMG ducts (Fig. 2E). Importantly, Ca2+ mobilization induced by l-Phe in these ducts was also severely reduced by adding Pyr3 in the medium (Fig. 2F). Together, these results suggest that CSR and TRPC3 converge on the same signaling pathway in the salivary gland ducts.
FIGURE 2.

Co-localization and interaction of TRPC3 and CSR in salivary gland duct. A, immunofluorescence detection of TRPC3 and AQP5 in SMG tissue using anti-TRPC3 and anti-AQP5 antibodies. Yellow arrows indicate labeling with TRPC3 (left), AQP5 (middle), and overlay (right); blue color shows DAPI-stained nucleus. Scale bar, 20 μm. B, confocal microscopy detection of TRPC3 and CSR in mouse SMG sections using anti-TRPC3 and anti-CSR antibodies. White arrows indicate TRPC3 (left; green), CSR (middle; red), and overlay of TRPC and CSR (right; yellow). Nuclei were stained with DAPI (blue). Scale bar, 20 μm. Using the Volocity software to threshold the signals, almost complete overlap was seen between green and red colors. In an unthresholded image the overlap was approximately 70%. C, Western blots (IB) showing co-immunoprecipitation of TRPC3 (*) and CSR (**) using anti-TRPC3 antibody for immunoprecipitation (IP). Anti-TRPC3 and anti-CSR antibodies were used to detect the two proteins in the IP fraction. Controls of co-IPs are shown using rabbit IgG instead of primary antibody. Input of TRPC3(x) and CSR (xx) are also shown (1/10–1/20 of the IP). D, freshly dispersed SMG primary cells were treated (+) with l-Phe (10 mm) plus 1.2 mm Ca2+, lysed with RIPA buffer, and then immunoprecipitation was performed using anti-CSR antibody. Untreated (−) cells were used as control. Western blotting (IB) using anti-TRPC3 antibody was used for detection of the protein in the IP fraction and lysates (input; 1/10 of IP). E and F, mouse SMG (freshly dispersed) cells were loaded with fura-2 for 30 min. Graphs are representative of mean fluorescence traces (3–4 separate experiments) of fura-2-loaded SMG duct (selected under microscope while imaging). E, OAG (100 μm) activation in cells treated (3 μm Pyr3; TRPC3 inhibitor) or not (control). F, Ca2+ entry in response to CSR activation by l-Phe (control; 20 mm) and block of this function in cells treated with Pyr3 (3 μm). All other additions are indicated in the figure.
TABLE 1.
Co-localization analysis using Volocity, version 6.0.1
This co-localization analysis was performed on three-dimensional image stacks of the image shown in Fig. 2B. After sequential imaging of two channels, images were analyzed in Volocity. Images were first deconvolved using iterative deconvolution and then analyzed for co-localization using Volocity. Deconvolved images were thresholded, then co-localization was performed. Both Pearson's correlation coefficient and Global Pearson's correlation coefficients were obtained after selecting a region on the scatter plot to minimize background (50). Data were verified by using the method as described by Costes et al. (13). Co-localization coefficient indicates the relative degree of overlap between the signals and shows a significant co-localization (overlap) between both of the signals (green and red). In the thresholded image 70% of the total pixels were yellow, indicated by Pearson's correlation. In a nonthresholded image there was 98.1% overlap as noted by the Global Pearson's correlation. Overlay of TRPC3 and CSR was validated by calculating Pearson's coefficients.
| Co-localization | Fig. 2C analysis | ||
|---|---|---|---|
| Pearson's correlation | 0.709 | Co-localization coefficient (Mx) | 1.000 |
| Overlap coefficient (R) | 0.981 | Co-localization coefficient (My) | 1.000 |
| Overlap coefficient (Kx) | 1.430 | Global Pearson's correlation | 0.979 |
| Overlap coefficient (Ky) | 0.672 | Voxel ratio Ch.X/Ch.Y | 1.000 |
Functional Association of CSR and TRPC3 in SMIE Cells
To delineate the mechanism of CSR-TRPC3 signaling, we used SMIE cells, a salivary gland cell line that exhibits key features of ductal cells as well as the ability to form a polarized monolayer when grown on membrane support (32). CSR function was first assessed in SMIE cells (nonpolarized) to test the role of different CSR agonists in [Ca2+]i mobilization. A rise in [Ca2+]i was seen when external [Ca2+] was increased to (1–5 mm), suggesting that Ca2+ itself serves as an agonist for CSR. Inhibition of CSR by NPS-2143 completely abolished this response (Fig. 3A). We examined the activation of CSR in SMIE cells by several other activating conditions and demonstrate here that presence of l-Phe can further modulate the CSR stimulation by Ca2+. Fig. 3B shows that 1.2 mm, but not 0.5 mm Ca2+, elicited an increase in [Ca2+]i in SMIE cells, which was further enhanced by including 10 mm l-Phe with Ca2+(1.2 mm). In contrast, activation of CSR by neomycin resulted an increase in [Ca2+]i even in the absence extracellular Ca2+ (Fig. 3C). A subsequent application of l-Phe together with 0.5 mm Ca2+ induced a second [Ca2+]i response in the same cells. It has been suggested that neomycin binds directly to the Ca2+ binding site of CSR, whereas l-Phe binds to an allosteric site and modulates the Ca2+ sensitivity of CSR (12). To confirm the signaling mechanism initiated by CSR stimulation, cells were treated with 1 μm U73122, a PIP2-PLC inhibitor. The almost complete inhibition of [Ca2+]i increase in response to 200 μm Gd3+ confirmed the involvement PIP2 hydrolysis as a proximal event in the signaling cascade (Fig. 3D). In contrast, the inactive analog (U73342) of U73122 did not inhibit the [Ca2+]i response.
FIGURE 3.

Properties of CSR-mediated signaling in SMIE cells. A, mean fluorescence traces (representative of data from 3–4 separate experiments) of fura-2-loaded SMIE cells. Cells were bathed in Ca2+-free medium, and then 1–5 mm Ca2+ was added to the external medium to activate CSR. 1 μm NPS-2143 was added where indicated (red) to block this response. B and C, individual fluorescence traces indicate the changes in response of Fluo-4-loaded SMIE cells. Cells were bathed in 0.5 mm Ca2+-containing medium and then applied to 10 mm l-Phe with 1.2 mm external Ca2+ (B) showing pattern of [Ca2+]i rise in response to both l-Phe and Ca2+. Similarly, application of neomycin (500 μm) in external Ca2+ free medium followed by l-Phe (10 mm) with 0.5 mm Ca2+ (C) resulted Ca2+ entry due to [Ca2+]i rise. D, Gd3+- (200 μm) induced activation of CSR in Fluo-4-loaded SMIE cells. Mean fluorescence traces (representative of 3–4 separate experiments) show an increase of [Ca2+]i in response to Gd3+ and a block of this response by application of PLC inhibitor, U73122 (500 μm). U73342, an inactive analog of U73122, was used as a control. Mean fluorescence traces represent data from 3–4 separate experiments.
The functional involvement of CSR in SMIE cells was further confirmed by examining the effect of the antagonist NPS-2143 on the receptor activation. Consistent with our findings in salivary gland ducts, the [Ca2+]i increase induced by l-Phe was completely suppressed by the addition of NPS-2143 (Fig. 4A). Furthermore, knockdown of TRPC3 expression in SMIE cells by shTRPC3 (27) demonstrated a loss of l-Phe-mediated [Ca2+]i increase in both 0.5 mm as well as 2 mm Ca2+. We confirmed the knockdown of TRPC3 in SMIE cells transfected with shTRPC3 by Western blot analysis, shown in the inset (Fig. 4B). To confirm the involvement of CSR in regulating TRPC3 function, siCSR was used to knock down the expression of CSR in SMIE cells. Our data (Fig. 4C) show that CSR-induced Ca2+ release and Ca2+ entry both are severely reduced due to the knockdown of CSR expression. Together, these data provide evidence that TRPC3 contributes to CSR-dependent [Ca2+]i increase in SMIE cells by mediating Ca2+ influx. Although CSR stimulation can be expected to lead to store-operated Ca2+ entry pathways, further studies will be required to determine whether TRPC3 is contributing to store-dependent or -independent Ca2+ entry mechanisms.
FIGURE 4.

Evidence for functional CSR and Ca2+ signaling in SMIE cells. A and B, mean fluorescence traces of Fluo-4-loaded SMIE cells. Cells bathed in 0.5 mm Ca2+ were treated with 10 mm l-Phe or l-Phe (10 mm) plus 1 μm NPS-2143 (A). l-Phe with 0.5 mm external Ca2+ was added to control or shTRPC3-transfected cells (B). Ca2+ (2 mm) addition is shown on the trace. B, inset, Western blots (IB) using anti-TRPC3 antibody, showing knockdown of TRPC3 protein by TRPC3 shRNA (31). β-Actin was used as a loading control for TRPC3 protein. C, mean fluorescence traces of Fura-2-loaded SMIE cells. Cells were transfected with scrambled (control) or siCSR. Both groups of cell were bathed in buffer with 0.5 mm external Ca2+ and then treated with 10 mm l-Phe. C, inset, Western blots using anti-CSR antibody showing knockdown of CSR protein by siRNA to CSR. β-Actin in the lower panel represents as loading control for CSR protein. Additions/removals of Ca2+ in the external medium are indicated in the figure.
Function of Spatially Localized CSR in SMIE Cells
To determine whether the CSR-TRPC3 signaling can have a role in Ca2+ absorption via the apical membrane of polarized salivary gland cells, SMIE cells were grown on permeable filter supports until they polarized and developed tight junctions. Fig. 5A shows that the TER increased with the growth of SMIE cell monolayer on a filter support. Further, although TRPC3 and ZO1 (indicating the location tight junctions) were localized apically in polarized SMIE cells (Fig. 5B), Na+,K+-ATPase was localized in the lateral region (Fig. 5B, note that the basal region cannot be imaged in these samples due to interference from the filter). Importantly, CSR was also localized in the apical region of these cells (Fig. 5C). The endogenous expression of CSR was assessed by Western blotting (Fig. 5D). Whereas SMIE cells endogenously express CSR, HEK cells do not. HEK cells overexpressing CSR were used as a control to confirm protein detection by the antibody.
FIGURE 5.

Localization of CSR and TRPC3 in polarized SMIE cells. A, bar diagram of TER showing the establishment of polarity while cells were growing onto Transwell filter. Error bars, S.D. B, representative images of confocal X-Z sections of polarized SMIE cells grown in Transwell filter support. Top panel represents the staining with Z01 (green signal), a tight junction marker at the junction of apical and lateral region. The middle panel shows Na+,K+-ATPase (NaK; basolateral marker), indicating lateral localization (green signal); nuclei are stained with DAPI (blue signal). Bottom panel shows apical localization of TRPC3 (green signal), indicating an apical expression of TRPC3; nuclei are stained with propidium iodide (red signal). C, confocal sections (XY and XZ) of polarized SMIE cells (grown onto a Transwell filter) labeled with anti-CSR antibody (rabbit, 1:100 dilution). D, Western blot (IB) using anti-CSR antibody (Rabbit, 1:500 dilution) showing the expression of endogenous CSR protein in SMIE cells and overexpression of CSR in typical HEK cells glycosylated bands (bands did not result in untransfected cells).
[Ca2+]i imaging was done with polarized SMIE cells to demonstrate the spatial pattern of the Ca2+ changes associated with CSR activation. Polarized SMIE cells were loaded with Fluo-4, and confocal microscopy was used to monitor fluorescence in an X-Z section of the cell (regions of interest were selected at the apical and basal regions of the cells as indicated in Fig. 6A). Addition of l-Phe (10 mm) to the apical side of the cells together with 0.5 mm Ca2+ induced a sharp increase in fluorescence, which was more rapid and pronounced in the apical region compared with the basal region of the cells (Fig. 6B). We also observed a dramatic increase in fluorescence intensity of Fluo-4 in the apical region (compared with basal region) with the increase in [Ca2+] from 0 to 1.2 mm in the apical chamber (Fig. 6C). The rate of increase of Ca2+ signal in the apical region was significantly higher than the basal region (inset in Fig. 6C), indicating the induction of Ca2+ mobilization from apical to basal region. The response to l-Phe, but not thapsigargin, was severely blunted when NPS-2143 was included in the apical medium (Fig. 6D). Further, thapsigargin, which uniformly releases Ca2+ from intracellular Ca2+ stores, induced simultaneous changes in Fluo-4 fluorescence in apical and basal regions of the cell (Fig. 6E).
FIGURE 6.

Domain-specific function of CSR in SMIE cells. A, representative images (Confocal on live cells; X-Z sections) of Fluo-4-loaded polarized SMIE cells on Transwell filter. Average intensity data of the demarked (equal) region of interest (dashed boxes) in the apical (white) and basal (yellow) were plotted showing changes in Fluo-4 ([Ca2+]i) signal. Graphical presentation of (mean fluorescence traces) of Fluo-4-loaded SMIE cells represents [Ca2+]i rise within the apical/basal region. All applications were made apically. B, addition of l-Phe (10 mm) and 0.5 mm external Ca2+. C, [Ca2+]i rise due to application of 1.2 mm external Ca2+. Inset, bar diagram showing the rate of increase in apical [Ca2+]i signal (p < 0.01) than basal signal in response to [Ca2+]o, indicating the induction of Ca2+ mobilization from apical to basal region. D, effect of l-Phe in the presence of CSR antagonist (NPS-2143; 1 μm) and thapsigargin (Tg; 1 μm). E, simultaneous changes in Fluo-4 fluorescence in apical and basal regions of the cells following application of thapsigargin (1 μm).
Dynamic CSR-TRPC3 Interaction Regulates TRPC3 Function
We have reported earlier that TRPC3 forms a signaling complex with key Ca2+-signaling proteins involved in muscarinic receptor signaling mechanism (20). As shown in Fig. 1, CSR and TRPC3 are associated in salivary gland cells. Here we examined the effect of stimulation on the CSR-TRPC3 complex in polarized SMIE cells (grown onto a Transwell filter). Activation of CSR by l-Phe addition to the apical medium induced an increase in the association of TRPC3 with CSR (Fig. 7Ai) which was decreased when cells were treated with l-Phe together with NPS-2143 (Fig. 7Aii, in both panels, input levels of the TRPC3 are also shown).
FIGURE 7.
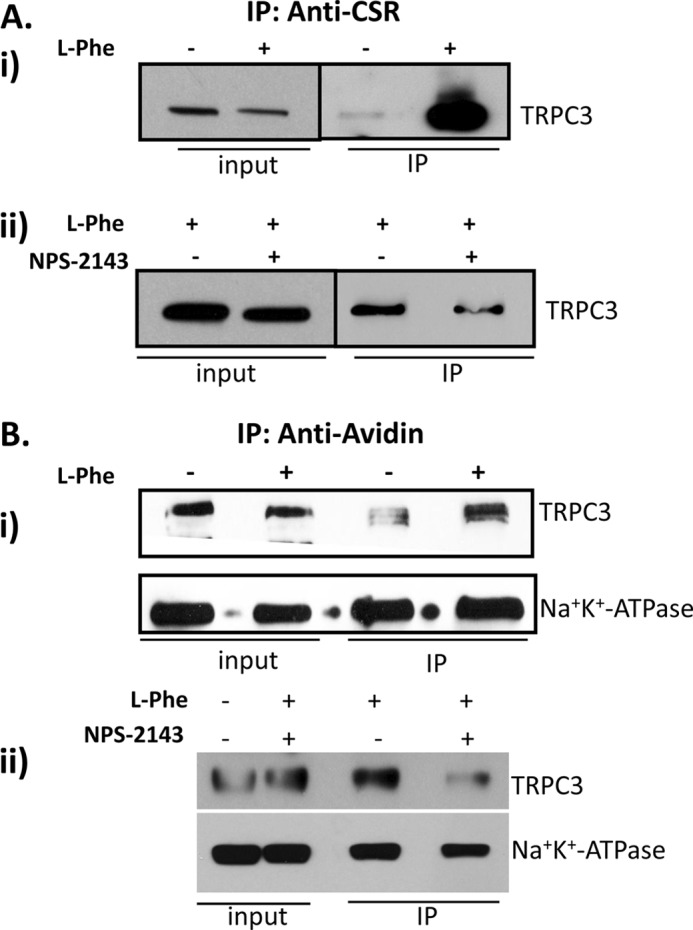
Activation of CSR in SMIE cells enhanced CSR-TRPC3 complex formation and plasma membrane expression of TRPC3. A, SMIE cells grown in Transwell filter treated with l-Phe and/or NPS-2143 were solubilized using RIPA buffer, and co-immunoprecipitation (IP) experiment was performed using anti-CSR antibody (1:100). i, Western blot using anti-TRPC3 antibody (1:500) shows changes in TRPC3 bands representing the increase in complex formation induced by the treatment with l-Phe. ii, complex formation is inhibited after SMIE cells are treated with l-Phe (10 mm) and/or NPS-2143 (1 μm). B, cell surface biotinylation experiment in SMIE cells was performed to determine plasma membrane expression of TRPC3 in response to CSR activation. SMIE cells grown in Transwell filter were treated with l-Phe (10 mm) and/or NPS-2143 (1 μm) then biotinylated using EZ-Link Sulfo-NHSSS-Biotin and solubilized using RIPA buffer. Co-IP experiments were performed using NeutrAvidin beads followed by Western blotting using anti-TRPC3 (1:500) and Na+,K+-ATPase (NaK; 1:1000) antibodies showing changes in TRPC3 bands representing an increase in surface expression induced by l-Phe (i). More importantly, treatment with l-Phe and/or NPS-2143 inhibited the complex formation (ii). Another plasma membrane protein did not change, indicating the specificity of the biotinylation experiment (lower panel).
Trafficking of TRPC3 to the membrane was previously shown to be regulated by stimulation of receptors leading to PIP2 hydrolysis, e.g. muscarinic receptor or EGF (33, 34). We therefore examined whether CSR can regulate surface expression of TRPC3. Biotinylation experiments with polarized SMIE cells demonstrate that the stimulation of CSR by apical application of 10 mm l-Phe plus 1.2 mm Ca2+ caused an increase in the surface expression of TRPC3 (Fig. 7Bi), which was again suppressed by simultaneous addition of NPS-2143 (Fig. 7Bii). Note that surface expression of Na+,K+-ATPase, a plasma membrane protein, did not change under these conditions (Fig. 7B, i and ii, lower panel). Together, these data suggest that plasma membrane expression of TRPC3 in SMIE cells depends on the apical stimulation of CSR-activated receptor-operated pathway. In aggregate, these data suggest that upon activation of CSR, a CSR-TRPC3 complex is formed in the apical membrane that is associated with an increase in the surface expression of TRPC3 and an activation of TRPC3-mediated Ca2+ entry via the apical membrane.
DISCUSSION
The study demonstrates that functional CSR are localized in SMG ducts. Further, we show using primary isolated salivary gland ducts as well as polarized salivary gland ductal cell line that CSR is co-localized and associated with TRPC3 in the apical region of these cells. Apical stimulation of CSR by physiologically relevant agonists leads to (i) an increase in the association of CSR with TRPC3, (ii) an increase in the surface expression of TRPC3, and (iii) Ca2+ entry via the apical membrane of cells. Together these studies reveal that CSR can be stimulated by Ca2+ as well as by agonists such as l-Phe, both of which are present in saliva, triggering Ca2+ entry via TRPC3. The apical localization of both CSR and TRPC3 in polarized salivary gland cells as well as the spatial patterning of the [Ca2+]i increase (apical > basal) strongly suggests that CSR can sense [Ca2+]i in the luminal side of the cell and regulate Ca2+ entry via TRPC3 across the luminal membrane. In aggregate, our findings are consistent with the suggestion that CSR regulates an apical Ca2+ absorption pathway in salivary gland ductal cells that is most likely mediated by TRPC3. Several PLC-coupled G-protein coupled receptors; Ca2+-sensing, bradykinin, and purinergic receptors, have been identified in the apical membrane of ductal cells, including those in the salivary glands (35, 36). These receptors have been described to regulate cellular functions such as protein and fluid secretion as well as ion reabsorption (23, 37). In polarized epithelial cells, strategic localization of Ca2+ channel proteins provides spatial specificity and compartmentalization of [Ca2+]i signals as well as effector proteins (38). Here we show that both CSR and TRPC3 are localized apically and lead to generation of [Ca2+]i increase in the apical region. Whereas most of the Ca2+ is pumped out by the activity of plasma membrane calcium pumps (see Fig. 8), Ca2+ could also be sequestered into the ER or other intracellular Ca2+ storage compartments. It is presently unclear whether the CSR-stimulated [Ca2+]i increase is also used for regulation of other cell functions, such as activation of ion channels localized within that region of the cell. Interestingly, such a function of TRPC3 has been recently demonstrated to involve in transepithelial (apical-to-basolateral) Ca2+ flux induced by a receptor operated purinergic G protein-coupled receptor pathway (39). Therefore, our finding of an enhanced mobilization of [Ca2+]i in the apical region (compared with the basal region) of polarized ductal cells induced by CSR supports the hypothesis of a transepithelial movement of Ca2+. Future experiments will address the details of this underlying mechanism of transepithelial Ca2+ transport.
FIGURE 8.

Proposed mechanism of CSR signaling in salivary duct. Diagram shows the proposed mechanism of CSR-regulated signaling in salivary ductal cells. The alteration in [Ca2+] in ductal lumen activates CSR, which in turn trigger TRPC3 activation though a PLC-dependent mechanism and causes Ca2+ entry into the ductal cell. This process could potentially initiate a transductal flow of Ca2+ from apical to basal end. Furthermore, Ca2+-sensing properties of CSR by [Ca2+]o could potentially be modulated by the presence of endogenous (saliva) CSR modulator(s).
CSRs have been described previously as key regulators of Ca2+ transport in epithelial cells (40). In the present study, luminal localization of CSR in the salivary duct suggests the functional significance of this receptor, i.e. as a sensor of changes in [Ca2+] in primary saliva. More interestingly, CSR is mainly expressed in granular convoluted ducts and striated ducts, the main regions of the duct where ion-reabsorption takes place (41). Our data also show that CSR in salivary gland ducts responds to physiologically relevant stimuli like Ca2+ aromatic amino acids such as l-Phe, both of which are present in saliva (14, 29). The physiological relevance of amino acid modulation of CSR activation has been demonstrated in the kidney and in the gastrointestinal tract, where amino acids, respectively, modulate calcium excretion and gastric acid secretion (18, 19). Amino acids also have been shown to have a physiological role as modulators of CSR activity in the brain (e.g. in the control of myelination by oligodendrocytes) (42). Further, toxic elevations of aromatic amino acids (e.g. in phenylketonuria or tyrosinemia) induce some of their effects by inappropriate activation and/or down-regulation of CSR in key sites (43). It is of interest that saliva contains several endogenous polyvalent cations which could potentially decrease threshold for activation of CSR (44). Thus, the CSR function can be positively or negatively regulated depending on the saliva constituents.
In the model shown in Fig. 8, we summarize our concept of the function of CSR-TRPC3 in the apical membrane of salivary gland ducts. We suggest that relatively high levels Ca2+ in primary saliva can potentially activate the CSR in the luminal membrane of striated and granular convoluted ducts resulting in Ca2+ influx via TRPC3 channel. The Ca2+ from the apical region is most likely pumped out of the cells by plasma membrane calcium ATPase that are localized in basolateral as well as apical membranes. Ca2+ can also be accumulated into intracellular stores. We propose that reabsorption of Ca2+ by salivary ducts is of potential importance for protection of salivary gland function. An uncontrolled increase in the [Ca2+] in ductal saliva could contribute to the formation of salivary calculi (8, 45). Relatively higher concentration of [Ca2+] could be causative in the formation of calcium deposits that consists of mainly calcium phosphate (8, 45–48). It is important to note that in tissues such as kidney, stone formation is associated with changes in calcium reabsorption (49). In salivary glands, such deposits cause ductal obstruction leading to decreased saliva flow (45, 48), a condition that often leads to infection (sialoadenitis). In conclusion, the data presented above reveal a novel and potentially important role for CSR and TRPC3 in Ca2+ reabsorption via the apical membrane of salivary gland ductal cells. Based on our findings we suggest that CSR senses the [Ca2+] in ductal saliva and regulates TRPC3 to mediate Ca2+ removal from saliva. More significantly, this mechanism is likely to be important for maintaining the Ca2+ balance and preventing salivary gland pathology associated with sialolithiasis.
Acknowledgments
We thank Drs. Gerard P. Ahern (Georgetown University), Robert S. Redman (DC VA Medical Center), and Sandeep C. Pingle (UCSD) for helpful discussions and input in the preparation of this manuscript; the Ahern laboratory members for help and providing some chemicals; and Mariyam Al-shatti for proofreading this manuscript.
This study was supported, in whole or in part, by National Institutes of Health Grant DE 019524 through the NIDCR (to B. C. B) and by NIDCR Intramural Research (to I. S. A.).
- CSR
- Ca2+-sensing receptor
- AM
- acetoxymethyl
- HEK
- human embryonic kidney
- OAG
- 1-oleoyl-2-acetyl-sn-glycerol
- PLC
- phospholipase C
- RIPA
- radioimmune precipitation assay
- SMG
- submandibular gland
- TER
- transepithelial resistance
- TRPC
- transient receptor potential canonical
- l-Try
- l-Tryptophan.
REFERENCES
- 1. Hoenderop J. G., Nilius B., Bindels R. J. (2005) Calcium absorption across epithelia. Physiol. Rev. 85, 373–422 [DOI] [PubMed] [Google Scholar]
- 2. Knauf H., Lubcke R., Kreutz W., Sachs G. (1982) Interrelationships of ion transport in rat submaxillary duct epithelium. Am. J. Physiol. 242, F132–139 [DOI] [PubMed] [Google Scholar]
- 3. Zentner M. D., Lin H. H., Wen X., Kim K. J., Ann D. K. (1998) The amiloride-sensitive epithelial sodium channel α-subunit is transcriptionally down-regulated in rat parotid cells by the extracellular signal-regulated protein kinase pathway. J. Biol. Chem. 273, 30770–30776 [DOI] [PubMed] [Google Scholar]
- 4. Dinudom A., Harvey K. F., Komwatana P., Young J. A., Kumar S., Cook D. I. (1998) Nedd4 mediates control of an epithelial Na+ channel in salivary duct cells by cytosolic Na+. Proc. Natl. Acad. Sci. U.S.A. 95, 7169–7173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Homann V., Kinne-Saffran E., Arnold W. H., Gaengler P., Kinne R. K. (2006) Calcium transport in human salivary glands: a proposed model of calcium secretion into saliva. Histochem. Cell Biol. 125, 583–591 [DOI] [PubMed] [Google Scholar]
- 6. Mangos J. A., Garrish M. T., Wells R., Farnham W., Bouchlas G. (1978) A micropuncture study of the handling of calcium by the rat parotid. J. Dent. Res. 57, 818–825 [DOI] [PubMed] [Google Scholar]
- 7. Mangos J. A., Boyd R. L., Loughlin G. M., Cockrell A., Fucci R. (1981) Handling of calcium by the ferret submandibular gland. J. Dent. Res. 60, 91–95 [DOI] [PubMed] [Google Scholar]
- 8. Grases F., Santiago C., Simonet B. M., Costa-Bauzá A. (2003) Sialolithiasis: mechanism of calculi formation and etiologic factors. Clin. Chim. Acta 334, 131–136 [DOI] [PubMed] [Google Scholar]
- 9. Thongon N., Nakkrasae L. I., Thongbunchoo J., Krishnamra N., Charoenphandhu N. (2008) Prolactin stimulates transepithelial calcium transport and modulates paracellular permselectivity in Caco-2 monolayer: mediation by PKC and ROCK pathways. Am. J. Physiol. Cell. Physiol. 294, C1158–1168 [DOI] [PubMed] [Google Scholar]
- 10. Khanal R. C., Nemere I. (2008) Endocrine regulation of calcium transport in epithelia. Clin. Exp. Pharmacol. Physiol. 35, 1277–1287 [DOI] [PubMed] [Google Scholar]
- 11. Brown E. M., MacLeod R. J. (2001) Extracellular calcium sensing and extracellular calcium signaling. Physiol. Rev. 81, 239–297 [DOI] [PubMed] [Google Scholar]
- 12. Bräuner-Osborne H., Jensen A. A., Sheppard P. O., O'Hara P., Krogsgaard-Larsen P. (1999) The agonist-binding domain of the calcium-sensing receptor is located at the amino-terminal domain. J. Biol. Chem. 274, 18382–18386 [DOI] [PubMed] [Google Scholar]
- 13. Barlow A. L., Macleod A., Noppen S., Sanderson J., Guérin C. J. (2010) Colocalization analysis in fluorescence micrographs: verification of a more accurate calculation of Pearson's correlation coefficient. Microsc. Microanal. 16, 710–724 [DOI] [PubMed] [Google Scholar]
- 14. Papanayotou P. H., Dozi-Vassiliades J., Kovatsis A. (1973) Free amino acids in human saliva. J. Dent. Res. 52, 418–419 [DOI] [PubMed] [Google Scholar]
- 15. Venza M., Visalli M., Cicciu D., Teti D. (2001) Determination of polyamines in human saliva by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B. Biomed. Sci. Appl. 757, 111–117 [DOI] [PubMed] [Google Scholar]
- 16. Rey O., Young S. H., Papazyan R., Shapiro M. S., Rozengurt E. (2006) Requirement of the TRPC1 cation channel in the generation of transient Ca2+ oscillations by the calcium-sensing receptor. J. Biol. Chem. 281, 38730–38737 [DOI] [PubMed] [Google Scholar]
- 17. Feng S. L., Sun M. R., Li T. T., Yin X., Xu C. Q., Sun Y. H. (2011) Activation of calcium-sensing receptor increases TRPC3 expression in rat cardiomyocytes. Biochem. Biophys. Res. Commun. 406, 278–284 [DOI] [PubMed] [Google Scholar]
- 18. Rey O., Young S. H., Jacamo R., Moyer M. P., Rozengurt E. (2010) Extracellular calcium sensing receptor stimulation in human colonic epithelial cells induces intracellular calcium oscillations and proliferation inhibition. J. Cell. Physiol. 225, 73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Conigrave A. D., Mun H. C., Brennan S. C. (2007) Physiological significance of l-amino acid sensing by extracellular Ca2+-sensing receptors. Biochem. Soc. Trans. 35, 1195–1198 [DOI] [PubMed] [Google Scholar]
- 20. Bandyopadhyay B. C., Swaim W. D., Liu X., Redman R. S., Patterson R. L., Ambudkar I. S. (2005) Apical localization of a functional TRPC3/TRPC6-Ca2+-signaling complex in polarized epithelial cells: role in apical Ca2+ influx. J. Biol. Chem. 280, 12908–12916 [DOI] [PubMed] [Google Scholar]
- 21. Liu X., Cheng K. T., Bandyopadhyay B. C., Pani B., Dietrich A., Paria B. C., Swaim W. D., Beech D., Yildrim E., Singh B. B., Birnbaumer L., Ambudkar I. S. (2007) Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1−/− mice. Proc. Natl. Acad. Sci. U.S.A. 104, 17542–17547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Romanenko V. G., Nakamoto T., Srivastava A., Begenisich T., Melvin J. E. (2007) Regulation of membrane potential and fluid secretion by Ca2+-activated K+ channels in mouse submandibular glands. J. Physiol. 581, 801–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao H., Xu X., Diaz J., Muallem S. (1995) Na+, K+, and H+/HCO3 transport in submandibular salivary ducts: membrane localization of transporters. J. Biol. Chem. 270, 19599–19605 [PubMed] [Google Scholar]
- 24. Bandyopadhyay B. C., Pingle S. C., Ahern G. P. (2011) Store-operated Ca2+ signaling in dendritic cells occurs independently of STIM1. J. Leukoc. Biol. 89, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tran S. D., Wang J., Bandyopadhyay B. C., Redman R. S., Dutra A., Pak E., Swaim W. D., Gerstenhaber J. A., Bryant J. M., Zheng C., Goldsmith C. M., Kok M. R., Wellner R. B., Baum B. J. (2005) Primary culture of polarized human salivary epithelial cells for use in developing an artificial salivary gland. Tissue Eng. 11, 172–181 [DOI] [PubMed] [Google Scholar]
- 26. Liu X., Bandyopadhyay B. C., Singh B. B., Groschner K., Ambudkar I. S. (2005) Molecular analysis of a store-operated and 2-acetyl-sn-glycerol-sensitive non-selective cation channel: heteromeric assembly of TRPC1-TRPC3. J. Biol. Chem. 280, 21600–21606 [DOI] [PubMed] [Google Scholar]
- 27. Bandyopadhyay B. C., Ong H. L., Lockwich T. P., Liu X., Paria B. C., Singh B. B., Ambudkar I. S. (2008) TRPC3 controls agonist-stimulated intracellular Ca2+ release by mediating the interaction between inositol 1,4,5-trisphosphate receptor and RACK1. J. Biol. Chem. 283, 32821–32830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Riccardi D., Hall A. E., Chattopadhyay N., Xu J. Z., Brown E. M., Hebert S. C. (1998) Localization of the extracellular Ca2+/polyvalent cation-sensing protein in rat kidney. Am. J. Physiol. 274, F611–622 [DOI] [PubMed] [Google Scholar]
- 29. Nakamura Y., Kodama H., Satoh T., Adachi K., Watanabe S., Yokote Y., Sakagami H. (2010) Diurnal changes in salivary amino acid concentrations. In Vivo 24, 837–842 [PubMed] [Google Scholar]
- 30. Nemeth E. F., Delmar E. G., Heaton W. L., Miller M. A., Lambert L. D., Conklin R. L., Gowen M., Gleason J. G., Bhatnagar P. K., Fox J. (2001) Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J. Pharmacol. Exp. Ther. 299, 323–331 [PubMed] [Google Scholar]
- 31. Kiyonaka S., Kato K., Nishida M., Mio K., Numaga T., Sawaguchi Y., Yoshida T., Wakamori M., Mori E., Numata T., Ishii M., Takemoto H., Ojida A., Watanabe K., Uemura A., Kurose H., Morii T., Kobayashi T., Sato Y., Sato C., Hamachi I., Mori Y. (2009) Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc. Natl. Acad. Sci. U.S.A. 106, 5400–5405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He X., Kuijpers G. A., Goping G., Kulakusky J. A., Zheng C., Delporte C., Tse C. M., Redman R. S., Donowitz M., Pollard H. B., Baum B. J. (1998) A polarized salivary cell monolayer useful for studying transepithelial fluid movement in vitro. Pflugers Arch. 435, 375–381 [DOI] [PubMed] [Google Scholar]
- 33. Singh B. B., Lockwich T. P., Bandyopadhyay B. C., Liu X., Bollimuntha S., Brazer S. C., Combs C., Das S., Leenders A. G., Sheng Z. H., Knepper M. A., Ambudkar S. V., Ambudkar I. S. (2004) VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Mol. Cell 15, 635–646 [DOI] [PubMed] [Google Scholar]
- 34. Smyth J. T., Lemonnier L., Vazquez G., Bird G. S., Putney J. W., Jr. (2006) Dissociation of regulated trafficking of TRPC3 channels to the plasma membrane from their activation by phospholipase C. J. Biol. Chem. 281, 11712–11720 [DOI] [PubMed] [Google Scholar]
- 35. Kiselyov K., Wang X., Shin D. M., Zang W., Muallem S. (2006) Calcium signaling complexes in microdomains of polarized secretory cells. Cell Calcium 40, 451–459 [DOI] [PubMed] [Google Scholar]
- 36. Ambudkar I. S., Bandyopadhyay B. C., Liu X., Lockwich T. P., Paria B., Ong H. L. (2006) Functional organization of TRPC-Ca2+ channels and regulation of calcium microdomains. Cell Calcium 40, 495–504 [DOI] [PubMed] [Google Scholar]
- 37. Futakuchi S., Ishiguro H., Naruse S., Ko S. B., Fujiki K., Yamamoto A., Nakakuki M., Song Y., Steward M. C., Kondo T., Goto H. (2009) High glucose inhibits HCO3− and fluid secretion in rat pancreatic ducts. Pflugers Arch. 459, 215–226 [DOI] [PubMed] [Google Scholar]
- 38. Kiselyov K., Shin D. M., Muallem S. (2003) Signalling specificity in GPCR-dependent Ca2+ signalling. Cell. Signal. 15, 243–253 [DOI] [PubMed] [Google Scholar]
- 39. Goel M., Schilling W. P. (2010) Role of TRPC3 channels in ATP-induced Ca2+ signaling in principal cells of the inner medullary collecting duct. Am. J. Physiol. Renal. Physiol. 299, F225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. VanHouten J. N., Neville M. C., Wysolmerski J. J. (2007) The calcium-sensing receptor regulates plasma membrane calcium adenosine triphosphate isoform 2 activity in mammary epithelial cells: a mechanism for calcium-regulated calcium transport into milk. Endocrinology 148, 5943–5954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Romanenko V. G., Nakamoto T., Catalán M. A., Gonzalez-Begne M., Schwartz G. J., Jaramillo Y., Sepúlveda F. V., Figueroa C. D., Melvin J. E. (2008) Clcn2 encodes the hyperpolarization-activated chloride channel in the ducts of mouse salivary glands. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G1058–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bandyopadhyay S., Tfelt-Hansen J., Chattopadhyay N.,(2010) Diverse roles of extracellular calcium-sensing receptor in the central nervous system. J. Neurosci. Res. 88, 2073–2082 [DOI] [PubMed] [Google Scholar]
- 43. Conigrave A. D., Mun H. C., Lok H. C. (2007) Aromatic l-amino acids activate the calcium-sensing receptor. J. Nutr. 137, 1524S-1527S [DOI] [PubMed] [Google Scholar]
- 44. Tanida T., Okamoto T., Okamoto A., Wang H., Hamada T., Ueta E., Osaki T. (2003) Decreased excretion of antimicrobial proteins and peptides in saliva of patients with oral candidiasis. J. Oral. Pathol. Med. 32, 586–594 [DOI] [PubMed] [Google Scholar]
- 45. Su Y. X., Zhang K., Ke Z. F., Zheng G. S., Chu M., Liao G. Q. (2010) Increased calcium and decreased magnesium and citrate concentrations of submandibular/sublingual saliva in sialolithiasis. Arch. Oral Biol. 55, 15–20 [DOI] [PubMed] [Google Scholar]
- 46. Tannenbaum P. J., Posner A. S., Mandel I. D. (1976) Formation of calcium phosphates in saliva and dental plaque. J. Dent. Res. 55, 997–1000 [DOI] [PubMed] [Google Scholar]
- 47. Westhofen M., Schäfer H., Seifert G. (1984) Calcium redistribution, calcification and stone formation in the parotid gland during experimental stimulation and hypercalcaemia: cytochemical and x-ray microanalytical investigations. Virchows Arch. A Pathol. Anat. Histopathol. 402, 425–438 [DOI] [PubMed] [Google Scholar]
- 48. Kasaboğlu O., Er N., Tümer C., Akkocaoğlu M. (2004) Micromorphology of sialoliths in submandibular salivary gland: a scanning electron microscope and x-ray diffraction analysis. J. Oral. Maxillofac. Surg. 62, 1253–1258 [DOI] [PubMed] [Google Scholar]
- 49. Grases F., Costa-Bauzá A., Ramis M., Montesinos V., Conte A. (2002) Simple classification of renal calculi closely related to their micromorphology and etiology. Clin. Chim. Acta 322, 29–36 [DOI] [PubMed] [Google Scholar]
- 50. Costes S. V., Daelemans D., Cho E. H., Dobbin Z., Pavlakis G., Lockett S. (2004) Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 86, 3993–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]