

Background: NF-κB and BRD4 control proinflammatory gene activation in HIV-associated kidney disease.
Results: Small molecule inhibition of BRD4 binding to NF-κB blocks target gene activation.
Conclusion: Targeting the proinflammatory activity of NF-κB may be a new therapeutic approach.
Significance: This study has broad implications as NF-κB-mediated inflammation represents the major pathology in chronic kidney and non-kidney diseases.
Keywords: Gene Transcription, HIV-1, Inflammation, NF-kappaB Transcription Factor, Small Molecules
Abstract
NF-κB-mediated inflammation is the major pathology in chronic kidney diseases, including HIV-associated nephropathy (HIVAN) that ultimately progresses to end stage renal disease. HIV infection in the kidney induces NF-κB activation, leading to the production of proinflammatory chemokines, cytokines, and adhesion molecules. In this study, we explored selective inhibition of NF-κB transcriptional activity by small molecule blocking NF-κB binding to the transcriptional cofactor BRD4, which is required for the assembly of the productive transcriptional complex comprising positive transcription elongation factor b and RNA polymerase II. We showed that our BET (Bromodomain and Extra-Terminal domain)-specific bromodomain inhibitor MS417, designed to block BRD4 binding to the acetylated NF-κB, effectively attenuates NF-κB transcriptional activation of proinflammatory genes in kidney cells treated with TNFα or infected by HIV. MS417 ameliorates inflammation and kidney injury in HIV-1 transgenic mice, an animal model for HIVAN. Our study suggests that BET bromodomain inhibition, targeting at the proinflammatory activity of NF-κB, represents a new therapeutic approach for treating NF-κB-mediated inflammation and kidney injury in HIVAN.
Introduction
Despite the decreased incidence of HIV-associated nephropathy (HIVAN)4 in the antiretroviral therapy era, the prevalence remains high due to aging of HIV-seropositive patients (1, 2). Many patients with HIVAN ultimately progress to end stage renal disease despite the antiretroviral therapy treatment (3). Other forms of renal disease are also increased in HIV-seropositive patients (4). Although it remains unclear how HIV induces a variety of kidney diseases, inflammation is the major pathology seen in HIVAN. Tubulointerstitial inflammation, tubular dilatation with formation of microcysts, and apoptosis of renal tubular epithelial cells (RTECs) are prominent findings in the kidney of HIVAN (5).
Infection of RTECs by HIV induces inflammatory response (6, 7). Using gene expression analysis, Ross et al. (8) identified differentially expressed genes in RTECs isolated from a HIVAN patient after infection with vesicular stomatitis virus-pseudotyped gag/pol-deleted HIV-1. The most prominent response of these cells to HIV-1 infection was the production of proinflammatory mediators, including chemokines, cytokines, and adhesion molecules. Many of these genes, as suggested by pathway analysis of the microarray data, are mediated by transcriptional activation of nuclear factor κB (NF-κB).
As a master transcription factor, NF-κB regulates expression of a host of cellular genes in immune response to infection (9, 10). Its proinflammatory activity contributes to the etiology and progression of many human diseases, including cancer, inflammatory and autoimmune diseases, and viral infection (11, 12). NF-κB-mediated proinflammatory response has been shown to play a key role in the pathogenesis of kidney disease, including diabetic nephropathy (13, 14) as well as in HIV-induced kidney cell injury (15–17). NF-κB also interacts with HIV-1 long terminal repeat (LTR) in the kidney (18). Thus, inhibition of the proinflammatory activity of NF-κB may attenuate HIV-induced inflammation and kidney injury. Given its protective role in cell immune response to infection, however, a complete inhibition of NF-κB might lead to adverse effects. A selective and temporal inhibition of NF-κB activity would maximize its beneficial effects.
It has been shown that NF-κB transcriptional activity is dependent upon its acetylation at lysine 310 and that Lys-310-acetylated NF-κB recruits the BET protein BRD4 in complex with positive transcription elongation factor b (p-TEFb) (consisting of CDK9 and cyclin T1) and RNA polymerase II that together form a productive transcriptional machinery complex (9). A recent study has reported that pharmacological treatment of bone marrow-derived macrophages with a BET bromodomain inhibitor (BrDi) blocks the innate immune response gene expression induced by lipopolysaccharide (LPS) (19). In this study, we sought to determine how a BET-specific BrD inhibitor might influence NF-κB transcriptional activity in control of the expression of proinflammatory genes in HIV-infected kidney cells and whether and how BrDi might attenuate kidney injury in HIV-1 transgenic mice (Tg26), which is an established animal model for HIVAN. Our study shows that BET bromodomain inhibition could represent an attractive therapeutic strategy for treating patients with HIVAN.
EXPERIMENTAL PROCEDURES
Sample Preparation
Expression and purification of the recombinant bromodomains from ASH1L, ATAD2, BAZ1B, BAZ2B, BPTF, BRD3, BRD4, BRD7, CBP, PCAF, PHIP-1, TAF1, TAF1L, and SMARCA4 in poly-His tag form were performed using a procedure described previously (20). Isotope-labeled protein samples of the BrDs were prepared from cells grown on a minimal medium containing 15NH4Cl with or without 13C6-glucose in either H2O or 75% 2H2O. The protein was purified by affinity chromatography on a nickel-IDA column (Invitrogen), followed by the removal of poly-His tag by thrombin cleavage.
Protein Structure Determination by NMR
The NMR data collection, analysis, and structure determination of the BRD4-BD2·NF-κB-K310ac peptide complex were performed using the procedures previously reported (20). Briefly, NMR samples contained a protein·ligand complex of 0.5 mm in a 100 mm phosphate buffer, pH 6.5, that contains 5 mm perdeuterated DTT and 0.5 mm EDTA in H2O/2H2O (9:1) or 2H2O. All NMR spectra were collected at 30 °C on NMR spectrometers of 800, 600, or 500 MHz. The 1H, 13C, and 15N resonances of a protein of the complex were assigned by triple-resonance NMR spectra collected with a 13C/15N-labeled and 75% deuterated protein bound to an unlabeled ligand (21). The distance restraints were obtained in three-dimensional 13C or 15N NOESY spectra. Slowly exchanging amides, identified in two-dimensional 15N HSQC spectra recorded after an H2O buffer was changed to a 2H2O buffer, were used with structures calculated with only NOE distance restraints to generate hydrogen bond restraints for final structure calculations. The intermolecular NOEs were detected in the 13C-edited (F1), 13C/15N-filtered (F3) three-dimensional NOESY spectrum. Protein structures were calculated with a distance geometry-simulated annealing protocol with X-PLOR (22). Initial structure calculations were performed with manually assigned NOE-derived distance restraints. Hydrogen bond distance restraints, generated from the hydrogen/deuterium exchange data, were added at a later stage of structure calculations for residues with characteristic NOEs. The converged structures were used for iterative automated NOE assignment by ARIA for refinement (23). Structure quality was assessed by Procheck-NMR (24). The structure of the protein·ligand complex was determined using intermolecular NOE-derived distance restraints.
Isothermal Titration Calorimetry
Experiments were carried out on a GE MicroCal auto-ITC200 instrument at 15 °C while stirring at 1,000 rpm in the isothermal titration calorimetry (ITC) buffer, pH 7.4, consisting of 50 mm sodium phosphate and 150 mm sodium chloride. Compound concentration was determined by NMR, and protein concentrations were determined by A280 measurements. The compound sample (40–50 μm) was placed in the cell, whereas the microsyringe (130 μl) was loaded with a solution of the correspondent protein sample (0.6 μm) in the ITC buffer. Injections were performed using protein in the syringe rather than the compound to overcome limitations of compound solubility. All titrations were conducted using 20 identical injections of 2 μl with a duration of 4 s/injection and a spacing of 150 s between injections. The heat of dilution was determined by independent titrations (protein into buffer) and was subtracted from the experimental data. The collected data were implicated in the MicroCalTM Origin software supplied with the instrument to yield enthalpies of binding (ΔH) and binding constants (KB). Thermodynamic parameters were calculated (ΔG = ΔH − TΔS = −RTlnKB, where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding, respectively). In all cases, a single binding site model was employed.
Fluorescence Anisotropy Binding Assay
The dissociation constant, Kd, for various BrD proteins to a fluorescein isothiocyanate (FITC)-labeled MS417 (named as MS574, or compound 9; see Schemes 1 and 2 in supplemental Materials and Methods) was measured in direct binding experiments using fluorescence anisotropy as a readout. The fluorescence anisotropy assay was carried out in polypropylene 96-well plates (Costar) with 80 nm FITC-MS417 and varying concentrations of a BrD protein in a PBS buffer (pH 7.4) in a total volume of 80 μl. Measurements were obtained after a 1-h incubation of the fluorescent ligand and the protein at 25 °C with a Safire 2 microplate reader (Tecan). The dissociation constant was determined by using the following one-site model equation using Prism software,
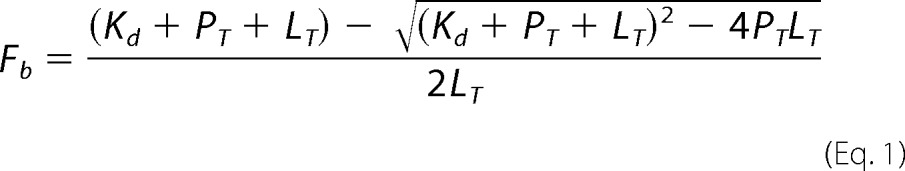 |
where Fb represents the fraction of bound labeled ligand, Kd is the dissociation constant, PT is total protein concentration, and LT is total fluorescent ligand concentration. Competition experiments were performed with a BrD protein (0.25–1 μm) and the fluorescent probe (80 nm) and increasing concentrations of unlabeled competing ligand. In a competition-binding assay, fluorescent ligand concentration was ≤2Kd, and protein concentration was set at which 50–80% of fluorescent ligand is bound. Dissociation constant of a competing ligand was calculated with the correction to the Cheng-Prussoff equation introduced by Nicolovska-Coleska et al. (25). Assuming a one-site competitive binding model, the following equation was used to calculate Ki values from IC50 values recovered from fitting data using Prism,
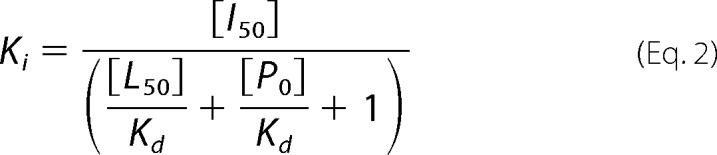 |
where [I50] represents the concentration of free inhibitor at 50% inhibition, [L50] is the concentration of free labeled ligand at 50% inhibition, and [P0] is the concentration of free protein at 0% inhibition. Note that Kd for each protein-probe pair is the limit of resolvable Ki in a competition assay.
Protein Crystallization and X-ray Diffraction Data Collection
Purified BRD4-BD1 protein (16 mg/ml) was mixed with MS417 at a 1:10 molar ratio of protein/ligand. The complex was crystallized using the sitting drop vapor diffusion method at 20 °C by mixing 1 μl of protein solution with 1 μl of the reservoir solution that contains 25% PEG 3350, 0.2 m Li2SO4, and 0.1 m Tris-HCl, pH 8.5. Crystals were soaked in the corresponding mother liquor supplemented with 20% ethylene glycerol as cryoprotectant before freezing in liquid nitrogen. X-ray diffraction data were collected at 100 K at beamline X6A of the National Synchrotron Light Source at Brookhaven National Laboratory. Data were processed using the HKL-2000 suite (26). The BRD4-BD1 structure was solved by molecular replacement using the program MOLREP (27), and the structure refinement was done using the program Refmac (28). The graphics program COOT (29) was used for model building and visualization. Crystal diffraction data and refinement statistics for the structure are displayed in supplemental Table 2.
Cell Cultures
Primary human RTECs were obtained from Promocell and cultured according to the manufacturer's protocol. RTECs were incubated with either MS417 or DMSO (0.1%) as the control. Cells were also stimulated with TNFα (10 ng/ml) to determine NF-κB activation. Cells were also infected with HIV or control vector as described below.
HIV Infection
For HIV infection, pNL4–3ΔG/P-EGFP, a gag/pol-deleted HIV-1 construct that contains EGFP in the gag open reading frame (HIV vector), and pHR′-IRES-EGFP (GFP control vector) were used to generate vesicular stomatitis virus-G-pseudotyped virus for infection of RTECs as described previously (8).
Western Blot
RTECs or kidney tissues were lysed with a buffer containing 1% Nonidet P-40, a protease inhibitor mixture (Fisher 78443), and acetylation inhibitors of 10 μm trichostatin A (Sigma T1952) and 5 mm nicotinamide (Sigma 72340). After protein concentration determination, protein lysates were subjected to Western blot analysis using specific antibodies for acetylated p65 or total p65 (Cell Signaling 3045). β-Actin (Sigma) was used as the control. Western blot was also performed for HIV Nef (Abcam 42355) in RTECs and kidney tissues of mice.
Reporter Gene Assay
The PGL-2 reporter vector containing the promoter of NF-κB or ISRE (gifts from Dr. Huabao Xiong) was used. 293T cells were transfected with luciferase expression plasmids (1 μg) using Lipofectamine 2000 (Invitrogen). 48 h after transfection, the cells were rinsed and cultured in serum-free medium containing TNFα (10 ng/ml) for 24 h. The cells were then lysed in luciferase assay buffer (Promega luciferase assay system). Luciferase was measured in a luminometer, and the data were analyzed after normalization for protein amount.
PCR Array and Microarray Studies
The PCR array for the Inflammatory Cytokines & Receptors PCR Array (PAHS-011, Qiagen) was used to examine the expression of NF-κB target genes. Human RETCs were treated with MS417 at the indicated concentrations for 24 h, and total RNA was extracted. PCR arrays were performed according to the manufacturer's instructions. -Fold changes for each gene were calculated by the 2−ΔΔCT method compared with GADPH. Human RTECs were infected with HIV pseudotyped virus or control vector with GFP for 4 days and then treated with either DMSO or MS417 at 1.0 μm for an additional 24 h. Total RNA was extracted from cells using the RNeasy minikit (Qiagen). Affymetrix gene expression microarrays were performed at the Mount Sinai Microarray Core Facility. Data were analyzed by t test controlled with false discovery rate, n = 3. Data were expressed as mean ± S.D. The unpaired t test was used to analyze data between two groups. Statistical significance was considered to be when p was <0.05.
Bioinformatics Analysis of Microarray Data
The raw intensity data were normalized across the chips by the log scale robust multiarray analysis (RMA) method (30, 31), and the quality control of each chip data was performed by investigating overall intensity values for all probes and negative and positive controls. The probe sets with low intensity values or outliers due to poor RNA quality or hybridization were excluded for further analysis. Significance Analysis of Microarray (SAM, version 2.1) (32) was used to identify significantly differentially expressed genes between groups (i.e. untreated versus BrD inhibitor-treated groups). SAM, a widely used microarray differential analysis tool, correlates gene expression data to a wide variety of clinical parameters and uses permutation to estimate the false discovery rate for multiple testing. The cut-off of the false discovery rate q value of 0.05 was applied to determine differentially expressed genes. The differentially expressed genes were queried against a well curated and experimentally verified NF-κB target database (see the Boston University Biology Web site) to identify NF-κB targets whose transcriptional expression was altered by HIV infection or the BrD inhibitor. The enrichment significance of NF-κB targets was calculated by comparing the percentage of NF-κB targets in differentially expressed gene with that of NF-κB targets in the whole gene list using Fisher's exact test. To understand how other genes were regulated by NF-κB targets upon HIV infection and BrDi, the genes that were up-regulated by HIV and subsequently down-regulated by BrDi or vice versa were identified and then subjected to association network analysis with the NF-κB target by the Ingenuity System (available on the World Wide Web). The significant biological functions of these genes were also determined by DAVID (33).
Chromatin Immunoprecipitation (ChIP)
Human RTECs were infected with HIV or GFP control vector for 72 h. The cells were then starved for 6 h and treated with DMSO or MS417 at 1 μm for 24 h in serum-free medium. Genomic DNA was precipitated, and ChIP was performed according to the manufacturer's protocol (Millipore). The following antibodies from Cell Signaling were used: anti-H3K9ac, anti-H3K18ac, anti-H3K4me3, and anti-H3K27me3 antibodies. Immunoprecipitated DNA was quantified by real-time PCR using the following primer sequences: CCL2-F, 5′-CCGGCCAAAGCTTGAGAGCTCC-3′; CCL2-R, 5′-GTCAGGCTCGCTGCCAGCTTAC-3′; CCL20-F, 5′-CCCCATGTGGCAACACGCCTT-3′; CCL20-R, 5′-GTACACAGAAGGCGTGTTGCCACAT-3′; CXCL5-F, 5′-CACCCTCCCCACCAGTTCCCA-3′; CXCL5-R, 5′-CTCATCTCTCCCCACTGACAGGAA-3′; TGM2-F, 5′-GCCGCCGTCCCTCCCTCGGG-3′; TGM2-R, 5′-AGCCCGCTTTGGGGCGG-3′.
All experiments were repeated three times. -Fold changes to the control are shown.
In Vivo Animal Model Study
As a well established transgenic model of HIV-associated nephropathy, the HIV-1 transgenic mouse (Tg26) bears a replication-defective form of the HIV-1 provirus lacking 3 kilobases of sequence overlapping the gag and pol sequences. The transgene is under the control of the viral LTR promoter and is expressed in most tissues (34). The Tg26 line is maintained as a heterozygous line as homozygous mice die prior to the age of breeding. Heterozygous mice appeared normal at birth but then develop wasting, heavy proteinuria, edema, hypoalbuminemia, and hypercholesterolemia between 4 and 8 months of age based on different penetration. Blood urea nitrogen is significantly elevated in these mice, and mice die probably from uremia (35). Pathologically, kidneys from animals with renal failure demonstrate diffuse segmental and global glomerulosclerosis, microcystic tubular dilatation with a monocytic interstitial infiltrate, and minimal interstitial fibrosis remarkably similar to HIVAN (35). Apart from kidney disease, these animals develop significant skin edema and cataract. However, these animals do not exhibit any other organ damage. For the in vivo animal model study, Tg26 mice and their corresponding littermates (n = 6/group, including three male and three female mice in each group) were fed with either control vehicle (DMSO) or MS417 by daily gavage at a concentration of 0.08 mg/kg. The mice were fed with this compound daily from the age of 4 weeks to 8 weeks. Unrestricted food and water were provided throughout the duration of the experiment. The mice were euthanized at 8 weeks of age for blood, urine, and tissue collection by exposure to carbon monoxide. Body weight was recorded. All animal studies were performed according to the protocols approved by the Institutional Animal Care and Use Committee at the Mount Sinai School of Medicine.
Measurement of Urine Protein and Creatinine
Urine albumin was quantified by ELISA using a kit from Bethyl Laboratory Inc. (Houston, TX). Urine creatinine levels were measured in the same samples using the urinary Creatinine Assay Kit (Cayman Chemical, Ann Arbor MI) according to the manufacturer's instructions. The urine albumin excretion rate was expressed as the ratio of albumin to creatinine.
Quantitative Histopathology
Mice were perfused with PBS containing 4% paraformaldehyde, and kidneys were further fixed in 4% paraformaldehyde for 2 h. Kidney tissue was embedded into paraffin by American Histolabs, Inc. (Gaithersburg, MD). Kidney histology was examined after periodic acid-Schiff staining. Glomerulosclerosis was scored as described previously by Dr. D'Agati (36). Briefly, each specimen received a score for three parameters: percentage of collapsing glomerular sclerosis, percentage of tubular cysts or casts, and podocyte hypertrophy. The percentage of collapsing glomerulosclerosis was obtained by identifying the total number of glomeruli with any sclerosis and dividing this number by the total number of glomeruli seen. The percentage of tubular cysts or casts score was obtained by the number of tubules with either microcystic dilatation or filled with casts divided by the total number of tubular cross-sections in a representative area. Finally, the degree of podocyte hypertrophy was scored as 0 (absent), 1+ (podocyte hypertrophy observed in <25% of all glomeruli), 2+ (podocyte hypertrophy observed in 25–50% of all glomeruli), and 3+ (podocyte hypertrophy in >50% of all glomeruli).
Real-time PCR
Total RNA was isolated from human RTECs or kidney cortex of mice using TRIzol (Invitrogen). Real-time PCR was performed with a Roche Applied Science Lightcycler and Qiagen QuantiTect One Step RT-PCR SYBR Green kit (Qiagen) according to the manufacturer's instructions. Predesigned primer sets were obtained from Qiagen (GeneGlobe). The sequences of RT-PCR primers were as follows: HsFAS-F, 5′-TATCACCACTATTGCTGGAGTCA-3′; HsFAS-R, 5′-GCTGTGTCTTGGACATTGTCA-3′; HsTRAF1-F, 5′-CCGGCCCCTGATGAGAATG-3′; HsTRAF1-R, 5′-TTCCTGGGCTTATAGACTGGAG-3′; MmCcl2-F, 5′-TTAAAAACCTGGATCGGAACCAA-3′; MmCcl2-R, 5′-GCATTAGCTTCAGATTTACGGGT-3′; MmCcl5-F, 5′-GCTGCTTTGCCTACCTCTCC-3′; MmCcl5-R, 5′-TCGAGTGACAAACACGACTGC-3′; MmCcl20-F, 5′-GCCTCTCGTACATACAGACGC-3′; MmCcl20-R, 5′-CCAGTTCTGCTTTGGATCAGC-3′; MmTlr2-F, 5′-GCAAACGCTGTTCTGCTCAG-3′; MmTlr2-R, 5′-AGGCGTCTCCCTCTATTGTATT-3′; MmTlr9-F, 5′-ATGGTTCTCCGTCGAAGGACT-3′; MnTlr9-R, 5′-GAGGCTTCAGCTCACAGGG-3′; HsGADPH-F, 5′-AATTGAGCCCCGCAGCCTCCC-3′; HsGADPH-R, 5′-CCAGGCGCCCAATACGACCA-3′; MmGadph-F, 5′-GCCATCCAACGAACCCCTTCAT-3′; MmGadph-R, 5′-ATGATGACCCGTTTGGCTCC-3′.
Light cycler analysis software was used to determine crossing points using the second derivative method. Data were normalized to housekeeping genes (tubulin) and presented as -fold increase compared with RNA isolated from WT animals using the 2−ΔΔCT method.
RESULTS
Molecular Basis of BRD4 Binding to Lysine 310-acetylated NF-κB
To determine the molecular basis of lysine acetylation-dependent NF-κB association with BRD4, we first characterized binding of the two bromodomains of BRD4 to a Lys-310-acetylated NF-κB peptide (NF-κB-K310ac) by NMR titration. As shown in two-dimensional 1H-15N HSQC spectra (Fig. 1A), the second bromodomain of BRD4 (BRD4-BD2) exhibited much more extended chemical shift perturbations upon binding to the NF-κB-K310ac peptide than that by the BRD4-BD1 or the BrDs of transcriptional co-activators CBP and PCAF, indicating a stronger interaction with the former. The differential NF-κB-K310ac binding by the bromodomains of BRD4 was further confirmed in a fluorescence anisotropy competition binding study using a fluorescein-labeled bromodomain inhibitor as an assay probe (see below), yielding a Ki of 274 μm and >2,000 μm for BRD4-BD2 and BRD4-BD1, respectively. We further observed that BRD4-BD1 or BD2 binds only very weakly if at all to other reported acetylation sites in NF-κB, including Lys-314, Lys-315, Lys-122, Lys-123, Lys-218, and Lys-221 in RelA-p65 and Lys-431, Lys-440, and Lys-441 in p50.
FIGURE 1.

Structural basis of K310ac NF-κB recognition by BRD4. A, two-dimensional 1H-15N HSQC spectra of BrDs of BRD4, CBP, and PCAF in the free form (black) and in the presence of a K310ac NF-κB peptide (red). Shown are ribbon (B) and surface-filled representations (C) of the three-dimensional solution structure of the BRD4-BD2 bound to the NF-κB-K310ac peptide. Side chains of key residues involved in intermolecular interactions are highlighted and color-coded by atom types.
We next solved the three-dimensional structure of BRD4-BD2 bound to the NF-κB-K310ac peptide by using NMR spectroscopy (supplemental Fig. 1 and Table 1), which explains the molecular basis of this selective interaction (Fig. 1, B and C). As shown in the structure of the BRD4-BD2·NF-κB-K310ac complex, the NF-κB-K310ac peptide is bound in the protein across an elongated cavity formed between the ZA and BC loops of this left-handed four-helical bundle structure. Specifically, the acetylated Lys-310 forms a hydrogen bond between its carbonyl oxygen and the side-chain nitrogen of the conserved Asn-433. In addition, Tyr-306 of NF-κB establishes aromatic and hydrophobic interactions with side-chain methyl groups of Ile-393, whereas Phe-309 binds to His-437, and Ile-312 is sandwiched between His-437 and Trp-374. Notably, in the conserved acetyl-lysine binding pocket, His-437 in BRD4-BD2 is unique, which corresponds to Asp-144 in BRD4-BD1. The latter cannot favorably interact with Phe-309 or Ile-312 of the NF-κB peptide, explaining markedly reduced binding of BRD4-BD1 to the K310ac site. The observation of direct recognition of Phe-309 (Kac−1) and Ile-312 (Kac+2) by the BRD4-BD2 explains why the K310ac site is selected over the other acetylation sites in NF-κB.
Blocking BRD4/NF-κB Association by a BET-specific BrD Inhibitor
To characterize the functional importance of acetylation-mediated BRD4/NF-κB binding in cells, we synthesized a small molecule bromodomain inhibitor MS417 (see supplemental Materials and Methods), which belongs to a group of thienodiazepine-based compounds that was first reported for their binding activity for BRD4 by the Mitsubishi Tanabe Pharma Corporation (37). MS417 shares the same thieno-triazolo-1,4-diazepine scaffold as a recently reported BET BrDi (+)-JQ1 (38) and is structurally related to I-BET (19, 39). The main difference between MS417 and (+)-JQ1 is that the former consists of a methyl ester, whereas the latter is substituted with a t-butyl ester moiety. Our 1.4 Å resolution crystal structure of the BRD4-BD1·MS417 complex (Fig. 2A and supplemental Fig. 2A and Table 2) reveals that MS417 is embedded in the acetyl-lysine binding pocket, forming a hydrogen bond between the triazole ring and the conserved Asn-140 in BRD4-BD1. Notably, the methyl ester interacts with Leu-95, Tyr-97, and Tyr-139 in a small hydrophobic cavity formed between the ZA and BC loops. The corresponding t-butyl group in (+)-JQ1 is too big to fit in this cavity such that it is rotated 180° with respect to the methyl ester in MS417, projecting outward on the protein surface (supplemental Fig. 2B). Similar to (+)-JQ1, MS417 is not in contact with Asp-144, which is unique in BRD4-BD1 (corresponding to His-437 in BRD4-BD2). This explains why MS417 binds BRD4-BD1 and BRD4-BD2 with similar affinity of Kd = 36.1 ± 7.8 and 25.4 ± 3.4 nm, respectively, as determined by isothermal titration calorimetry measurement (Fig. 2B). Notably, the tight binding of MS417 by the BRD4 bromodomains was confirmed by simultaneous observation of both the free and MS417-bound NMR resonances of the protein in the presence of substoichiometry concentrations of the ligand as those illustrated in two-dimensional 1H-15N HSQC spectra of 15N-labeled BRD4-BD1 (Fig. 2C). This is due to slow exchange of the protein conformations between the free and ligand-bound states on the NMR time scale, which is characteristic of submicromolar affinity of the binding ligand. Further, the structure explains that the BRD4-BD1 prefers MS417 over its enantiomer (i.e. MS566, with R-configuration of the methyl ester as supposed to S-configuration in MS417) (Fig. 2D and supplemental Fig. 6 and Table 4) because the methyl ester moiety in the latter would have steric clash with the protein residues, such as Leu-92, Leu-95, and Tyr-139. Finally, we demonstrated in a fluorescence anisotropy binding assay using a fluorescein-conjugated-MS417 as an assay probe that MS417 is highly specific for the BET bromodomains, and it binds to CBP, BRD7, or BPTF with a 100–200-fold weaker affinity or does not bind at all to many other bromodomains tested, including BAZ1B, BAZ2B, ATAD2, ASH1L, SMARCA4, PCAF, PHIP-1, TAF1L, and TAF1 (Fig. 2D and supplemental Fig. 3).
FIGURE 2.

Molecular basis of MS417 binding to the BRD4 bromodomains. A, x-ray crystal structure of MS417 bound to BRD4-BD1, depicted in a stereo view (left) and surface-filled representation (right). Side chains of key protein residues involved in ligand recognition are color-coded by atom types. The chemical structures of MS417 and its enantiomer MS566 are shown. B, ITC measurement of BRD4-BD1 binding MS417. C, two-dimensional 1H-15N HSQC spectra of BRD4-BD1 in the free form (black) and in the presence of the BrDi MS417 with a protein/ligand molar ratio of 1:0.5 (green) and 1:1 (red). D, affinity measurements of MS417 or its enantiomer MS566 binding to the BrDs of BRD4, BRD3, and CBP, as assessed in a fluorescence anisotropy competition assay using an FITC-labeled MS417 (i.e. MS574) as an assay probe.
We next evaluated the ability of MS417 to modulate NF-κB transcription activity in cells. Using a luciferase reporter gene assay, we found that MS417 suppressed TNFα-induced NF-κB transcription activation in human embryonic kidney 293T cells in a dose-dependent manner, and a nearly complete suppression of the NF-κB activation was observed at 1.0 μm MS417 (Fig. 3A). Little effect was seen on ISRE as a negative control. The inhibitory effect of MS417 on NF-κB activity correlates with a dose-dependent reduction of NF-κB acetylation (Fig. 3B). We next evaluated the NF-κB-inhibitory effects of MS417 in human primary RTECs that were infected with HIV pseudovirus or control GFP vector for 3 days followed by stimulation with MS417 in a serum-free medium for an additional 24 h. The Western blot analysis showed a similar inhibitory effect of MS417 on reducing NF-κB p65 acetylation in the HIV-infected RTECs as compared with TNFα-treated RTECs (Fig. 3C). These results suggest that MS417 can effectively suppress the NF-κB function through inhibition of its acetylation and transcriptional activation.
FIGURE 3.

Modulation of BRD4 binding to K310ac NF-κB. A, effects of MS417 on TNFα-induced NF-κB activation in 293T cells, as measured by a NF-κB luciferase reporter assay. 293T cells were transfected with a NF-κB luciferase reporter vector for 2 days and then incubated with MS417 at the indicated doses in serum-free medium with or without TNFα for 24 h. As a negative control, the cells were also transfected with an ISRE luciferase reporter vector and treated with MS417 and TNFα in the same condition as above. The cells were harvested for determination of luciferase activity (n = 5; **, p < 0.01). B, Western blot analysis assessing effects of MS417 on acetylated p65 in 293T cells. Representative Western blots of three independent experiments are shown. C, effects of MS417 on NF-κB activation by HIV pseudovirus infection in human RTECs. RTECs were infected with HIV pseudovirus or control GFP vector for 3 days and then incubated with MS417 at the indicated doses in serum-free medium for 24 h. The cells were harvested for determination of acetylated p65 (p65-Kac), p65, and β-actin as well as HIV Nef protein by Western blot. Representative Western blots of four independent experiments are shown. The densitometric analysis of the Western blots in B and C was performed and the ratio of acetylated p65 to total p65 was calculated, *, p < 0.05; n = 4 (lower panels of Fig. 3, B and C).
MS417 Down-regulates Genes That Were Up-regulated in HIV-infected RTECs
To gain mechanistic insights into the role of NF-κB/BRD4 binding in HIV-induced chronic inflammation in the kidney cells, we infected human RTECs with HIV pseudovirus for 4 days and then treated the cells with MS417 (1.0 μm) or DMSO for an additional 24 h. We then performed Affymatrix microarrays to characterize the effects of MS417 on genome-wide gene expression profiles of RTECs. Of 33,298 probe sets on the Human Gene 1.0 ST Array chip, gene expression significance analysis between groups by SAM showed that MS417 treatment significantly altered the transcriptional expression of 326 and 537 genes in the RTECs that were infected with either HIV or control GFP vector, respectively (with 2-fold cut-off, q < 0.05). To understand the functional relevance of these differentially expressed genes, we performed Ingenuity pathway analysis, which revealed several gene networks enriched in MS417-treated RTECs with or without HIV infection (Fig. 4A). In particular, genes related to inflammation, oxidative stress, and IL-17 proinflammatory pathways were among those highly influenced by MS417. Notably, of a set of 134 genes that were up-regulated by HIV infection and then down-regulated by MS417 are 11 NF-κB target genes, which were significantly enriched in this differentially expressed gene set over those in the whole gene list (see below; Fig. 4B).
FIGURE 4.

MS417 Modulation of gene transcription in HIV-infected human primary renal tubular epithelial cells. A, Ingenuity pathway analysis revealing the top gene networks enriched in the down-regulated genes by MS417 treatment in the HIV-infected human RTECs. The RTECs were infected with control GFP vector or HIV vector for 4 days and then treated with DMSO or MS417 (1.0 μm) for an additional 24 h. RNA was isolated from these cells for microarray studies. B, gene heat-map of 11 NF-κB targets that were both up-regulated by HIV infection and down-regulated by the MS417 treatment. C, MS417 inhibits NF-κB target gene activation by HIV infection in human RTECs. RTECs were infected with GFP or HIV for 72 h, and then the cells were starved for 6 h and treated with DMSO control or MS417 (1.0 μm) for an additional 24 h. The cells were harvested for real-time PCR analysis of cytokine and chemokine (n = 3; *, p < 0.01 when compared between HIV-infected cells treated with DMSO and MS417). D, some of the NF-κB target genes by HIV infection in the RTECs were not inhibited by MS417. E, chromatin immunoprecipitation analysis of the MS417-sensitive and non-sensitive genes probing changes of the corresponding histone modifications, such as H3K9ac, H3K18ac, H3K4me3, and H3K27me3, at the promoter sites of these genes (n = 3; *, p < 0.01 when compared between HIV-infected cells treated with DMSO and MS417).
To understand how MS417 modulates NF-κB transcriptional activity in the RTECs, we analyzed these NF-κB target genes. As shown in supplemental Fig. 4A, NF-κB target genes were significantly enriched among MS417 down-regulated genes in GFP or HIV-infected RTECs and HIV-up-regulated genes. As illustrated by Venn diagram (supplemental Fig. 4A), MS417 down-regulated 49 NF-κB target genes in the HIV-infected RTECs and 69 in the control, of which 38 are common. Most notably, MS417 down-regulated 11 of the 30 NF-κB genes in RTECs that were up-regulated by HIV infection. On the other hand, of the 14 and 15 genes up-regulated by MS417 in the RTECs infected with or without HIV, respectively, 13 are common, and none of the 18 HIV down-regulated NF-κB targets was up-regulated by MS417 in the HIV-infected RTECs. Given the low enrichment fold of these latter gene sets, we focused our study on these 11 NF-κB target genes, which were up-regulated by HIV and down-regulated by MS417 treatment, as illustrated in a gene expression heat map (Fig. 4B). Among these genes are well known NF-κB-dependent cytokine and chemokine genes, such as CCL2, CCL20, and IL-8, which are known to be up-regulated in RTECs by HIV infection as shown previously (8). These genes are at the core of a well interlinked expression network of genes centered on NF-κB that were mostly up-regulated upon HIV infection and reversely affected by subsequent MS417 treatment (supplemental Fig. 4B). Collectively, we concluded from our microarray data that MS417 inhibits the expression of the inflammation-related genes in human RTECs, induced by HIV infection.
MS417 Suppresses NF-κB-targeted Cytokines and Chemokines
We confirmed with RT-PCR analysis that MS417 suppressed the HIV-induced transcriptional activation of NF-κB-targeted proinflammatory chemokine genes, such as CCL2 and CCL20, in human RTECs (Fig. 4C). Notably, several genes, such as CXCL5 and TGM2, that underwent HIV-induced transcriptional activation were not affected significantly by the MS417 treatment in the RTECs (Fig. 4D). We further performed a ChIP study to determine effects of MS417 on chromatin modifications at the promoter site of these cytokine and chemokine genes. For the MS417-sensitive genes (i.e. CCL2 and CCL20), we observed a markedly increase of acetylation at histone H3 lysine 9 (H3K9ac) and lysine 18 (H3K18ac) and trimethylation at H3 lysine 4 (H3K4me3) and a concomitant decrease in trimethylation at H3 lysine 27 (H3K27me3) at their promoter site due to HIV infection and then a dramatic decrease of H3K9ac, H3K18ac, and H3K4me3 as well as an increase of H3K27me3 upon the MS417 treatment (Fig. 4E). On the other hand, for the MS417-insensitive genes CXCL5 and TGM2, we detected similar histone modification changes upon HIV infection (i.e. an increase of activation marks (i.e. H3K9ac, H3K18ac, and H3K4me3) and a decrease of repression marks (H3K27me3). However, very small changes if any, particularly for H3K4me3 and H3K27me3, were observed upon the MS417 treatment. These results are consistent with the effects of MS417 on transcriptional activation of these genes. In addition, by PCR array, we observed that MS417 suppressed TNFα-induced transcriptional activation of proinflammatory cytokines and chemokines in human RTECs, including IL1A, IL1B, LTA, LTB, CCL2, CCL3, CCL20, CXCL3, and CXCL11 (supplemental Fig. 5A). Interestingly, several genes, including CXCL5 and CXCL6, were not affected significantly by the MS417 treatment (supplemental Fig. 5B). Furthermore, we found that MS417 inhibited expression of proapoptotic genes targeted by NF-κB in the RTECs, such as BCL2 (Fig. 4B) and FAS and TRAF1 (supplemental Fig. 5C). Because RTEC apoptosis and inflammation are the major pathogenic mechanisms in HIVAN as well as in other kidney disease (16, 40–42), our findings suggest that the BRD4 BrD inhibitor may function as an anti-inflammatory agent to reduce inflammation and/or RTEC apoptosis in kidney disease.
MS417 Attenuates Proteinuria and Kidney Injury in HIV-1 Transgenic Mice (Tg26)
Given that MS417 can block the expression of both primary response genes (i.e. CCL2 and CCL3) and secondary response genes (i.e. CXCL11, IL1A, and IL1B), which are proinflammatory genes that are regulated by NF-κB, we postulated that BET BrD inhibition may represent a therapeutic strategy for HIVAN treatment. To test this hypothesis, we carried out an in vivo study to evaluate anti-inflammatory effects of MS417 in HIV-1 transgenic mice, an established mouse model of human HIVAN with kidney tubulointerstitial inflammation and RTEC apoptosis (43). The Tg26 mice are known to develop proteinuria starting at the age of 4 weeks and peaking at the age of 8 weeks associated with glomerular and tubular injuries (44). Accordingly, we treated a group of the Tg26 mice with MS417 or vehicle (0.1% DMSO) and compared them with their age- and sex-matched littermates in a control group starting at the age of 4 weeks for a total of 4 weeks. No significant side effects were noticed in these mice treated with MS417 at a dose of 0.08 mg/kg daily. Instead, we found that MS417 markedly improved renal function in the Tg26 mice as assessed by the measurement of blood urea nitrogen (BUN) without substantial changes of liver function and body weight (Fig. 5A and supplemental Table 3). MS417 treatment also resulted in reduced proteinuria in the Tg26 mice as compared with the DMSO-treated mice (Fig. 5B), and histology analysis showed significantly reduced glomerular and tubular injury (supplemental Table 3). The infiltration of inflammatory cells was also drastically reduced in the kidney of the Tg26 mice treated with MS417 as compared with the Tg26 mice treated with DMSO (Fig. 5C). NF-κB acetylation level (Fig. 5D) as well as the expression of NF-κB target genes, including CCL2, CCL5, CCL20, TLR-2, and TLR-9 (Fig. 5E) were reduced in kidney cortices of the MS417-treated Tg26 mice as compared with the control group. Finally, it appears that both mRNA and protein levels of HIV Nef in the MS417-treated mice were reduced by over 20% as compared with the control group, but this did not reach statistical significance (Fig. 5, D and F). Taken together, these data indicate that MS417 is capable of attenuating kidney injury in the Tg26 mice, probably through inhibition of NF-κB-mediated inflammation.
FIGURE 5.

In vivo study of MS417 effects in HIV-1 transgenic mice (Tg26). A, MS417 improves renal function in Tg26 mice as measured by blood urea nitrogen (BUN) (n = 6; *, p < 0.05 as compared with DMSO-treated Tg26 mice). B, MS417 reduces proteinuria in the Tg26 mice as determined by urinary albuminuria/creatinine ratio (n = 6; *, p < 0.05 as compared with DMSO-treated Tg26 mice). C, MS417 reduces glomerulosclerosis, tubular injury, and infiltration of inflammatory cells in the kidney of Tg26 mice. Tg26 mice were treated with vehicle (0.1% DMSO) or MS417 at 0.08 mg/kg from the age of 4 weeks for a total duration of 4 weeks. The mice were sacrificed at the age of 8 weeks. The kidney sections from these mice were stained for periodic acid-Schiff, and the representative pictures from six mice in each group are shown here. D, MS417 inhibits acetylation of p65 in the kidney of the Tg26 mice. The kidney cortices from these mice were used for Western blot analysis of acetylated and total p65. The expression of HIV Nef and β-actin was also assessed by Western blot. The densitometric analysis of these Western blots is shown in the lower panels (n = 6; *, p < 0.05 as compared with DMSO-treated Tg26 mice). E, NF-κB target genes were suppressed in kidneys of the Tg26 mice treated with MS417. The kidney cortices from these mice were used for total RNA isolation and real-time PCR analysis for NF-κB target genes (n = 6; *, p < 0.05 as compared with DMSO-treated Tg26 mice). F, effects of MS417 on the expression of HIV Nef in the kidneys of Tg26 mice. All mRNA levels of the testing genes were normalized to that of GAPDH, a housekeeping gene.
DISCUSSION
Several lines of evidence suggest that HIV directly infects renal tubular epithelial cells, leading to the development of HIV-associated nephropathy. The effect of HIV expression on the development of HIVAN was first revealed by kidney transplantation between normal and HIV transgenic mice. This study demonstrated that HIVAN was developed in HIV transgenic kidneys when transplanted into nontransgenic littermates, whereas normal kidneys remained disease-free when transplanted into HIV transgenic littermates, suggesting that the renal disease in the HIV transgenic murine model is intrinsic to the kidney (45). HIV can also infect directly kidney glomerular and tubular epithelial cells, as shown by in situ PCR or hybridization or by PCR of laser capture microdissected kidney biopsy samples from HIV-positive patients (6, 46, 47). Recently, the mechanisms of HIV entry into kidney epithelial cells have been described (48, 49). Taken together, these studies suggest that HIV infection probably occurs locally in kidney cells, leading to the development of HIVAN.
In this study, we have shown that the BET bromodomain-specific inhibitor MS417 down-regulates HIV infection-induced and NF-κB-mediated transcriptional activation of proinflammatory cytokine and chemokine genes in human renal tubular epithelial cells. MS417 reduces proteinuria, improves renal function, and attenuates NF-κB acetylation and expression of its target genes in the kidney of the Tg26 mouse, a model for HIVAN. Our findings, therefore, strongly suggest that BET bromodomain inhibition, targeting NF-κB proinflammatory activity in the pathogenesis of HIVAN, represents a new therapeutic strategy for treating patients with HIVAN. This is the first study that demonstrates BET bromodomain inhibitors as potentially new therapeutic agents in a kidney disease model.
Transcription factor NF-κB/Rel proteins govern cellular immune and inflammatory responses in the form of cell apoptosis, proliferation, or differentiation through regulation of gene expression (9, 10). The prototypical NF-κB is a heterodimer of p50 and RelA and is sequestered in the cytoplasm when bound to inhibitor IκBα in resting cells. Stimulation of the cells activates IKKs, phosphorylation, and degradation of IκBα, leading to nuclear translocation and activation of NF-κB. Transcriptional activity of NF-κB requires its association with the lysine acetyltransferase p300/CBP and PCAF and transcriptional protein BRD4. The cofactor binding results in phosphorylation and acetylation of NF-κB, promoting the formation of the transcriptional complexes that bridges the sequence-specific activators to the basal transcription machinery, thereby facilitating its target gene activation.
BRD4 belongs to the BET protein family, which consists of four members, BRD2, BRD3, BRD4, and BRDT, each of which contains two bromodomains in tandem (50). Recent studies show that BET proteins play an important role in coordinating gene transcriptional activation in a highly ordered fashion in chromatin through their ability to facilitate chromatin targeting and assembly of a productive transcriptional machinery complex at a target gene transcriptional start site in an acetylation-sensitive manner. In the case of NF-κB, BRD4 recruitment by the transcription factor to a target gene represents a preinitiation commitment step in transcriptional activation, which involves chromatin structure opening through histone lysine acetylation as well as BRD4 interaction with acetylated histone H3 and H4 at the H3K14ac, H4K5ac, H4K8ac, H4K12ac, and H4K16ac sites. In the next step of transcriptional initiation/postinitiation, BRD4 functions in the recruitment of the initiation factor mediator, which leads to Ser-5 phosphorylation of the carboxyl-terminal domain of the RNA polymerase II (9). Subsequently, BRD4 facilitates the recruitment of the elongation cofactor p-TEFb, consisting of CDK9 and cyclin T1, to paused RNA polymerase II, which results in Ser-2 phosphorylation of the carboxyl-terminal domain, thereby enabling polymerase II to resume transcriptional elongation.
The mechanism of action of our BET BrDi MS417 probably involves its modulation of BRD4 functions in multiple steps during gene transcriptional activation. As shown by the current study, BRD4 binding to K310ac NF-κB is achieved by its second bromodomain (BRD4-BD2); blocking of this interaction by MS417 resulted in a reduced acetylation level of NF-κB at Lys-310 and down-regulation of NF-κB target gene transcriptional activation. Further, inhibition of both bromodomains of BRD4 by MS417 may blunt the ability of BRD4 to interact with lysine-acetylated H3 and H4 for chromatin targeting at the NF-κB target gene transcriptional start site or to promote the formation of a productive transcriptional machinery with p-TEFb and RNA polymerase II, which is important for transcriptional elongation (9).
NF-κB activity is known to be elevated in HIV-infected renal tubular epithelial cells and podocytes as well as in kidneys of HIV transgenic mice, mediating HIV-induced kidney injury (8, 15, 16). NF-κB has also been shown to be recruited to interact with HIV-1 LTR to facilitate HIV transcription and replication in the kidney (18). Targeting NF-κB would be a logical approach to attenuate both HIV-induced inflammation and kidney injury. Indeed, we have shown that both primary response and secondary response genes directed by NF-κB activation that is triggered by HIV infection were down-regulated by the treatment of MS417. Furthermore, the changes of their transcriptional expression are consistently correlated to the alteration of histone modifications at the transcriptional start sites of these genes. Interestingly, we have also noticed that the transcriptional expression of a subset of NF-κB-inducible genes encoding cytokines or chemokines, such as CXCL5 and CXCL6, was not affected by MS417, suggesting that BRD4 is not involved in their gene transcriptional activation. Further investigation is warranted to dissect the molecular mechanistic details of BRD4 functions during transcriptional activation of these genes.
Although suppression of viral replication with antiretroviral therapy alters the course of kidney disease, many patients with HIVAN still unfortunately progress to end stage renal disease (3). No drugs are available to specifically stop or reverse this disease process. Because NF-κB is a key pathway in the pathogenesis of HIVAN, MS417 could be developed as a new class of drugs to treat patients with HIVAN. In addition, NF-κB-mediated inflammation is also a major pathology in other kidney disease, such as diabetic nephropathy and lupus nephritis (51), as well as other non-kidney inflammatory diseases, such as rheumatoid arthritis (52). Because of the protective role of NF-κB in acute inflammatory response, complete inhibition of NF-κB may lead to harmful side effects. Small molecules for selective and transient inhibition of NF-κB proinflammatory activity may allow us to maximize their therapeutic index. Therefore, we believe that BET bromodomain-specific inhibitors could be developed as more effective and safer treatments for many inflammatory diseases.
Supplementary Material
Acknowledgments
We acknowledge the staff at the X6A beamline of the National Synchrotron Light Source at the Brookhaven National Laboratory for facilitating x-ray data collection. We thank the Rockefeller University High-Throughput Screening Resource Center for the ITC data collection.
This work was supported, in whole or in part, by National Institutes of Health Grants HG004508 (to M.-M. Z.) and DK088541 (to J. C. H.). This work was also supported by Veterans Affairs Merit Award 1I01BX000345 (to J. C. H.).
The atomic coordinates and structure factors (codes 2LSP and 4F3I) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The chemical shifts were deposited in BioMagResBank under code 18439.

This article contains supplemental Materials and Methods, Tables 1–4, Figs. 1–6, and Schemes 1 and 2.
- HIVAN
- HIV-associated nephropathy
- RTEC
- renal tubular epithelial cell
- BET
- Bromodomain and Extra-Terminal domain
- BrD
- bromodomain
- BrDi
- bromodomain inhibitor
- p-TEFb
- positive transcription elongation factor b
- CBP
- CREB-binding protein
- CREB
- cAMP-response element-binding protein
- RMA
- robust multiarray analysis
- ITC
- isothermal titration calorimetry
- ISRE
- interferon-sensitive response element
- PCAF
- p300/CBP-associated factor
- K310ac
- lysine 310-acetylated
- H3K9ac and H3K18ac
- acetylation at histone H3 lysine 9 and 18, respectively
- H3K4me3 and H3K27me3
- trimethylation at H3 lysine 4 and 27, respectively.
REFERENCES
- 1. Wyatt C. M., Klotman P. E. (2007) HIV-1 and HIV-associated nephropathy 25 years later. Clin. J. Am. Soc. Nephrol. 2, Suppl. 1, S20–S24 [DOI] [PubMed] [Google Scholar]
- 2. Atta M. G., Fine D. M., Kirk G. D., Mehta S. H., Moore R. D., Lucas G. M. (2007) Survival during renal replacement therapy among African Americans infected with HIV type 1 in urban Baltimore, Maryland. Clin. Infect. Dis. 45, 1625–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lucas G. M., Eustace J. A., Sozio S., Mentari E. K., Appiah K. A., Moore R. D. (2004) Highly active antiretroviral therapy and the incidence of HIV-1-associated nephropathy. A 12-year cohort study. AIDS 18, 541–546 [DOI] [PubMed] [Google Scholar]
- 4. Wyatt C. M., Winston J. A., Malvestutto C. D., Fishbein D. A., Barash I., Cohen A. J., Klotman M. E., Klotman P. E. (2007) Chronic kidney disease in HIV infection. An urban epidemic. AIDS 21, 2101–2103 [DOI] [PubMed] [Google Scholar]
- 5. D'Agati V., Suh J. I., Carbone L., Cheng J. T., Appel G. (1989) Pathology of HIV-associated nephropathy. A detailed morphologic and comparative study. Kidney Int. 35, 1358–1370 [DOI] [PubMed] [Google Scholar]
- 6. Bruggeman L. A., Ross M. D., Tanji N., Cara A., Dikman S., Gordon R. E., Burns G. C., D'Agati V. D., Winston J. A., Klotman M. E., Klotman P. E. (2000) Renal epithelium is a previously unrecognized site of HIV-1 infection. J. Am. Soc. Nephrol. 11, 2079–2087 [DOI] [PubMed] [Google Scholar]
- 7. Mikula M., Fuchs E., Huber H., Beug H., Schulte-Hermann R., Mikulits W. (2004) Immortalized p19ARF null hepatocytes restore liver injury and generate hepatic progenitors after transplantation. Hepatology 39, 628–634 [DOI] [PubMed] [Google Scholar]
- 8. Ross M. J., Fan C., Ross M. D., Chu T. H., Shi Y., Kaufman L., Zhang W., Klotman M. E., Klotman P. E. (2006) HIV-1 infection initiates an inflammatory cascade in human renal tubular epithelial cells. J. Acquir. Immune Defic. Syndr. 42, 1–11 [DOI] [PubMed] [Google Scholar]
- 9. Huang B., Yang X. D., Zhou M. M., Ozato K., Chen L. F. (2009) Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol. Cell. Biol. 29, 1375–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin Y., Bai L., Chen W., Xu S. (2010) The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 14, 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pasparakis M. (2009) Regulation of tissue homeostasis by NF-κB signaling. Implications for inflammatory diseases. Nat. Rev. Immunol. 9, 778–788 [DOI] [PubMed] [Google Scholar]
- 12. Chen L. F., Greene W. C. (2004) Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 5, 392–401 [DOI] [PubMed] [Google Scholar]
- 13. Rangan G., Wang Y., Harris D. (2009) NF-κB signaling in chronic kidney disease. Front. Biosci. 14, 3496–3522 [DOI] [PubMed] [Google Scholar]
- 14. Schmid H., Boucherot A., Yasuda Y., Henger A., Brunner B., Eichinger F., Nitsche A., Kiss E., Bleich M., Gröne H. J., Nelson P. J., Schlöndorff D., Cohen C. D., Kretzler M., and European Renal cDNA Bank (ERCB) Consortium (2006) Modular activation of nuclear factor-κB transcriptional programs in human diabetic nephropathy. Diabetes 55, 2993–3003 [DOI] [PubMed] [Google Scholar]
- 15. Martinka S., Bruggeman L. A. (2006) Persistent NF-κB activation in renal epithelial cells in a mouse model of HIV-associated nephropathy. Am. J. Physiol. Renal Physiol. 290, F657–F665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ross M. J., Martinka S., D'Agati V. D., Bruggeman L. A. (2005) NF-κB regulates Fas-mediated apoptosis in HIV-associated nephropathy. J. Am. Soc. Nephrol. 16, 2403–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ross M. J., Wosnitzer M. S., Ross M. D., Granelli B., Gusella G. L., Husain M., Kaufman L., Vasievich M., D'Agati V. D., Wilson P. D., Klotman M. E., Klotman P. E. (2006) Role of ubiquitin-like protein FAT10 in epithelial apoptosis in renal disease. J. Am. Soc. Nephrol. 17, 996–1004 [DOI] [PubMed] [Google Scholar]
- 18. Bruggeman L. A., Adler S. H., Klotman P. E. (2001) Nuclear factor-κB binding to the HIV-1 LTR in kidney. Implications for HIV-associated nephropathy. Kidney Int. 59, 2174–2181 [DOI] [PubMed] [Google Scholar]
- 19. Nicodeme E., Jeffrey K. L., Schaefer U., Beinke S., Dewell S., Chung C. W., Chandwani R., Marazzi I., Wilson P., Coste H., White J., Kirilovsky J., Rice C. M., Lora J. M., Prinjha R. K., Lee K., Tarakhovsky A. (2010) Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zeng L., Zhang Q., Gerona-Navarro G., Moshkina N., Zhou M. M. (2008) Structural basis of site-specific histone recognition by the bromodomains of human coactivators PCAF and CBP/p300. Structure 16, 643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clore G. M., Gronenborn A. M. (1994) Multidimensional heteronuclear nuclear magnetic resonance of proteins. Methods Enzymol. 239, 349–363 [DOI] [PubMed] [Google Scholar]
- 22. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system. A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 23. Nilges M., O'Donoghue S. (1998) Ambiguous NOEs and automated NOE assignment. Prog. NMR Spectrosc. 32, 107–139 [Google Scholar]
- 24. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR. Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 25. Nikolovska-Coleska Z., Wang R., Fang X., Pan H., Tomita Y., Li P., Roller P. P., Krajewski K., Saito N. G., Stuckey J. A., Wang S. (2004) Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 332, 261–273 [DOI] [PubMed] [Google Scholar]
- 26. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 27. Vagin A., Teplyakov A. (1997) MOLREP. An automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 28. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 29. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 30. Irizarry R. A., Bolstad B. M., Collin F., Cope L. M., Hobbs B., Speed T. P. (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Irizarry R. A., Hobbs B., Collin F., Beazer-Barclay Y. D., Antonellis K. J., Scherf U., Speed T. P. (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 [DOI] [PubMed] [Google Scholar]
- 32. Tusher V. G., Tibshirani R., Chu G. (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dennis G., Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A. (2003) DAVID. Database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 34. Dickie P., Felser J., Eckhaus M., Bryant J., Silver J., Marinos N., Notkins A. L. (1991) HIV-associated nephropathy in transgenic mice expressing HIV-1 genes. Virology 185, 109–119 [DOI] [PubMed] [Google Scholar]
- 35. Kopp J. B., Klotman M. E., Adler S. H., Bruggeman L. A., Dickie P., Marinos N. J., Eckhaus M., Bryant J. L., Notkins A. L., Klotman P. E. (1992) Progressive glomerulosclerosis and enhanced renal accumulation of basement membrane components in mice transgenic for human immunodeficiency virus type 1 genes. Proc. Natl. Acad. Sci. U.S.A. 89, 1577–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. D'Agati V. (2003) Pathologic classification of focal segmental glomerulosclerosis. Semin. Nephrol. 23, 117–134 [DOI] [PubMed] [Google Scholar]
- 37. Miyoshi S., Ooike S., Iwata K., Hikawa H., Sugaraha K. (December 26, 2008) International Patent PCT/JP2008/073864 (US2010/0286127 A1)
- 38. Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W. B., Fedorov O., Morse E. M., Keates T., Hickman T. T., Felletar I., Philpott M., Munro S., McKeown M. R., Wang Y., Christie A. L., West N., Cameron M. J., Schwartz B., Heightman T. D., La Thangue N., French C. A., Wiest O., Kung A. L., Knapp S., Bradner J. E. (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dawson M. A., Prinjha R. K., Dittmann A., Giotopoulos G., Bantscheff M., Chan W. I., Robson S. C., Chung C. W., Hopf C., Savitski M. M., Huthmacher C., Gudgin E., Lugo D., Beinke S., Chapman T. D., Roberts E. J., Soden P. E., Auger K. R., Mirguet O., Doehner K., Delwel R., Burnett A. K., Jeffrey P., Drewes G., Lee K., Huntly B. J., Kouzarides T. (2011) Inhibition of BET recruitment to chromatin as an effective treatment for MLL fusion leukemia. Nature 478, 529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Snyder A., Alsauskas Z. C., Leventhal J. S., Rosenstiel P. E., Gong P., Chan J. J., Barley K., He J. C., Klotman M. E., Ross M. J., Klotman P. E. (2010) HIV-1 viral protein r induces ERK and caspase-8-dependent apoptosis in renal tubular epithelial cells. AIDS 24, 1107–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Havasi A., Borkan S. C. (2011) Apoptosis and acute kidney injury. Kidney Int. 80, 29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Susztak K., Raff A. C., Schiffer M., Böttinger E. P. (2006) Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55, 225–233 [PubMed] [Google Scholar]
- 43. Lu T. C., He J. C., Klotman P. (2006) Animal models of HIV-associated nephropathy. Curr. Opin. Nephrol. Hypertens. 15, 233–237 [DOI] [PubMed] [Google Scholar]
- 44. Ratnam K. K., Feng X., Chuang P. Y., Verma V., Lu T. C., Wang J., Jin Y., Farias E. F., Napoli J. L., Chen N., Kaufman L., Takano T., D'Agati V. D., Klotman P. E., He J. C. (2011) Role of the retinoic acid receptor-α in HIV-associated nephropathy. Kidney Int. 79, 624–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bruggeman L. A., Dikman S., Meng C., Quaggin S. E., Coffman T. M., Klotman P. E. (1997) Nephropathy in human immunodeficiency virus-1 transgenic mice is due to renal transgene expression. J. Clin. Invest. 100, 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marras D., Bruggeman L. A., Gao F., Tanji N., Mansukhani M. M., Cara A., Ross M. D., Gusella G. L., Benson G., D'Agati V. D., Hahn B. H., Klotman M. E., Klotman P. E. (2002) Replication and compartmentalization of HIV-1 in kidney epithelium of patients with HIV-associated nephropathy. Nat. Med. 8, 522–526 [DOI] [PubMed] [Google Scholar]
- 47. Tanji N., Ross M. D., Tanji K., Bruggeman L. A., Markowitz G. S., Klotman P. E., D'Agati V. D. (2006) Detection and localization of HIV-1 DNA in renal tissues by in situ polymerase chain reaction. Histol. Histopathol. 21, 393–401 [DOI] [PubMed] [Google Scholar]
- 48. Chen P., Chen B. K., Mosoian A., Hays T., Ross M. J., Klotman P. E., Klotman M. E. (2011) Virological synapses allow HIV-1 uptake and gene expression in renal tubular epithelial cells. J. Am. Soc. Nephrol. 22, 496–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mikulak J., Teichberg S., Arora S., Kumar D., Yadav A., Salhan D., Pullagura S., Mathieson P. W., Saleem M. A., Singhal P. C. (2010) DC-specific ICAM-3-grabbing nonintegrin mediates internalization of HIV-1 into human podocytes. Am. J. Physiol. Renal Physiol. 299, F664–F673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chiang C. M. (2009) Brd4 engagement from chromatin targeting to transcriptional regulation. Selective contact with acetylated histone H3 and H4. F1000 Biol. Rep. 1, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zheng L., Sinniah R., Hsu S. I. (2006) Renal cell apoptosis and proliferation may be linked to nuclear factor-κB activation and expression of inducible nitric-oxide synthase in patients with lupus nephritis. Hum. Pathol. 37, 637–647 [DOI] [PubMed] [Google Scholar]
- 52. Criswell L. A. (2010) Gene discovery in rheumatoid arthritis highlights the CD40/NF-κB signaling pathway in disease pathogenesis. Immunol. Rev. 233, 55–61 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.