

Background: HDC catalyzes the rate-limiting step in histamine biosynthesis.
Results: A mutation based on the crystal structure of HDC changed the substrate selectivity from l-histidine to l-DOPA.
Conclusion: The molecular basis for substrate specificity and recognition of group II PLP-dependent decarboxylases were clarified.
Significance: Knowledge of the HDC tertiary structure now makes it possible to design novel drugs that prevent histamine biosynthesis.
Keywords: Crystal Structure, Enzyme Kinetics, Enzyme Structure, Mutagenesis, Pyridoxal Phosphate, X-ray Crystallography, Decarboxylase, Histamine
Abstract
Histamine is an important chemical mediator for a wide variety of physiological reactions. l-Histidine decarboxylase (HDC) is the primary enzyme responsible for histamine synthesis and produces histamine from histidine in a one-step reaction. In this study, we determined the crystal structure of human HDC (hHDC) complexed with the inhibitor histidine methyl ester. This structure shows the detailed features of the pyridoxal-5′-phosphate inhibitor adduct (external aldimine) at the active site of HDC. Moreover, a comparison of the structures of hHDC and aromatic l-amino acid (l-DOPA) decarboxylase showed that Ser-354 was a key residue for substrate specificity. The S354G mutation at the active site enlarged the size of the hHDC substrate-binding pocket and resulted in a decreased affinity for histidine, but an acquired ability to bind and act on l-DOPA as a substrate. These data provide insight into the molecular basis of substrate recognition among the group II pyridoxal-5′-phosphate-dependent decarboxylases.
Introduction
Histamine has an important role in a number of physiological reactions. It acts as a neurotransmitter in the central nervous system (1), chemical mediator of allergic reactions (2), and activator of various biological processes including gastric acid secretion (3), capillary dilatation, and smooth muscle contraction (2). Histidine decarboxylase (HDC3; EC 4.1.1.22) catalyzes the rate-limiting reaction in histamine biosynthesis. HDC gene knock-out mice lack histamine almost completely (4). Inhibition of histamine synthesis in mice, by treatment with an HDC inhibitor or by an HDC gene knock-out, alters the histaminergic neuron-mediated physiological responses, including arousal (5, 6), appetite (7), and locomotor activity (8, 9). Tourette syndrome, a developmental neuropsychiatric disorder, is related to an HDC gene mutation (10). Outside of the central nervous system, HDC inhibitors are expected to be useful for treatment of type I allergic reactions, gastric ulcers, and inflammation.
Mammalian HDC is a pyridoxal-5′-phosphate (PLP)-dependent decarboxylase. Aromatic amino acid decarboxylase (AroDC) is also a PLP-dependent enzyme (11) and functions in the biosynthesis of two important neurotransmitters: dopamine and serotonin. Synthesis of γ-aminobutyric acid (GABA), another important neurotransmitter, is dependent on two PLP-dependent glutamate decarboxylase (GAD) isoforms: GAD65 (65 kDa) and GAD67 (67 kDa) (12). PLP-dependent decarboxylases are classified into four groups on the basis of their evolutionary origin (13), and HDC, AroDC, and GAD are all members of the same group (group II). When compared with AroDC and GAD, the structural study of HDC has been limited because of the difficulty in handling the enzyme and the low amount of HDC in examined tissues.
Mammalian HDC cDNA encodes a 74-kDa polypeptide. Taguchi et al. (13) and Ohmori et al. (14) purified HDC from fetal rat liver and mouse mastocytoma P-815 cells, respectively. The molecular mass of both HDCs was estimated to be 53–54 kDa, which is ∼20 kDa smaller than the size estimated from the full-length cDNA. The difference in the molecular mass was explained by post-translational processing that truncates a C-terminal region of ∼20 kDa. The C-terminal truncated 54-kDa form of human recombinant HDC possessed sufficient catalytic activity to support histamine synthesis (15). Therefore, the C-terminal truncated HDC is considered as an active form. Purification of the active HDC has been investigated by several research groups; however, large scale preparation is difficult even for the recombinant protein (16).
The mechanisms underlying substrate recognition and high substrate specificity at the active site as well as the catalytic mechanism of HDC remain to be clarified. The existence of a loop region in HDC has been suggested, which is thought to play an important role in catalysis (17). A site-directed mutagenesis study focusing on the loop region showed that the conformation of the loop significantly affects enzyme activity (17). Further structural analysis is required to clarify the role of the loop region in HDC catalysis.
Many histidine derivatives have been tested to identify potent inhibitors of HDC (18, 19). α-Fluoromethyl histidine (18) and histidine methyl ester (HME) (19) were both found to be potent HDC inhibitors more than three decades ago. However, neither agent has been clinically used. Epigallocatechin gallate from green tea (20) and pyridoxyl histidine methyl ester (21) were recently found to act as potent HDC inhibitors; however, the possibility of side effects, in vivo efficiency, and specificity for HDC of these inhibitors have not been assessed, making them unavailable for clinical use. Detailed information on the structure of HDC would facilitate the development of HDC inhibitors with high affinity and specificity.
Recently, we identified two cysteine residues that induce oligomerization of HDC (22). The replacement of these cysteine residues with serine residues prevented the oligomerization, enabling us to prepare a homogeneous sample appropriate for crystallization (22). Here, we have determined the crystal structure of the active PLP-dependent human HDC (hHDC) complexed with HME. Our results provide the first insight into the substrate specificity of PLP-dependent HDC.
EXPERIMENTAL PROCEDURES
Preparation of C-terminal Truncated hHDC and Its Mutants
hHDC cDNA was obtained from a plasmid previously constructed for the expression of hHDC in Escherichia coli (23). An active form of recombinant human HDC, i.e. a C-terminal truncated form (23, 24), was prepared as described in our previous study (22). The amino acids of the C-terminal truncated HDC were from 2 to 477 (hHDC2–477). hHDC2–477 mutants were made using standard molecular biology procedures and verified by sequencing. Each mutant enzyme was expressed as a glutathione S-transferase (GST) fusion protein according to the procedures described in our previous study (22). After digestion by PreScission Protease (GE Healthcare) to cleave off the GST tag, the untagged enzymes were purified using anion-exchange chromatography. Absorption spectra were recorded on a V-660 spectrophotometer (Jasco International Co., Tokyo, Japan) with a 10-mm path length quartz cuvette, at room temperature.
Structure Determination and Refinement
Protein crystallization protocols have been previously described (22). hHDC was mixed with a 10-fold molar excess of the inhibitor HME. Crystals were obtained at 20 °C with a reservoir solution containing 0.1 m Tris-HCl (pH 8.5), 28% (w/v) PEG 3350, and 0.2 m lithium sulfate. Protein droplets were prepared by mixing 1 μl of protein solution containing 10 mg × ml−1 hHDC, 0.1 mm PLP, and 2 mm HME with 1 μl of reservoir solution and were equilibrated against 100 μl of reservoir solution at 20 °C. X-ray diffraction data sets were collected with an x-ray CCD detector (Rayonix MX225HE), using a synchrotron radiation source at the BL44XU beamline at SPring-8 (Harima, Japan). Diffraction images were processed with the HKL2000 (25) and CCP4 (26) programs. The structure was solved by the molecular replacement method implemented in the PHASER crystallographic software (27) by using the structure of AroDC (Protein Data Bank (PDB) code: 1JS6) as the primary search model. The model was completed manually using COOT (28), interspersed with reciprocal space refinement cycles in REFMAC5 (29). Further model building and refinement of the structure were carried out with the COOT (28) and REFMAC5 (29) programs. Progress and validity of the refinement process were checked by monitoring the Rfree value for 5% of the total reflections (30). Model geometry analysis with the MolProbity server (31) showed 97.6% of all residues in Ramachandran-favored regions. The data collection and refinement statistics are summarized in Table 1. The figures were prepared with PyMOL (44) using the coordinates from PDB files 1JS6 and 1JS3 for the pig AroDC-carbiDOPA complex and 2OKJ for human GAD (GAD67), respectively.
TABLE 1.
Summary of x-ray crystallographic data of hHDC-HME complex
Values in parentheses are for the outer resolution shell.
| Data collection | |
| Space group | C2 |
| Unit cell parameters | |
| a, b, c (Å), | 215.16, 112.72, 171.39 |
| β (°) | 110.3 |
| Refinement | |
| Resolution (Å) | 50.00–1.80 (1.83–1.80) |
| No. of unique reflections | 354,047 |
| Multiplicity | 3.8 (3.8) |
| Completeness (%) | 100.0 (100.0) |
| 〈I/σ(I)〉 | 16.0 (2.6) |
| Rmerge (%) | 6.5 (68.0) |
| R-value (%) | 16.3 (22.6) |
| Rfree (%) | 18.1 (25.0) |
| No. of non-hydrogen atoms | 25,270 |
| No. of amino acid residues | 2869 |
| No. of solvent molecules | 2039 |
| Average B-factor (Å2) | |
| Protein | 13.8 |
| Solvent | 20.2 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.054 |
| Ramachandran plot | |
| Favored residues (%) | 97.6 |
| Allowed regions (%) | 2.4 |
Activity Assay
Enzyme activity was assayed on the basis of previous studies with minor modifications (14). The assay mixture (0.4 ml) contained l-histidine or l-DOPA, 0.1 mm dithiothreitol, 0.01 mm PLP, and enzyme in 0.1 m potassium phosphate (pH 6.8). The reaction was carried out at 37 °C and terminated by adding 0.02 ml of 60% perchloric acid. Histamine or dopamine formed during the incubation was separated from the substrate on a TSKgel Histaminepak column (Tosoh, Tokyo, Japan) by HPLC and then fluorometrically measured using the o-phthalaldehyde method (32). The enzyme concentration in the solution was determined spectrophotometrically. The apparent molar extinction coefficient used was 7.83 × 104 m−1 × cm−1 at 280 nm. This value was determined on the basis of the amino acid sequence of hHDC from residues 2–477. For the LC-MS/MS sample, 50 μl of the assay mixture contained 0.1 mm l-DOPA, 0.1 mm dithiothreitol, 0.01 mm PLP, and enzyme (S354G mutant) in 0.1 m potassium phosphate (pH 6.8). The reaction was carried out at 37 °C for 10 min and terminated by adding 150 μl of H2O and 800 μl of acetonitrile with 1% (v/v) formic acid.
LC-MS/MS Analysis
Dopamine was analyzed using an electrospray ionization quadruple tandem mass spectrometer (API-3000, Applied Biosystems) connected to an Agilent HP 1100 HPLC system. A ZIC-HILIC column (150 × 2.1 mm, 5-μm particle size) (Merck) was used to separate dopamine from l-DOPA. To reduce possible contamination of the ion source, the flow was separated by a Valco switching valve using the following program: 0 min (waste), 10 min (mass), 15 min (waste). The gradient condition and other parameters used for MS/MS experiments are shown in supplemental Table S1.
Limited Proteolysis
Purified hHDC2–477 (25 μl, 2.5 mg × ml−1) was incubated at 25 °C with trypsin (0.4 μg × μl−1) either in the absence or in the presence of 5 mm HME. Before and at different times after trypsin addition, aliquots of the proteolysis reaction were withdrawn and monitored by polyacrylamide gel electrophoresis in the presence of SDS on 5–20% gels, which were stained with Coomassie Brilliant Blue.
RESULTS
Overall Structure
We constructed an E. coli system that expresses a C-terminal truncated form, specifically, amino acid residues 2–477 of hHDC (hHDC2–477) (supplemental Fig. S1). Mutation of two cysteine residues (C180S and C418S) facilitated the purification and crystallization of hHDC (22). From the aspect of enzyme kinetics, both hHDC2–477 and the double mutated hHDC2–477 showed similar Km and kcat values (see Table 2), indicating that mutation of these two cysteine residues did not affect enzyme activity. This truncated mutant (hHDC) was crystallized with HME. The final model contained all 476 amino acid residues of hHDC and had acceptable R-factors and stereochemistry (Table 1). The asymmetric unit contained three hHDC dimers with almost identical structures, although some disordered regions were present.
TABLE 2.
Kinetic parameters of hHDC2–477 and mutant hHDC proteins
Human rHDC, mouse HDC, rat HDC, and rat rHDC represent human recombinant HDC of amino acids 1–477, HDC purified from mouse mastocytoma P-815 cells, HDC purified from fetal rat liver and rat recombinant HDC of amino acids 1–512, respectively. Human rAroDC, pig rAroDC, and rat rAroDC represent human recombinant AroDC, pig recombinant AroDC, and rat recombinant AroDC, respectively. All these were full-length AroDC.
| Enzyme activitya | Km | kcat | kcat/Km | |
|---|---|---|---|---|
| nmol × min−1 × mg−1 | mm | s−1 | s−1 × mm−1 | |
| Histidine | ||||
| hHDC2–477 | 1880 ± 150 | 0.10 ± 0.01 | 1.73 ± 0.07 | 17.3 ± 1.9 |
| Double mutant (C180S & C418S) hHDC2–477 | 1950 ± 150 | 0.09 ± 0.02 | 1.87 ± 0.12 | 20.8 ± 4.8 |
| S354G hHDC2–477 | 620 ± 10 | 1.45 ± 0.21 | 2.01 ± 0.04 | 1.4 ± 0.2 |
| Y334F hHDC2–477 | <1 | NDb | ND | ND |
| K305G hHDC2–477 | <1 | ND | ND | ND |
| Human rHDC (15) | 1120 | NRc | NR | NR |
| Mouse HDC (14) | 800 | 0.26 | 0.95 | 3.7 |
| Rat HDC (13) | 128 | NR | NR | NR |
| Rat rHDC (16) | NR | 0.4 | 0.077 | 0.19 |
| DOPA | ||||
| hHDC2–477 | 10 ± 1 | ND | ND | ND |
| S354G hHDC2–477 | 1160 ± 20 | 0.13 ± 0.00 | 1.29 ± 0.01 | 9.9 ± 0.1 |
| Human rAroDC (33) | NR | 0.11 ± 0.01 | 7.6 ± 0.1 | 70.6 |
| Pig rAroDC (34) | NR | 0.07 ± 0.01 | 4.26 ± 0.21 | 60.9 |
| Rat rAroDC (35) | NR | 0.086 | 5.0 | 58.1 |
a 0.8 mm histidine or 0.5 mm DOPA.
b ND, not determined.
c NR, not reported.
The overall structure of hHDC is shown in Fig. 1. The hHDC structure was obtained as a homodimer, and the two monomers of the dimer were related by a noncrystallographic two-fold axis. Each hHDC monomer could be divided into three structural domains: an N-terminal (2–71) domain, a large (71–371) domain, and a small (372–477) domain. The large domain was connected to the small domain by a long α-helix. The dimer interface was formed by the N-terminal and large domains, producing a stable dimeric structure. The N-terminal regions formed extensive hydrophobic interactions with the other subunit, whereas the large domains formed mainly water-mediated electrostatic interactions with the other subunit. On the basis of the information from the crystal structure, the PLP-binding site and loop region of hHDC were investigated.
FIGURE 1.

Overall structure of the hHDC dimer in a complex with PLP and inhibitor (HME). The N-terminal, large, and small domains of Molecule A are shown in dark green, green, and light green, respectively, whereas those of Molecule B are shown in dark blue, blue, and light blue, respectively. The atoms in PLP and HME are shown in orange balls.
PLP-binding Site
The cofactor PLP was located in the large domain. The pyridine ring of PLP was sandwiched between the methyl group of Ala-275 and the imidazole ring of His-194. Asp-273 and Thr-248 seemed to interact with the protonated nitrogen and O3 atoms of the pyridine ring of PLP (supplemental Fig. S2). The phosphate group of PLP was involved in a number of hydrogen bonds with Ser-151, Asn-302, and Ser-354 (Fig. 2). In addition, the negative charge of the phosphate group interacted with the dipole of helix α5, which was positioned with its N terminus close to the phosphate group.
FIGURE 2.

Stereo view of the PLP-binding site. Molecules A and B are shown in green and cyan with residue names and numbers, respectively. PLP-HME are shown in gray. Fo − Fc omit map for PLP-HME contoured at 4σ is shown in orange. Hydrogen bonds are depicted as black.
The electron density map clearly showed the atom position for HME (Fig. 2). hHDC and HME appeared to interact at two sites: at the imidazole ring- and carboxyl ester-binding regions. The imidazole ring of HME pointed toward the si-face of the PLP-HME aldimine. Nδ and Nϵ of the imidazole ring appeared to participate in hydrogen-bonding interactions with the backbone amino group of Tyr-81 and a water molecule, respectively. Except for two hydrogen bonds, the imidazole ring was surrounded by hydrophobic residues (Trp-72, Tyr-80, Leu-102, and Phe-104). The orientation of the imidazole ring of HME was tentatively assigned by their hydrogen-bonded neighbors. OG of Ser-354 was located within van der Waals proximity to the imidazole ring of HME (Fig. 3A). Interestingly, flipping of the imidazole ring allowed the formation of hydrogen bonds with Ser-354 and the carbonyl group of Tyr-81, instead of the water molecule and amino group of Tyr-81, respectively. The carboxyl ester of HME was located at the hydrophobic pocket (Tyr-80, Phe-104, Thr-248, Tyr-334, Leu-335, and Leu-353). His-194 was found within hydrogen-bonding distance of the carboxyl oxygen.
FIGURE 3.

Stereo views of the substrate-binding site. A, molecules A and B are shown in green and cyan with residue names and numbers, respectively. Hydrogen bonds and van der Waals contacts are depicted as black and gray dashed lines. B, superposition of the structure around hHDC-HME (green) onto the AroDC-carbiDOPA structure (gray, PDB ID: 1JS3). Ser-354 in hHDC is labeled.
AroDC, a group II decarboxylase similar to HDC, has been crystallized as a complex with carbiDOPA (11). The structure around the substrate-binding pocket of hHDC-HME was compared with that of AroDC-carbiDOPA, and the conformation of the inhibitors in the substrate-binding pockets was quite similar (Fig. 3B) except for the residue at position 354 in the substrate-binding pocket. This is a serine residue found in hHDC and glycine in AroDC. The hydrophobic residues found in the substrate-binding pocket of hHDC were well conserved in AroDC. The Ser-354 residue was expected to affect substrate selectivity of hHDC. A mutagenesis study was performed for further investigations.
The S354G mutant hHDC2–477 was prepared for enzyme kinetic analysis. hHDC2–477 and S354G mutant enzymes were purified to apparent homogeneity with molecular masses of ∼50 kDa by SDS-PAGE (supplemental Fig. S3). hHDC2–477 and the S354G mutant exhibited similar absorption spectra (Fig. 4) with two absorption bands at 335 and 425 nm, which are typically observed for PLP-dependent enzymes. The kinetic analysis showed notable differences between hHDC2–477 and the S354G mutant. When l-histidine was used as the substrate, the Km of the S354G mutant increased ∼10-fold when compared with that of hHDC2–477, whereas kcat values for both enzymes were similar, resulting in a decrease of kcat/Km values for the S354G mutant (Table 2). These data indicate that mutation of Ser-354 decreased the affinity between the enzyme and histidine.
FIGURE 4.

Absorption spectra of purified hHDCs under the conditions of 20 mm HEPES buffer (pH 7.0) with 100 mm NaCl in the absence of exogenous PLP. hHDC2–477, double-cysteine mutant hHDC2–477, K305G hHDC2–477, Y334F hHDC2–477, and S354G hHDC2–477 are shown in black, blue, red, orange, and cyan. Two bands at 335 and 425 nm are generally ascribed to enolimine and ketoenamine tautomers, respectively, of the Schiff base (aldimine) formed between PLP and the active site lysine residue.
LC-MS/MS ion chromatograms were obtained to confirm dopamine production for the S354G mutant (Fig. 5). Dopamine was produced during the reaction of the S354G mutant with l-DOPA. The enzyme kinetic parameters of dopamine production by hHDC2–477 or the S354G mutant were examined (Table 2). Low activity of hHDC2–477 prevented us from determining the Km and kcat values. In contrast, the activity of the S354G mutant was high enough to determine the Km and kcat values. The Km and kcat values for the S354G mutant of hHDC2–477 were similar to those of human AroDC (0.11 mm and 7.6 s−1, respectively) (33), pig AroDC (0.070 mm and 4.26 s−1, respectively) (34), and rat AroDC (0.086 mm and 5.0 s−1, respectively) (35). kcat/Km values for l-DOPA were ∼7 times larger than those for l-histidine (Table 2), indicating that the S354G mutant showed higher specificity for l-DOPA than l-histidine.
FIGURE 5.

LC-MS/MS ion chromatograms of 1 μm dopamine (a), 10-min reacted S354G mutant assay sample (b), and 0-min reacted S354G mutant assay sample (blank) (c). MRM, Multiple Reaction Monitoring.
Loop Region
The loop region (amino acid residues 330–340), including a short 310-helix, was shown to connect helices α11 and η9 and protruded from the large domain to the active site of the other subunit of the dimer (Fig. 6). This loop also appeared to be located proximal to the neighboring active site, enabling residues in the loop to participate in PLP binding and the formation of the active site entrance. Tyr-334 on the short 310-helix in the loop region formed a hydrogen bond with the backbone amino nitrogen of Ser-195 of the other subunit (Fig. 2). Except for Tyr-334 and water-mediated interactions, no direct electrostatic interactions were found between the loop and the large domain.
FIGURE 6.

Stereo view around of the catalytic loop (amino acid residues 330–340), which covers the substrate-binding site. The residues on the catalytic loop are shown in blue with residue names and numbers, whereas PLP-HME is shown in orange. The other parts of the molecule are shown in a transparent gray surface model.
Limited proteolysis was performed for hHDC2–477 in the presence or absence of an inhibitor, which showed that the inhibitor-free enzyme was more easily degraded than the inhibitor-bound complex (Fig. 7). The molecular masses of the proteolytic fragments, estimated by SDS-PAGE, were 37 and 15 kDa. These fragments correspond to proteolysis around the loop region (residues 328–339). The estimated molecular masses of residues 2–336 and 337–477 are 37.5 and 16.2 kDa, respectively. The degradation probably occurred in the loop region of the inhibitor-free enzyme.
FIGURE 7.
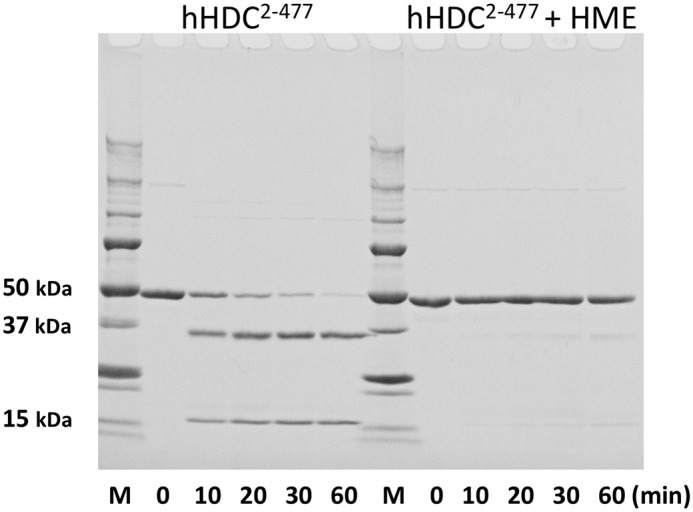
Time courses of the limited proteolysis of HDC in the absence or presence of inhibitor (HME) by electrophoretic analysis. Purified hHDC2–477 was treated with trypsin in the absence or presence of inhibitor, HME. Incubation times are indicated at the bottom. At the indicated times, aliquots were taken and proteolysis was stopped with trypsin inhibitor. Molecular mass marker proteins are shown on the left as follows: 250, 150, 100, 75, 50, 37, 25, 20, 15, and 10 kDa.
The role of Tyr-334, a characteristic residue in the loop, was investigated by mutagenesis. The Y334F mutant of hHDC2–477 was prepared, and its purity and molecular mass are shown in supplemental Fig. S3. The Y334F mutant exhibited two absorption bands at 335 and 425 nm (Fig. 4), suggesting the formation of a Schiff base between PLP and the active-site lysine residue. The Schiff base is considered critical for catalysis by PLP-dependent enzymes. Despite this, the enzyme activity of Y334F was far less than that of hHDC2–477. The histamine level produced by the Y334F mutant was below the detection limit of the instrument (Table 2). The K305G mutant was prepared for comparison with the Y334F mutant. The K305G mutant did not exhibit absorption bands ∼330–430 nm (Fig. 4). As expected, the enzyme activity of the K305G mutant was negligible; the histamine level produced by the K305G mutant was also below the detection limit of the instrument (Table 2). These data indicate that the effect of Tyr-334 on histamine synthesis was comparable with Lys-305. Tyr-334 plays a critical role in the catalytic reaction of hHDC2–477.
DISCUSSION
In this study, we performed x-ray crystallographic and enzyme kinetics analyses on hHDC, the active C-terminal truncated double-mutated human HDC. Our data showed that 1) Ser-354 is a key residue for substrate specificity and 2) the flexible loop (amino acid residues 330–340) participates in the catalytic reaction of histamine synthesis.
Substrate Specificity
The imidazole ring of HME is surrounded by hydrophobic residues (Trp-72, Tyr-80, Leu-102, and Phe-104) and participates in hydrogen-bonding interactions with the backbone amino group of Tyr-81 and a water molecule (Fig. 3A). This conformation was similar to the PLP-carbiDOPA adduct observed in the crystal structure of AroDC (Fig. 3B). CarbiDOPA is an anti-Parkinson drug and is an inhibitor of AroDC. Because carbiDOPA has an α-hydrazino group with an extra nitrogen atom, it was expected to adopt a significantly different conformation from that of the substrate-PLP reactant. However, the two different types of inhibitors shared the same conformation (Fig. 3B). The position and orientation of the imidazole ring and the catechol ring of the substrate were almost identical in the crystal structures of hHDC-HME and AroDC-carbiDOPA (Fig. 3B).
One notable difference in the substrate-binding pockets of hHDC and pig AroDC was Ser-354. To elucidate the effect of the mutation at Ser-354 on catalytic activity, the enzymatic kinetic parameters of the mutant and original enzymes were determined. We observed that when serine 354 was substituted with glycine, the affinity between the enzyme and substrate decreased ∼10-fold according to Km values (Table 2); these results suggested that Ser-354 is critical for forming a part of the substrate-binding pocket. Furthermore, decarboxylase activity of the S354G mutant on l-DOPA, an aromatic substrate, was higher than that of hHDC2–477 (Table 2). This large increase in the mutant activity is attributable to the reduced steric hindrance between the enzyme and the bulky aromatic substrate (six-membered ring). The substrate-binding pocket of AroDC can accommodate substrates with variable side chains consisting of a six-membered ring, whereas that of HDC can provide a pocket that favors binding of histidine, an amino acid with a five-membered ring side chain. With this specificity, the enzyme might be able to exclude amino acids such as l-DOPA and tyrosine that contain a six-membered ring.
The change in substrate specificity of the S354G mutant strongly suggests that the orientation of HME in the present crystal structure reflects the orientation of histidine in the PLP-binding site during catalysis. The side chain of histidine would be buried in the interior pocket (si-face of the PLP aldimine), and the carboxyl ester group of histidine would be on the other side of the pocket (re-face of the PLP aldimine). The carboxyl ester group would be nearly perpendicular to the pyridoxal ring of PLP,4 facilitating the reaction according to the Dunathan hypothesis (36). PLP in this conformation may act as an electron sink that can stabilize the negative charge produced during the transition state of decarboxylation (Fig. 8). Previous simulation studies based on rat HDC suggested that histidine binds to PLP with the side chain on the re-face of the PLP cofactor (37). The reason for the difference between the present study and the previous simulation is not clear, but this difference might explain why the catalytic efficiency was remarkably different between hHDC2–477 and rat HDC as shown in Table 2.
FIGURE 8.

Schematic diagram of the substrate-binding site of hHDC2–477. The key residues Lys-305, Ser-354, Leu-102, Trp-72, Phe-104, Tyr-80, Leu-353, Leu-335, and Tyr-334 are shown. The mutated residues in this study Lys-305, Ser-354, and Tyr-334 are shown in red.
Role of the Loop
Another notable finding in the present crystal structure is that the entrance of the PLP-HME-binding site was covered by the loop region, which restricts access of the substrate to the PLP cofactor. Only weak interactions were observed between the loop and the substrate-binding region of the large domain, thus making the loop inherently flexible. A limited proteolysis experiment (Fig. 7) suggested that the loop is flexible in the inhibitor-free form of the enzyme (38). The entrance of the PLP-HME-binding site is formed by five hydrophobic residues (Tyr-80, Phe-104, Tyr-334, Leu-335, and Leu-353). The carboxyl ester of HME is located at the entrance of the substrate-binding site. Residues Tyr-334 and Leu-335 are within the flexible loop, which appears to provide a solvent-shielded cavity. This kind of solvent shielding facilitates the conversion of carboxylate to the less hydrophilic CO2 product.
In addition to this solvent-shielding activity, the hydroxyl group of Tyr-334 in the flexible loop, which is highly conserved among group II decarboxylases (39, 40), appears to act as a possible proton donor to the negatively charged Cα atom in the transition state (Figs. 2 and 8). The hydroxyl group of Tyr-334 can be positioned close enough to the Cα atom of HME by rotating around the Cα–Cβ bond. The substitution of tyrosine with phenylalanine at position 334 resulted in loss of decarboxylation activity (Table 2). A previous enzymatic assay of rat HDC has shown that not only Tyr-334 but also the surrounding loop region affects catalytic activity (17). Pro-332, Tyr-334, and Leu-335 are located in the flexible loop that takes part in hydrophobic interactions. These mutations are likely to disrupt hydrophobic interactions and consequently destabilize the loop structure and the position of Tyr-334. The hHDC nonsense mutation W317X, which causes a deletion of the loop region, was found to be associated with Tourette syndrome (10).
The flexible loop in the inhibitor-bound form of hHDC is well ordered and clearly assigned in the electron density map, whereas amino acids 328–339 (corresponding to the flexible loop) of AroDC were mostly disordered and lacked electron density (11). In the case of GAD, the mobility of the flexible loop of GAD65 promoted a side reaction, resulting in auto-inactivation (41). The position of an important tyrosine (corresponding to Tyr-334 in hHDC) has not been identified in the AroDC structure, and the substitution of the corresponding tyrosine with phenylalanine resulted in suppression of decarboxylation activities (42). Taken together, these findings indicate that the loop of PLP-dependent decarboxylases should be positioned near the substrate-binding site when this loop is involved in the catalytic reaction as a proton donor and lid for solvent shielding.
Possible Drug Design
HDC may become a drug target for the development of allergy treatments. Understanding the structure of HDC is an essential starting point for the design of potent and specific inhibitors of hHDC. Here, we presented a crystal structure of hHDC. Our results showed detailed structural information on the substrate-binding site. Furthermore, the flexible loop region was found to be important for the reaction catalyzed by HDC like other decarboxylases in this family. Despite the lack of structural similarity between histidine and epigallocatechin gallate, epigallocatechin gallate has been reported to inhibit HDC activity (20, 43). Although the mechanism of this inhibition remains unclear, epigallocatechin gallate might inhibit histamine synthesis by binding to HDC, possibly via the flexible catalytic loop. Inhibitors designed to block the flexibility of the catalytic loop might be potential candidates for drugs that can block histamine synthesis.
Supplementary Material
Acknowledgments
We thank the staff at beamline BL44XU at SPring-8 (Harima, Japan). The MX225-HE (Rayonix) CCD detector at BL44XU was financially supported by Academia Sinica and by the National Synchrotron Radiation Research Center (Hsinchu City, Taiwan).
This work was partly supported by the Kawanishi Memorial ShinMaywa Education Foundation and a grant-in-aid for young scientists (B) from the Japan Society for the Promotion of Science.

This article contains supplemental Figs. S1–S3 and Table S1.
The atomic coordinates and structure factors (code 4E1O) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
We compared the structure of α-oxamine synthase (AONS) to that of HDC because both HDC and AONS share a similar orientation of the substrate carboxylate group to PLP and interaction with a corresponding His residue, but still catalyze different reactions. In the case of AONS, belonging to the same fold type as group II decarboxylases, the surrounding environment around the carboxylate group is completely different from that of HDC. In addition, AONS lacks the loop region. Thus, the catalysis mechanism of HDC will be different from that of AONS (45).
- HDC
- l-histidine decarboxylase
- hHDC
- C-terminal truncated double mutant HDC
- rHDC
- recombinant HDC
- AroDC
- aromatic l-amino acid decarboxylase
- rAroDC
- recombinant AroDC
- GAD
- glutamate decarboxylase
- PLP
- pyridoxal-5′-phosphate
- HME
- histidine methyl ester
- AONS
- α-oxamine synthase.
REFERENCES
- 1. Nuutinen S., Panula P. (2010) Histamine in neurotransmission and brain diseases. Adv. Exp. Med. Biol. 709, 95–107 [DOI] [PubMed] [Google Scholar]
- 2. Galli S. J., Tsai M., Piliponsky A. M. (2008) The development of allergic inflammation. Nature 454, 445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andersson K., Chen D., Mattsson H., Sundler F., Håkanson R. (1998) Physiological significance of ECL-cell histamine. Yale J. Biol. Med. 71, 183–193 [PMC free article] [PubMed] [Google Scholar]
- 4. Ohtsu H., Tanaka S., Terui T., Hori Y., Makabe-Kobayashi Y., Pejler G., Tchougounova E., Hellman L., Gertsenstein M., Hirasawa N., Sakurai E., Buzás E., Kovács P., Csaba G., Kittel A., Okada M., Hara M., Mar L., Numayama-Tsuruta K., Ishigaki-Suzuki S., Ohuchi K., Ichikawa A., Falus A., Watanabe T., Nagy A. (2001) Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett. 502, 53–56 [DOI] [PubMed] [Google Scholar]
- 5. Kiyono S., Seo M. L., Shibagaki M., Watanabe T., Maeyama K., Wada H. (1985) Effects of α-fluoromethylhistidine on sleep-waking parameters in rats. Physiol. Behav. 34, 615–617 [DOI] [PubMed] [Google Scholar]
- 6. Parmentier R., Ohtsu H., Djebbara-Hannas Z., Valatx J. L., Watanabe T., Lin J. S. (2002) Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. J. Neurosci. 22, 7695–7711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morimoto T., Yamamoto Y., Mobarakeh J. I., Yanai K., Watanabe T., Watanabe T., Yamatodani A. (1999) Involvement of the histaminergic system in leptin-induced suppression of food intake. Physiol. Behav. 67, 679–683 [DOI] [PubMed] [Google Scholar]
- 8. Sakai N., Onodera K., Maeyama K., Yanai K., Watanabe T. (1992) Effects of (S)-α -fluoromethylhistidine and metoprine on locomotor activity and brain histamine content in mice. Life Sci. 51, 397–405 [DOI] [PubMed] [Google Scholar]
- 9. Kubota Y., Ito C., Sakurai E., Sakurai E., Watanabe T., Ohtsu H. (2002) Increased methamphetamine-induced locomotor activity and behavioral sensitization in histamine-deficient mice. J. Neurochem. 83, 837–845 [DOI] [PubMed] [Google Scholar]
- 10. Ercan-Sencicek A. G., Stillman A. A., Ghosh A. K., Bilguvar K., O'Roak B. J., Mason C. E., Abbott T., Gupta A., King R. A., Pauls D. L., Tischfield J. A., Heiman G. A., Singer H. S., Gilbert D. L., Hoekstra P. J., Morgan T. M., Loring E., Yasuno K., Fernandez T., Sanders S., Louvi A., Cho J. H., Mane S., Colangelo C. M., Biederer T., Lifton R. P., Gunel M., State M. W. (2010) L-histidine decarboxylase and Tourette syndrome. N. Engl. J. Med. 362, 1901–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burkhard P., Dominici P., Borri-Voltattorni C., Jansonius J. N., Malashkevich V. N. (2001) Structural insight into Parkinson disease treatment from drug-inhibited DOPA decarboxylase. Nat. Struct. Biol. 8, 963–967 [DOI] [PubMed] [Google Scholar]
- 12. Ueno H. (2000) Enzymatic and structural aspects on glutamate decarboxylase. J. Mol. Catalysis B: Enzymatic 10, 67–79 [Google Scholar]
- 13. Taguchi Y., Watanabe T., Kubota H., Hayashi H., Wada H. (1984) Purification of histidine decarboxylase from the liver of fetal rats and its immunochemical and immunohistochemical characterization. J. Biol. Chem. 259, 5214–5221 [PubMed] [Google Scholar]
- 14. Ohmori E., Fukui T., Imanishi N., Yatsunami K., Ichikawa A. (1990) Purification and characterization of l-histidine decarboxylase from mouse mastocytoma P-815 cells. J. Biochem. 107, 834–839 [DOI] [PubMed] [Google Scholar]
- 15. Yatsunami K., Tsuchikawa M., Kamada M., Hori K., Higuchi T. (1995) Comparative studies of human recombinant 74- and 54-kDa l-histidine decarboxylases. J. Biol. Chem. 270, 30813–30817 [DOI] [PubMed] [Google Scholar]
- 16. Olmo M. T., Sánchez-Jiménez F., Medina M. A., Hayashi H. (2002) Spectroscopic analysis of recombinant rat histidine decarboxylase. J. Biochem. 132, 433–439 [DOI] [PubMed] [Google Scholar]
- 17. Fleming J. V., Sánchez-Jiménez F., Moya-García A. A., Langlois M. R., Wang T. C. (2004) Mapping of catalytically important residues in the rat l-histidine decarboxylase enzyme using bioinformatic and site-directed mutagenesis approaches. Biochem. J. 379, 253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kollonitsch J., Perkins L. M., Patchett A. A., Doldouras G. A., Marburg S., Duggan D. E., Maycock A. L., Aster S. D. (1978) Selective inhibitors of biosynthesis of aminergic neurotransmitters. Nature 274, 906–908 [DOI] [PubMed] [Google Scholar]
- 19. Kelley J. L., Miller C. A., White H. L. (1977) Inhibition of histidine decarboxylase: derivatives of histidine. J. Med. Chem. 20, 506–509 [DOI] [PubMed] [Google Scholar]
- 20. Rodríguez-Caso C., Rodríguez-Agudo D., Sánchez-Jiménez F., Medina M. A. (2003) Green tea epigallocatechin-3-gallate is an inhibitor of mammalian histidine decarboxylase. Cell Mol. Life Sci. 60, 1760–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu F., Yu J., Gehring H. (2008) Inhibitory and structural studies of novel coenzyme-substrate analogs of human histidine decarboxylase. FASEB J. 22, 890–897 [DOI] [PubMed] [Google Scholar]
- 22. Komori H., Nitta Y., Ueno H., Higuchi Y. (2012) Purification, crystallization and preliminary X-ray analysis of human histidine decarboxylase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 675–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nitta Y., Ohshita J., Liu H., Kuronuma Y., Ueno H. (2010) Expression of recombinant human histidine decarboxylase with full length and C-terminal truncated forms in yeast and bacterial cells. J. Biol. Macromol. 10, 73–82 [Google Scholar]
- 24. Furuta K., Nakayama K., Sugimoto Y., Ichikawa A., Tanaka S. (2007) Activation of histidine decarboxylase through post-translational cleavage by caspase-9 in a mouse mastocytoma P-815. J. Biol. Chem. 282, 13438–13446 [DOI] [PubMed] [Google Scholar]
- 25. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 26. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 27. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61, 458–464 [DOI] [PubMed] [Google Scholar]
- 28. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 29. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 30. Brünger A. T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 31. Davis I. W., Murray L. W., Richardson J. S., Richardson D. C. (2004) MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shore P. A., Burkhalter A., Cohn V. H., Jr. (1959) A method for the fluorometric assay of histamine in tissues. J. Pharmacol. Exp. Ther. 127, 182–186 [PubMed] [Google Scholar]
- 33. Montioli R., Cellini B., Borri Voltattorni C. (2011) Molecular insights into the pathogenicity of variants associated with the aromatic amino acid decarboxylase deficiency. J. Inherit. Metab. Dis. 34, 1213–1224 [DOI] [PubMed] [Google Scholar]
- 34. Bertoldi M., Borri Voltattorni C. (2000) Reaction of DOPA decarboxylase with l-aromatic amino acids under aerobic and anaerobic conditions. Biochem. J. 352, 533–538 [PMC free article] [PubMed] [Google Scholar]
- 35. Hayashi H., Mizuguchi H., Kagamiyama H. (1993) Rat liver aromatic l-amino acid decarboxylase: spectroscopic and kinetic analysis of the coenzyme and reaction intermediates. Biochemistry 32, 812–818 [DOI] [PubMed] [Google Scholar]
- 36. Dunathan H. C. (1966) Conformation and reaction specificity in pyridoxal phosphate enzymes. Proc. Natl. Acad. Sci. U.S.A. 55, 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moya-García A. A., Ruiz-Pernía J., Martí S., Sánchez-Jiménez F., Tuñón I. (2008) Analysis of the decarboxylation step in mammalian histidine decarboxylase. A computational study. J. Biol. Chem. 283, 12393–12401 [DOI] [PubMed] [Google Scholar]
- 38. Pino-Angeles A., Morreale A., Negri A., Sánchez-Jiménez F., Moya-García A. A. (2010) Substrate uptake and protein stability relationship in mammalian histidine decarboxylase. Proteins 78, 154–161 [DOI] [PubMed] [Google Scholar]
- 39. Ishii S., Mizuguchi H., Nishino J., Hayashi H., Kagamiyama H. (1996) Functionally important residues of aromatic l-amino acid decarboxylase probed by sequence alignment and site-directed mutagenesis. J. Biochem. 120, 369–376 [DOI] [PubMed] [Google Scholar]
- 40. Okigami H., Ueno H. (2006) Bioinformatic study on histidine decarboxylase. J. Biol. Macromol. 6, 11–27 [Google Scholar]
- 41. Fenalti G., Law R. H., Buckle A. M., Langendorf C., Tuck K., Rosado C. J., Faux N. G., Mahmood K., Hampe C. S., Banga J. P., Wilce M., Schmidberger J., Rossjohn J., El-Kabbani O., Pike R. N., Smith A. I., Mackay I. R., Rowley M. J., Whisstock J. C. (2007) GABA production by glutamic acid decarboxylase is regulated by a dynamic catalytic loop. Nat. Struct. Mol. Biol. 14, 280–286 [DOI] [PubMed] [Google Scholar]
- 42. Bertoldi M., Gonsalvi M., Contestabile R., Voltattorni C. B. (2002) Mutation of tyrosine 332 to phenylalanine converts DOPA decarboxylase into a decarboxylation-dependent oxidative deaminase. J. Biol. Chem. 277, 36357–36362 [DOI] [PubMed] [Google Scholar]
- 43. Nitta Y., Kikuzaki H., Ueno H. (2007) Food components inhibiting recombinant human histidine decarboxylase activity. J. Agric. Food Chem. 55, 299–304 [DOI] [PubMed] [Google Scholar]
- 44. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 45. Webster S. P., Alexeev D., Campopiano D. J., Watt R. M., Alexeeva M., Sawyer L., Baxter R. L. (2000) Mechanism of 8-amino-7-oxononanoate synthase: spectroscopic, kinetic, and crystallographic studies. Biochemistry 39, 516–528 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.