

Abstract
CD5 activates CK2, a serine/threonine kinase that constitutively associates with the CK2-binding domain at the end of its cytoplasmic tail. To determine the physiological significance of CD5-dependent CK2 activation in T-cells we generated a knock-in mouse that expresses a CD5 protein containing a microdeletion with selective inability to interact with CK2 (CD5ΔCK2BD). The levels of CD5 on developing and mature T-cell populations from CD5ΔCK2BD mice and CD5WT mice were similar. The thymus of CD5ΔCK2BD mice contained fewer double positive (DP) thymocytes than that of both CD5WT and CD5KO mice, though the numbers of all other immature and mature T-cell populations were unaltered. CD5ΔCK2BD T-cells hypoproliferated and exhibited enhanced AICD when stimulated with anti-CD3 or cognate peptide in comparison to CD5WT T-cells. We also found that functional CD5-dependent CK2 signaling was necessary for efficient differentiation of naïve CD4+ T-cells into Th2 and Th17 cells, but not Th1 cells. We previously showed that experimental autoimmune encephalomyelitis (EAE) in CD5KO mice was less severe and delayed in onset than in CD5WT mice. Remarkably, CD5ΔCK2BD mice recapitulated both EAE severity and disease onset of CD5KO mice. Increasing the immunization dose of myelin oligodendrocyte (MOG35-55) peptide, a model that mimics high dose tolerance, led to decreased severity of EAE in CD5 WT mice but not in CD5KO or CD5ΔCK2BD mice. This property was recapitulated in in vitro re-stimulation assays. These results demonstrate that CD5-CK2 signaling sets the threshold for T-cell responsiveness and it is necessary for efficient generation of Th2 and Th17 cells.
Introduction
The cell surface glycoprotein CD5 has a well-recognized function as a negative regulator of antigen receptor activation in lymphocytes (1, 2). In mice, the receptor is constitutively expressed on developing and mature T-cells, B-1a B-cells, and the recently described CD1dhi regulatory B-cells (B10 cells) (3-5). Structurally, CD5 is closely related and linked in the genome to CD6 with an extracellular domain comprised of three group B scavenger receptor cysteine-rich extracellular domains (6-10). Although several ligands have been proposed for CD5, none have been independently verified (9). In developing and mature T-cells, expression levels of CD5 correlate with avidity and/or affinity of T-cell antigen receptor and are dynamically altered by changes in threshold of antigen receptor activation (11, 12). Reciprocally, changes in CD5 expression levels alter T-cell activation thresholds. Other mechanisms that control CD5 expression are GATA3 levels during thymocyte selection, TCR signaling and p56lck expression levels in peripheral T-cells, and a NFAT-dependent cd5 enhancer linked with BCR engagement in B-1a B-cells (1, 11, 13-16).
The cytoplasmic tail of CD5 contains three phosphorylatable tyrosines, two of which are in a configuration resembling an ITAM/ITIM domain (9). The CD5-mediated negative regulation of antigen receptor activation is primarily ascribed to its ITIM domain (3, 17). The regulatory activity of CD5 raises the threshold for T-cell activation to control response to antigen and discourage autoreactivity (18). While CD5 is generally regarded as an attenuator of lymphocyte activation, it also serves to enhance T-cell function with its unique role in supporting prosurvival signaling. Increased surface expression of CD5 protects autoreactive CD4+ T-cells from Fas-mediated AICD and represents a mechanism through which T-cells, otherwise destined for death following activation by a strong antigenic stimulus, can survive (19, 20). Although there is not yet a comprehensive understanding of the CD5-mediated pathways resulting in prosurvival signaling in T-cells, an important emerging player in this process is casein kinase 2 (CK2), which constitutively associates with a CK2 binding domain located in the distal portion of the CD5 cytoplasmic tail (21, 22). CK2 is a serine/threonine kinase that is commonly expressed in all cell types and phosphorylates a large number of substrates to participate in a variety of cell regulatory and survival pathways (21-26).
The first evidence that a major biological activity exerted by CD5 is prosurvival in activated T-cells came from the study of experimental autoimmune encephalomyelitis (EAE)3 in the CD5 knock-out (CD5KO) mouse (27, 28). Although CD4+ T-cells in CD5KO mice responded more vigorously to immunization with myelin oligodendrocyte glycoprotein (MOG35-55) peptide, the onset and severity of EAE in these mice was less severe than in CD5WT mice. The decreased severity in CD5KO mice was at least in part associated with enhanced AICD. This finding provided an insight into the mechanisms underlying the absence of spontaneous autoreactivity in the CD5KO mouse in spite of T-cell hyperactivity. To determine if the prosurvival activity was associated with the ability of CD5 to activate a CK2 regulated pathway, we reconstituted the CD5KO mouse with a T-cell expression-restricted CK2 binding/activation-deficient CD5 transgene (CD5ΔCK2BD-Tg) (27). Remarkably CD5ΔCK2BD-Tg mice developed EAE with lower incidence and severity than CD5WT mice and CD5KO mice reconstituted with a CD5WT transgene. T-cells from CD5ΔCK2BD-Tg mice also exhibited elevated AICD.
The previous study clearly established the CD5-dependent CK2 signaling pathway is important for survival of activated CD4+ cells and can impact the outcome of EAE in mice. However, a major limitation of the CD5ΔCK2BD-Tg mouse was that the transgene was under the control of the CD2 promoter and enhancer; therefore the expression of CD5 could not be physiologically regulated by the threshold of antigen receptor activation. To resolve this problem and to study the biological activities of CD5-CK2 signaling pathway, in this study we generated a CD5 knock-in mouse in which the nucleotides encoding the four amino acids (S458-D-S-S461) necessary for CK2 interacting with CD5 were deleted in the genome. Using this novel mouse model, we report that the CD5-CK2 signaling pathway plays a critical role in regulating the threshold for the development of TCR non-responsiveness in vivo and in vitro. We further determined that the CD5-CK2 signaling pathway is essential for efficient CD4+ and CD8+ T-cell activation and differentiation of naïve CD4+ T-cells to Th2 and Th17, but not Th1, cells. In summary, our results reveal the CD5-CK2 signaling pathway represents a major signaling cascade initiated by CD5 that regulates the threshold of T-cell activation and Th differentiation.
Materials and Methods
Mice
C57BL/6 mice (CD5WT) were purchased from NCI-Frederick Cancer Research. CD5−/− backcrossed greater than 12 generations into C57BL/6 (CD5KO) mice were from our colony (28). The 2D2 TCRMOG (2D2) transgenic mouse has been described previously (29). All animals were housed and treated in accordance with National Institutes of Health and University of Alabama at Birmingham Institutional Animal Care and Use Committee guidelines.
Generation of the Cd5Δck2bd/Δck2bd knock-in mouse
In order to generate the Cd5Δck2bd/Δck2bd mouse (CD5ΔCK2BD), we used the recombineering protocol described by Lee et al. (30). Briefly, homology arms “A” and “B” were used to retrieve the region of Cd5 gene that contains exons 6-10 and several kb of the 3′ non-coding region into the pSK-M-TK1 targeting plasmid from the C57BL/6 BAC clone RP24-424J1 (accession # AC132247) (Fig 1A). This BAC clone contains the entire Cd5 gene. Using a combination of bacterial recombineering and Quikchange mutagenesis (Stratagene), we targeted a loxP-flanked Neo selection cassette between exons 9 and 10 and deleted the codons encoding the four amino acids necessary for CK2 binding (CK2BD), S458-S461 (21, 22). The targeting construct was transfected into Bruce-4 (C57BL/6) embryonic stem cells and screened by Southern blot analysis of BamH1-restricted DNA using a screening probe 5′ of the targeting construct (Fig 1A). Insertion of the loxP-flanked Neo cassette created a novel BamH1 site between exons 9 and 10 that allowed for the identification of correctly targeted clones. The embryonic stem cells were microinjected into C57BL/6 albino blastocysts and implanted into pseudopregnant females. A male chimera that successfully transmitted the targeted allele was bred with a C57BL/6.E2a-Cre mouse (The Jackson Laboratory) to delete the Neo cassette (Fig. 1A). The targeted allele could be readily distinguished from the Cd5wt allele by PCR using a forward primer 5′ of exon 9 (5′-atggactcccacgaagtgctg-3′) and a reverse primer 3′ of the loxP site, but 5′ of exon 10 (5′-cttgtagaggatggtgcca-3′) (Fig 1B).
FIGURE 1.

Generation and phenotypic characterization of CD5ΔCK2BD mice. A, Targeting strategy to generate the Cd5Δck2bd/Δck2bd knock-in mouse. The targeting construct was generated with a region of Cd5 gene containing exons 6-10 retrieved from the BAC clone RP24-424J1 (C57BL/6). Nucleotides encoding the four amino acids necessary for binding of CK2 with CD5 (S458-S461) in exon 10 (*) of the mouse Cd5 gene were deleted in the targeting construct and founder chimeras were developed using Bruce-4 targeted embryonic stem cells. The Neo selection cassette was deleted by breeding with C57BL/6.E2a-Cre mice to generate the Cd5Δck2bd/Δck2bd Cre-deleted mouse. The 5′ probe used for screening embryonic stem cells based on the insertion of a novel BamH1 site (boxed) is represented in the 5′ Arm A that was used to retrieve the Cd5 gene from the BAC clone. B, PCR-based screening strategy for knock-in mice. Photomicrograph of an agarose gel showing PCR product from tail DNA of Cd5Δck2bd/Δck2bd, Cd5wt/wt, and Cd5Δck2bd/wt mice amplified with specific forward (5′-atggactcccacgaagtgctg-3′) and reverse (5′-cttgtagaggatggtgcca-3′) primers targeting the 5′ region of exon 9 and the 3′ region of the lox-P site. C, CD5 expression levels on T-cell subsets in the thymus and lymph node of 6-8 week old CD5WT (red), CD5KO (green) andCD5ΔCK2BD (blue) male mice. D, Frequencies of CD4+ and CD8+ lymphocytes in the thymus, lymph node, and spleen of 7-8 week old CD5WT, CD5KO and CD5ΔCK2BD male mice. Numbers represent the average percentage of cells is each gate ±SEM. Arrows point to the CD4loCD8lo thymocytes. Data is representative from one mouse of 6-9 mice per group. DN, double negative. DP, double positive. SP, single positive.
Induction of Active EAE
EAE in male (8-10 week-old) mice was induced and evaluated as described previously (28). MOG35-55 peptide was obtained from CPC Scientific (San Jose, CA) and pertussis toxin came from List Biological Laboratories (Campbell, CA).
Flow Cytometry
Staining was performed on peripheral lymphoid populations and spinal cord cells prepared as described previously (27). For characterization of cellular populations the following antibodies were used: anti-CD4-PerCP (GK1.5), anti-CD8a-PE (53-6.7), anti-CD5-PE (53-7.3), anti-CD25-biotin (PC61.5), anti-CD25-PE (PC61.5), anti-CD5-PE-Cy7 (53-7.3), anti-CD4-APC-Alexa750 (RM4-5), anti-B220-PE Cy5 (RA3-6B2), anti-Foxp3-APC (FJK-16s), anti-RORγ(t)-PE (AFKJS-9) (eBioscience, San Diego, CA), anti-IL23R (R&D Systems, Minneapolis, MN), anti-goat IgG (H+L)-Alexa488 (Invitrogen, Carlsbad, CA), anti-pSTAT1(Ser727), anti-pSTAT3 (Tyr705) (Cell Signaling Technology, Danvers, MA), anti-rabbit IgG (H+L)-Alexa488 (Jackson ImmunoResearch, West Grove, PA), anti-CD69-biotin (H1.2F3) (BD Biosciences Pharmingen), anti-B220-PerCP (RA3-6B2), anti-CD8a-Alexa700 (53-6.7), anti-mouse-IFNγ-PE (XMG1.2), anti-mouse-IL-17a-Alexa488 (TC11-18H10.1), and anti-mouse-IL-4-Alexa647 (11B11) (BioLegend, San Diego, CA). In some cases we used the AlexaFluor647 or AlexaFluor488 protein labeling kits (Invitrogen) to label anti-CD4 (GK1.5), anti-CD8a (53-6.7), or anti-CD5 (53-7.3) antibodies produced from hybridomas obtained from ATCC (Rockville, MD). For all immunofluorescent analyses, Fc receptors were blocked with anti-CD16/32 (2.4G2, FcR block; ATCC) before any staining procedure and live cells were gated after staining with the LIVE/DEAD Fixable Far Red Dead Cell Staining Kit or LIVE/DEAD Aqua Dead Cell Staining Kit (Invitrogen). All stained samples were analyzed using the FACSCalibur and LSRII flow cytometers (BD Biosciences). Intracellular cytokine staining in PMA and ionomycin-stimulated cultures were performed as reported previously (27). Intracellular staining for phosphorylated STAT1 and STAT3 was conducted using the protocol described by Cell Signaling Technology (http://www.cellsignal.com/support/protocols/Flow.html).
In vitro T-cell differentiation
CD4+CD25− T-cells were purified from lymph nodes and spleens of naïve mice by two-step magnetic chromatography using cell purification reagents from Invitrogen (Dynabeads) and StemCell (EasySep; Vancouver, Canada). The CD4+ T-cells were stimulated with anti-CD3 (1.0 μg/ml) or MOG35-55 peptide (10 μg/ml) in culture medium (IMDM, 10% FBS, 1% sodium pyruvate, 1% non-essential amino acids and 0.5 mM β-mercaptoethanol) with no added cytokines or antibodies for unpolarized, 10 ng/ml IL-12 + 5 μg/ml anti-IL-4 (11B11) for Th1-polarized, 10 ng/ml IL-4 + 10 μg/ml anti-IFNγ (XMG1.2) for Th2-polarized, or 5 ng/ml TGFβ + 20 ng/ml IL-6 + 10 ng/ml IL-23 + 10 μg/ml anti-IFNγ + 5 μg/ml anti-IL-4 for Th17-polarized cultures in the presence of irradiated APCs. All cytokines were purchased from eBioscience or BioLegend. Irradiated Thy1.2+cell-depleted splenocytes or six day bone marrow derived dendritic cells (BMDC) were used as APCs.
Cell proliferation, apoptosis measurement, lymphocyte activation, and calcium mobilization
For these experiments “untouched” CD4+CD25− T-cells and CD8+ T-cells were purified from spleens of naïve mice or from mice immunized with 150 MOG35-55 peptide seven days previously as described above. Proliferation was measured by two independent approaches, [3H]thymidine incorporation and 5-ethynyl-2′-deoxyuridine (EdU) incorporation. The [3H]thymidine incorporation proliferation assay was performed with CD4+CD25− T-cells stimulated with varying concentrations of MOG35-55 peptide or anti-CD3 in the presence of irradiated APCs as described previously or with CD8+ T-cells stimulated with varying concentrations of anti-CD3 and 1μg/ml anti-CD28 (37.51; BioLegend) (27). To measure proportion of dividing cells, EdU (10μM) was added for the last 1 h of the proliferation assay and its incorporation was measured using the Click-iT EdU Flow Cytometry Assay Kit (Invitrogen) following the manufacturer’s recommended protocol. In some experiments, EdU incorporation or CFSE dilution was determined after co-culturing WT-CD5 or ΔCK2BD-CD5 expressing CD4+ T-cells and stimulating with anti-CD3 mAb or MOG35-55 peptide as appropriate. To distinguish between the two different CD5 expressing T-cells, T-cells from one of the groups was labeled with CFSE (CFDA-SE - Molecular Probes, Eugene, OR) just prior to co-culturing. Dilution of CFSE in labeled, dividing CD4+ T-cells was also utilized to track cell division.
For the measurement of apoptosis, CD4+CD25− or CD8+ T-cells were stimulated with varying concentrations of MOG35-55 peptide or anti-CD3 for different time periods, and in some instances under Th17 polarizing conditions. The cells were then stained with AnnexinV-FITC (BD Biosciences) and 7-aminoactinomycin D (7-AAD; BioLegend) to quantitate apoptotic cells (AnnexinV+7-AAD−) or 7-AAD alone to quantitate dying cells by flow cytometry. To evaluate upregulation of CD25 and CD69, splenic cells were stimulated with varying concentrations of anti-CD3 for different time periods and then immunofluorescently stained with antibodies to CD4, CD25 and CD69. The upregulation of CD25 and CD69 in CD4+ gated T-cells was determined by flow cytometry.
Intracellular Ca2+ levels in CD4+ T-cells were measured in fluo-4 AM (Invitrogen)-loaded spleen cells stimulated with anti-CD3. Single cell suspensions of splenocytes were surface stained with anti-CD4 and resuspended to a concentration of 5×106 cells/ml in HBSA++ + 3% FBS. Cells were incubated with fluo-4 AM for 30 min at 250C, washed, and placed on ice. Samples were loaded onto the FACSCalibur and events were collected at a rate of 200-400 events/sec. After establishing a 1 min baseline, 0.1-10 μg/ml anti-CD3 (145-2C11) was added to the samples and analysis continued for 7 min. At the end of this time period, 10μM ionomycin was added to the samples and data collection was continued for an additional 2 min.
Quantitative Real Time RT-PCR
RNA was collected from cultured cells using TRIzol reagent and cDNA was generated with the SuperScript VILO cDNA synthesis (Invitrogen) kit following the manufacturer’s instructions. The EXPRESS SYBR GreenER qPCR Supermix Universal (Invitrogen) was used to detect Rorc and Il23ra mRNA expression. Expression of both mRNAs was calculated by normalizing data to Gapdh mRNA expression. Primers for Rorc (Forward: 5′-CCGCTGAGAGGGCTTCAC-3′; Reverse: 5′-TGCAGGAGTAGGCCACATTACA-3′), Il23ra (Forward: 5′-GCCAAGAGAACCATTCCCGA-3′; Reverse: 5′-TCAGTGCTACAATCTTCAGAGGACA-3′), and GAPDH (Forward: 5′-TGGTGAAGGTCGGTGTGAAC-3′; Reverse: 5′-CCATGTAGTTGAGGTCAATGAAGG-3′) were obtained from Eurofins MWG Operon (Huntsville, AL). Relative gene expression was calculated using the comparative CT method and examining fold differences in target gene expression for CD5ΔCK2BD samples compared to those observed in CD5WT samples.
Induction of EAE by Passive Transfer of Th1 or Th17 cells
Passive transfer studies were performed as reported previously (31). Briefly, male mice (8-10 weeks old) were immunized with 150 μg MOG35-55 peptide without any pertussis toxin injection. Twelve days following immunization, mononuclear cells were harvested from the spleens and draining lymph nodes of these mice and CD8+ and CD25+ T-cells were depleted using magnetic beads. The cells were then restimulated with MOG35-55 peptide and cultured under Th1 or Th17 polarizing conditions as described above. After three days of restimulation, the cells were harvested and dead cells were eliminated using Ficoll-Paque (GE Healthcare, Piscataway, NJ) density (1.084 g/ml) gradient centrifugation. Cells were injected (4×106 cells/mouse) into naïve mice that were sublethally irradiated 18h earlier with 350 rads of X-irradiation. The recipient mice were injected with 200 ng of pertussis toxin on day of transfer and two days post-transfer.
Cytokine ELISAs
CD4+CD25− T-cells cultures were stimulated with MOG35-55 peptide or anti-CD3 for various time periods, and culture supernates were collected and analyzed for the expression of IFNγ, IL-17a, IL-6, IL-2, and IL-10 by ELISA. ELISA kits were purchased from eBioscience or BioLegend.
Statistical Analysis
Results were analyzed for statistical significance using the Student t test (*, p< 0.05; **, p< 0.01; ***, p< 0.001).
Results
CD5 expression and T-lymphocyte populations in CD5ΔCK2BD mouse
To determine the biological role of CD5-dependent CK2 activation in T-cell function we generated a “knock-in” mouse in which the nucleotides encoding the four amino acids necessary for binding of CK2 with CD5 (S458-S461; CK2 binding domain or CK2BD) were deleted in exon 10 of the mouse Cd5 gene (Fig. 1A) (22). The gene targeted mouse was bred with the C57BL/6.E2a-Cre mouse to delete the loxP flanked Neo selection cassette to generate the Cd5Δck2bd/Δck2bd mouse, hereafter referred to as the CD5ΔCK2BD mouse (Fig. 1B). The strength of this novel model is that the expression of CD5 remains under the control of the endogenous promoter and regulatory elements. This is a very important consideration because CD5 level on T-cells is dynamically regulated by changes in strength of T-cell receptor signals both during early development in the thymus and later in the periphery (9, 11). In our previous study the CD5-dependent CK2 binding deficient mouse was generated by reconstituting the CD5KO mouse with a mutant CD5 expressing transgene under the control of the CD2 promoter (27).
The mean expression levels of CD5 on all T-cell populations in the thymus, lymph nodes and spleens of CD5WT and CD5ΔCK2BD mice were similar (Fig. 1C, and data not shown). We next examined the effect of loss of CD5-dependent CK2 signaling on developing T-cell populations in the thymus and in mature CD4+ and CD8+ T-cells in the spleen and lymph node. As previously reported, the proportion and absolute numbers of CD4+- and CD8+-expressing double positive (DP) and single positive (SP) T-cells in CD5WT and CD5KO mice were similar (Fig.1D and Table I) (32). In contrast, the thymus of CD5ΔCK2BD mice contained significantly fewer DP cells compared to both CD5WT and CD5KO mice (Table I). The DP population in CD5ΔCK2BD mice contained an expanded population of cells that were CD4loCD8lo, possibly representing pre-apoptotic cells. During negative selection, DP thymocytes go through a transitional CD4loCD8lo step just before undergoing apoptosis (33). Double negative (DN) thymocytes were higher in the thymus of CD5ΔCK2BD mice compared to that in CD5WT or CD5KO mice (Fig 1D). The proportion and numbers of CD4+ or CD8+ SP cells in the thymus of CD5ΔCK2BD mice were not significantly different from those in the thymus of CD5WT or CD5KO C57BL/6 mice (Fig. 1D, Table I, and data not shown). We also observed significantly fewer CD4+ T-cells in the lymph nodes and B220+ cells in spleens of CD5ΔCK2BD mice compared to CD5WT and CD5KO mice. The lower numbers of peripheral CD4+ T-cells might be related to loss of survival as a result of inability to activate the CD5-dependent CK2 activation pathway. However, the presence of fewer follicular B-cells in the spleen was unexpected as they do not express detectible levels of CD5. We observed that the frequency of nTreg (CD4+CD25+Foxp3+) cells in lymph nodes and spleens of CD5WT CD5ΔCK2BD mice were similar (Supplemental Fig. 1A). However, these tissues in CD5KO mice consistently contained greater number of Tregs than that in either CD5WT or CD5ΔCK2BD mice. This observation is consistent with that previously reported in Balb/c mice (34). In contrast, Dasu et al. reported equal numbers of nTreg cells in CD5WT and CD5KO in the C57BL/6 strain (35). The reason for difference in the finding between the current study and that of Dasu et al. is currently unknown.
Table I.
Total numbers of lymphocyte populations in different tissues of CD5WT, CD5KO, and CD5ACK2BD mice.
| Mouse Strain | Thymus | Lymph Nodes | Spleen | |||||
|---|---|---|---|---|---|---|---|---|
| CD4CD8+ | CD4+ | CD8+ | CD4+ | CD8+ | B220+ | CD4+ | CD8+ | |
| CD5WT | 43.3±2.1 | 4.6±0.7 | 1.5±0.2 | 31.2±2.2 | 24.0±3.2 | 43.1±3.0 | 14.8±1.5 | 7.7±0.8 |
| CD5KO | 39.4±3.4 | 6.2±0.8 | 2.3±0.5 | 30.3±2.4 | 34.1±2.0b | 41.0±2.1 | 15.5±2.1 | 10.2±1.6b |
| CD5ΔCK2BD | 33.0±2.7a | 5.2±0.9 | 1.7±0.6 | 24.5±1.4a | 24.4±3.1 | 36.4±2.0a | 12.8±2.2 | 7.4±1.4 |
Data are presented as mean ± SEM × 106(n= 4-5 mice).
p<0.05; CD5ΔCK2BD vs. CD5WT or CD5KO mice
p<0.05; CD5KO vs. CD5WT or CD5ΔCK2BD mice
CD5-dependent CK2 signaling pathway regulates threshold for severity of EAE
We previously showed that both severity and incidence of MOG35-55 peptide-induced EAE in CD5KO mice reconstituted with a CD5ΔCK2BD transgene was significantly reduced compared to CD5WT mice, CD5KO mice and CD5KO mice reconstituted with CD5WT transgene (27). The finding demonstrated that the CD5-CK2 binding dependent signaling pathway altered pathogenesis in a disease that is T-cell dependent. We now wanted to test if this observation was recapitulated in the CD5ΔCK2BD knock-in mouse where the expression of CD5 is physiologically regulated. We found that EAE in CD5ΔCK2BD mice and CD5KO mice was significantly delayed in onset and less severe than that in CD5WT mice (Fig. 2A and Table II). The incidence of disease in both of the CD5 mutant mice was also lower but the difference was not statistically significant. Since CD5 regulates threshold of T-cell activation, we asked if the severity of EAE also correlated with increase in MOG35-55 peptide immunization dose. We observed that in CD5WT mice immunized with 150 μg MOG35-55 peptide, the onset of EAE was significantly earlier and more severe than those immunized with 50 μg MOG35-55 peptide (Table II). Remarkably, EAE in CD5WT mice immunized with a higher immunization dose of 450 μg MOG35-55 peptide was significantly less severe (accumulative score) than that observed with 150 μg of peptide, a characteristic that resembles the generation of high dose tolerance. In contrast, the severity of EAE in both CD5KO and CD5ΔCK2BD mice correlated with immunization dose with no indication of high dose anergy (Table II). Furthermore, with increase in immunization dose the severity and time of onset of EAE in CD5KO mice and CD5ΔCK2BD mice approached and equaled that of CD5WT mice (Fig. 2B, 2C and Table II). A striking finding is that with respect to EAE disease severity, the mere deletion of the four amino acid CK2 binding domain within CD5 recapitulated the phenotype of the CD5KO mouse.
FIGURE 2.

CD5-dependent CK2 signaling pathway regulates the threshold for EAE severity. A-C, Clinical course of EAE in CD5WT, CD5KO, and CD5ΔCK2BD mice induced with 50 μg/ml MOG35-55 peptide (A), 150 μg/ml MOG35-55 peptide (B), and 450 μg/ml MOG35-55 peptide (C). Data are mean ± SEM, n=10-20 mice/group). Bars above plots denote regions of statistical significance. **p<0.01 with Student t test. D, Intracellular cytokine staining of infiltrating CD4+ T-cell populations in spinal cords from CD5WT, CD5KO, and CD5ΔCK2BD mice analyzed 14 d post-immunization with 150 μg MOG35-55 peptide. Unbracketed numbers in each quadrant represent the average frequency of CD4+ T-cells expressing IFNγ and/or IL-17a ±SEM. Bracketed numbers represent average absolute cell counts for each subpopulation x104 ±SEM. Data are representative of one of 2-3 experiments, with 2-3 mice per experiment/group.
Table II.
Analysis of disease parameters for active EAE in CD5WT, CD5KO, and CD5ΔCK2BD mice induced with different doses of MOG35-55 peptide
| MOG35-55 Peptide Dose |
Mouse Strain | Day of Onset | Time to Peak | Accumulative Score |
Incidence | Mortality |
|---|---|---|---|---|---|---|
| 50 μg | CD5WT | 15.0±1.9a | 18.6±4.0e | 44.5±10.4e | 20/20 | 1/20 |
| CD5KO | 19.0±5.0b | 20.2±4.4 | 29.8±11.7b | 12/15 | 0/15 | |
| 1 CD5ΔCK2BD | 19.2±5.2b | 20.0±4.2 | 35.1±14.3b | 14/17 | 2/17 | |
| 150 μg | CD5WT | 12.5±1.5 | 15.8±3.6 | 59.5±8.7f | 19/19 | 4/19 |
| CD5KO | 17.2±4.2b | 18.6±3.2b | 34.6±14.8b | 15/15 | 1/15 | |
| CD5ΔCK2BD | 12.9±2.9 | 17.2±6.9 | 48.8±4.9b,d | 15/15 | 2/15 | |
| 450 μg | CD5WT | 13.1±3.2 | 19.0±6.2 | 45.4±18.5 | 11/12 | 0/12 |
| CD5KO | 14.7±2.7c | 18.9±4.3 | 44.3±9.7 | 11/12 | 0/12 | |
| CD5ΔCK2BD | 13.4±1.7d | 17.7±2.7 | 52.5±11.7d | 9/10 | 0/10 |
Data are presented as mean ± SEM (n=10-20 mice).
No scores were assigned following day of death.
p<0.05; CD5WT 50 μg dose vs. CD5WT 150 μg dose or CD5WT 450 μg dose
p<0.05; vs. CD5WT mice at same dose
p<0.05; CD5KO 450 μg dose vs. CD5KO 50 μg dose
p<0.05; vs. CD5ACK2 50 μg dose
p<0.05; CD5WT 50 μg dose vs. CD5WT 150 μg dose
p<0.05; CD5WT 150 μg dose vs. CD5WT 450 μg dose
We analyzed the CNS cell infiltration in mice of all three CD5 strains to evaluate the correlation between disease severity and Th cell populations. The results showed that the proportion and absolute number of Th1 cells in the CNS of CD5WT, CD5KO and CD5ΔCK2BD mice were similar. With respect to Th17 cells and Th-IFNγ+IL17a+cells (double producers), the spinal cords of CD5ΔCK2BD mice with EAE contained fewer cells of this population than both CD5WT and CD5KO mice (Fig. 2D). Our results suggest that the CD5-dependent CK2 activation pathway has an impact preferentially on the generation of IL-17a-expressing Th cells, an observation also made in our previous study in the CD5ΔCK2BD transgenic mouse (27).
CD5-dependent CK2 signaling pathway is necessary for efficient generation of antigen-specific Th2 and Th17 cells
In order to test if the CD5-CK2 signaling pathway has an impact on differentiation of naïve CD4+ T-cells to Th1 or Th17 cells under non-polarizing and polarizing conditions, we bred the C57BL/6 MOG35-55 specific TCR transgenic mouse, 2D2TCRMOG, with both the CD5KO mouse and CD5ΔCK2BD mouse. Under non-polarizing conditions, the generation of Th1 cells following stimulation with MOG35-55 peptide was 2D2.CD5WT> 2D2.CD5ΔCK2BD> 2D2.CD5KO (Fig. 3A). Consistently, the proportion of Th1 cells in 2D2.CD5WT CD4+ T-cell cultures was approximately 50% greater than in 2D2.CD5ΔCK2BD and three fold greater than in 2D2.CD5KO CD4+ T-cell cultures. Under Th1 polarizing conditions, the proportion of Th1 cells generated was equivalent in CD4+ T-cell cultures from 2D2.CD5KO mice and 2D2.CD5ΔCK2BD mice but marginally greater than that in 2D2.CD5WT mice (Fig. 3A). However, under Th17 polarizing conditions, the proportion of Th17 cells in 2D2.CD5ΔCK2BD CD4+ T-cell cultures was less than half of that in CD4+ T-cell cultures from 2D2.CD5WT and 2D2.CD5KO mice (Fig. 3A). This ex vivo result largely recapitulates the observation in spinal cords of mice with EAE (Fig. 2D).
FIGURE 3.

Contribution of the CD5-CK2 signaling pathway to the generation of Th1, Th2, and Th17 cells from naïve CD4+ T-cells. Naïve CD4+ T-cells were co-cultured with irradiated APCs and stimulated with MOG35-55 peptide (A) or anti-CD3 (B) under non-polarizing (unpolarized) or polarizing conditions and generation of Th subpopulations was evaluated. Numbers in each quadrant represent the average frequency of CD4+ T-cells expressing IFNγ, IL-17a and/or IL-4±SEM from four independent experiments performed with at least three mice per experiment. C, Percent 7-AAD+CD4+ T-cell at 24 h and 120 h and D, RORγt+CD4+ T-cells at 120 h following stimulation with MOG35-55 peptide under Th17 polarizing conditions. Data is mean±SEM of three independent experiments with at least two mice/group. E, pSTAT1 (Ser727) and F, pSTAT3 (Tyr705) in CD4+ T-cells stimulated for 72h with MOG35-55 peptide under Th17 polarizing conditions. The filled shaded histograms are unstimulated and the dashed histograms are isotype immunoglobulin stained cell controls. For pSTAT1 (E), the MFI of 2D2.CD5ΔCK2BD is 52±4.8 and 2D2.CD5WT is 30±4.3. For pSTAT3 (F) the MFI for 2D2.CD5ΔCK2BD is 32±2.9 and 2D2.CD5WT is 21±3.5. Histograms represent data from one representative mouse per group from two mice/experiment, n=3 experiments. *p<0.05, **p<0.01, ***p<0.001.
The stimulation with anti-CD3 mAb (antigen non-specific) had a different outcome than stimulation with MOG35-55 peptide (cognate peptide). The generation of Th1 cells was similar in T-cells cultures under non-polarizing conditions from all three strains of mice (Fig. 3B). Following anti-CD3 stimulation under Th1 polarization conditions, greater proportions of Th1 cells were obtained from CD5ΔCK2BD CD4+ T-cell cultures than from CD5WT and CD5KO CD4+ T-cell cultures. Unlike cognate peptide stimulation, the generation of Th17 cells from anti-CD3 mAb stimulated Th17 polarization cultures was generally equivalent in CD4+ T-cell cultures from the three strains of mice (Fig. 3A, 3B). We posited that this divergent outcome reflects direct engagement of TCR with anti-CD3 mAb vs cognate stimulation (peptide:MHC) rather than differences in CD5 signaling between T-cells from C57Bl/6 mice vs 2D2 mice. This was supported by our observation that the generation of Th1 and Th17 cells following stimulation of CD4+ T-cells from the different CD5 genotypes of 2D2 mice with anti-CD3 mAb was similar to anti-CD3 mAb stimulated C57BL/6 mice T-cells. (Supplemental Fig. 2A and Fig 3B).CD5 is recruited the immunological synapse (36). It is possible that direct stimulation of TCR vs cognate peptide:MHC induces a different type of receptor assembly that alters positioning of CK2. Such differences in receptor assembly may affect the manner in which CK2 activates Jak1 or Jak2 (37). We are now beginning to address these questions.
We also evaluated the contribution of CD5-CK2 signaling pathway in the generation of Th2 T-cells following stimulation with MOG35-55 peptide or anti-CD3 under Th2 polarization conditions. In MOG35-55 peptide stimulated Th2 polarized cultures, the proportion of Th2 cells in CD4+ T-cell cultures from 2D2.CD5ΔCK2BD mice was lower than that in T-cell cultures from both 2D2.CD5WT and 2D2.CD5KO mice (Fig.3A). As in Th17 polarization cultures, the generation of Th2 cells in anti-CD3 stimulated cultures was similar between CD4+ T-cell cultures from the three strains of mice (Fig. 3B and Supplemental Fig. 2A). Considering that stimulation of T-cells with cognate peptide rather than anti-CD3 more closely represents normal physiology, the data indicates that loss of the CD5-CK2 signaling pathway results in preferential diminution in the generation of Th17 and Th2 cells but not Th1 cells.
Cell death, RORγT expression and activation of STAT1 and STAT3 in CD5ΔCK2BD T-cells during Th17 polarization
The CD5-CK2 signaling pathway regulates survival of activated T-cells and could represent a mechanism for the diminished Th17 cells in Th17 polarized cultures containing T-cells from 2D2.ΔCD5CK2BD mice (27). Consistent with this possibility we observed greater proportion of dying T-cells in Th17 polarization cultures of 2D2.ΔCD5CK2BD mice than 2D2.CD5WT mice (Fig. 3C). The increased cell death was not compensated by greater cell division. The proportion of Th17 cells in cell cycle as measured by uptake of EdU during a one hour pulse labeling was similar for both 2D2.CD5WT and 2D2.ΔCK2BD T-cells Supplemental Fig 2D). The diminished Th17 generation with naïve 2D2.ΔCD5CK2BD T-cells was also associated with fewer T-cells that have upregulated RORγT than that in 2D2.CD5WT cultures (Fig 3D). However, the expression of Rorc (gene for RORγT) at a per-cell level were almost identical between 2D2.CD5WT and 2D2.ΔCK2BD T-cells indicating that loss of CD5-CK2 signaling pathway did not alter intrinsic RORγT expression (Supplemental Fig 2C).
A recent report showed that CD5 costimulation enhanced differentiation of naïve human T-cells to Th17 cells that was associated with upregulation of IL-23R and elevated activation of STAT3 (38). We observed that loss of CD5-CK2 signaling pathway had no effect on IL-23R expression on Th17 cells (Supplemental Fig. 2B, 2C). However, 2D2.ΔCK2BD Th17 cells did contain greater levels of activated STAT1 and STAT3 compared to 2D2.CD5WT Th17 cells (Fig. 3E, 3F). Overall the data indicates that the loss of CD5-CK2 signaling attenuates Th17 generation by enhancing cell death and enhanced activation of STAT1 and STAT3.
CD5ΔCK2BD T-cells hypoproliferate in response to antigen-receptor activation
CD5KO T-cells hyperproliferate following stimulation of the antigen receptor, a property associated with the loss of CD5-dependent regulation (9). The inhibitory activity of CD5 is ascribed to the ITAM/ITIM domain of CD5, a domain that is intact in the CD5ΔCK2BD mouse. This mouse model now offers the opportunity to selectively determine the contribution of the CD5-CK2 signaling pathway to T-cell activation. Ca2+mobilization and upregulation of CD25/CD69 are parameters of early T-cell activation. In response to TCR stimulation, CD4+T-cells from CD5WT mice and CD5ΔCK2BD mice behaved similarly for both of these parameters (Supplemental Fig. 1B, 1C, and data not shown). In contrast, CD5KO CD4+ T-cells exhibited a lower threshold for Ca2+flux and more accelerated kinetics for upregulation of CD25 and CD69 (Supplemental Fig. 1B, 1C). This observation is similar to that previously reported for thymic T-cells (1). The results indicate that the CD5-CK2 signaling pathway does not have an impact on proximal events of TCR signaling.
CK2 is a major promoter of cell proliferation, cell survival, and inhibitors of this kinase attenuate cell-cycle progression (26). We therefore tested the prediction that loss of CD5-dependent CK2 signals attenuates cell proliferation. Consistent with our prediction we observed that at all concentrations of anti-CD3, CD4+ T-cells from CD5ΔCK2BD mice significantly hypoproliferated in comparison to CD4+ T-cells from CD5WT mice (Fig. 4A). As expected, CD4+ T-cells from CD5KO mice hyperproliferated following stimulation with anti-CD3. A similar observation was noted when antigen-specific T-cells were stimulated with MOG35-55 peptide (Fig. 4B). 2D2.CD5ΔCK2BD T-cells significantly hypoproliferated and 2D2.CD5KO T-cells hyperproliferated in comparison to 2D2.CD5WT T-cells following stimulation with cognate peptide. Since CK2 also promotes cell survival, we wanted to test if the apparent diminished proliferation of CD5-CK2 signaling deficient T-cells reflected enhanced activation induced cell death (AICD). When anti-CD3 was used for activation, we found that the proportion of CD4+ T-cells undergoing apoptosis (Annexin V+7-AAD−) was similar in cultures from CD5ΔCK2BD mice and CD5WT mice (Fig. 4C). However, in MOG35-55 peptide-stimulated cultures, apoptosis was significantly greater when CK2 binding to CD5 was selectively abrogated (Fig. 4D and Table III). CD5KO T-cells consistently exhibited enhanced AICD following stimulation with anti-CD3 or cognate peptide (Fig. 4C, 4 D). Taken together, the above results indicate that decreased survival in CD5-CK2 signaling-deficient T-cells contributes only partially to the diminished proliferation that follows T-cell activation.
FIGURE 4.

CD5ΔCK2BD T-cells hypoproliferate and exhibit increased AICD. Proliferation of CD4+ T-cells following 72 h stimulation with anti-CD3 (A) or MOG35-55 peptide (B) in the presence of Thy 1.2+ cell-depleted splenocytes serving as APCs as measured by the [3H]thymidine incorporation assay. [3H]thymidine was added for the final 18 h of culture. Data ± SEM are from a representative experiment with n =2-3 experiments and 2-3 mice per experiment. C and D, Frequency of early apoptotic events (AnnexinV+7-AAD−) following 24 h of stimulation with anti-CD3 (C) or MOG35-55 peptide (D). Data± SEM are the average from n =3 experiments and 2-3 mice per experiment. E and F, Frequencies of CD4+ T-cells in cell cycle measured by the EdU incorporation assay after stimulation with anti-CD3 (E) or MOG35-55 peptide (F). G and H, Frequencies of CD4+ T-cells in cell cycle in co-cultures of G, CFSE-labeled CD5WT and unlabeled CD5ΔCK2BD CD4+ T-cells stimulated with anti-CD3 mAb or H, CFSE-labeled 2D2.CD5WT and unlableled 2D2.CD5ΔCK2BD T-cells stimulated with MOG35-55 peptide for the indicated time periods. In E, F, G, and H, the cells were pulse labeled with EdU for 1 h prior to analysis. Data ± SEM represent the average of 2-3 mice per group per experiment, n= at least 3 experiments. I and J, Proliferation of CD8+ T-cells measured by the [3H]thymidine incorporation assay (I) or EdU incorporation (J). Purified CD8+ T-cells were stimulated with varying concentrations of anti-CD3 for 72 h. The [3H]thymidine incorporation assay and EdU incorporation assay were performed as described above for CD4+ T-cells. Data ± SEM represent the average of 3 mice per group per experiment, n= 3 experiments. Bars above plots denote regions of statistical significance. *p<0.05, **p<0.01, ***p<0.001 with Student t test. EdU, 5-ethynyl-2′-deoxyuridine.
Table III.
CD5-CK2 signaling pathway regulates TCR-induced apoptosis in T-cells
| Anti- CD3 (μs/ml) |
%Annexin V+7-AAD− | MOG35-55 (μg/ml) |
%Annexin V+7-AAD− | ||||
|---|---|---|---|---|---|---|---|
| CD5WT | CD5KO | CD5ΔCK2BD | 2D2.CD5WT | 2D2.CD5KO | 2D2.CD5 ACK2BD | ||
| 0 | 9.7±1.3 | 10.7±1.8 | 9.8±0.3 | 0 | 15.0±1.1 | 27.0±3.6 | 26.2±2.8b |
| 0.1 | 9.6±0.7 | 10.9±0.1 | 9.5±0.7 | 1 | 19.6±0.6 | 39.7±2.1c,d | 35.9±1.5c |
| 1 | 8.8±0.6a | 14.0±1.3 | 9.6±0.7a | 10 | 33.1±0.3e | 43.1±2.6f | 46.9±7.6 |
| 100 | 31.4±0.5g | 39.0±2.2g,h | 71.4±4.4 | ||||
Cells were stimulated with varying concentrations of anti-CD3 or MOG35-55 peptide in the presence of irradiated APCs for 24 h. Apoptosis was evaluated by staining cells with Annexin V and 7-AAD.
Data are presented as % of events ± SEM.
p<0.01; vs. CD5KO mice
p<0.05; 2D2.CD5ΔCK2BD vs. 2D2.CD5WT mice
p<0.001; vs. 2D2.CD5WT mice
p<0.05; 2D2.CD5KO mice vs. 2D2.CD5ΔCK2BD mice
p<0.01; 2D2.CD5WT mice vs. 2D2.CD5ΔCK2BD mice
p<0.05; 2D2.CD5KO mice vs. 2D2.CD5ΔCK2BD mice
p<0.001; vs. 2D2.CD5CK2BD mice
p<0.01; 2D2.CD5KO mice vs. 2D2.CD5WT mice
We directly interrogated if loss of CD5-CK2 signaling impairs entry into the cell cycle. To address this question we used EdU to pulse label cells for the last 1 h following 72h of stimulation with anti-CD3 or cognate peptide. We then quantitated the amount of EdU uptake by a flow cytometric assay. This approach enumerates the proportion of cells progressing through the cell cycle during the short pulse labeling and therefore overcomes confounders such as cell death. We found that the proportion of EdU+CD4+ CD5-CK2 signaling deficient T-cells stimulated with either anti-CD3 or MOG35-55 peptide was lower than CD5WT T-cells stimulated similarly (Fig. 4E and 4F). In contrast, the proportion of EdU+CD4+ CD5-deficient T-cells was always greater than that in stimulation cultures containing CD5WT T-cells. The production of several cytokines by activated CD4+ T-cells is different between cells that are able and unable to engage the CD5-CK2 signaling pathway (Fig 6). Therefore, the hypoproliferation of CD5ΔCK2BD or 2D2.ΔCK2BD T-cells may not be intrinsic to the activation threshold but rather influenced by cytokines. To address this possibility, we co-cultured WT-CD5 expressing CD4+ T cells with ΔCK2BD-CD5 expressing CD4+ T-cells and stimulated with anti-CD3 or MOG35-55 peptide for 48 h or 72 h. To distinguish between the two T-cell populations for analysis WT-CD5 T-cells were labeled with CFSE. Proliferation was assessed by measuring EdU uptake. We observed that the proportion of dividing CD5-CK2 signaling deficient CD4+ T-cells was significantly less than total dividing WT-CD5 expressing CD4+ T-cells even when cultured under same environmental conditions (Fig 4G, 4H). Similar results were obtained with co-cultures containing labeled ΔCK2BD-CD5 expressing T-cells with CFSE and unlabeled WT-CD5 expressing T-cells demonstrating that CFSE labeling did not introduce any artifact (data not shown). These results demonstrate that CD5-CK2 signaling deficient CD4+ T-cells are intrinsically hypoactive to antigen receptor induced proliferation.
FIGURE 6.
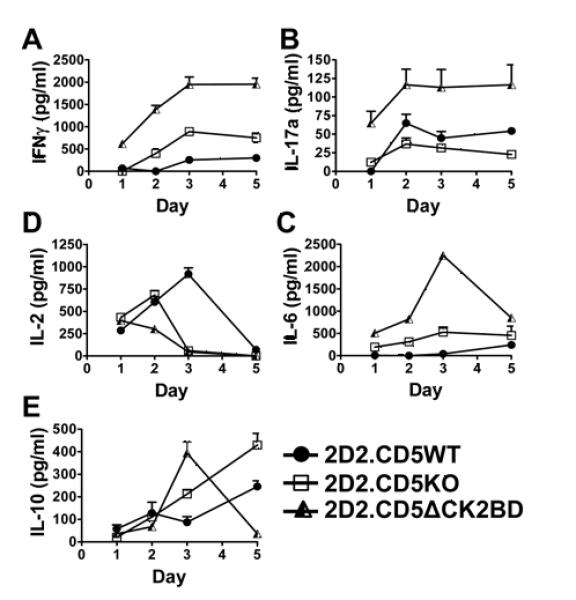
CD5-CK2 signaling pathway regulates antigen-receptor induced cytokine production. Naïve 2D2.CD5WT, 2D2.CD5KO, and 2D2.CD5ΔCK2BD CD4+ T-cells were co-cultured with irradiated APCs and stimulated with 100 μg/ml MOG35-55 peptide for 1-5 d. Supernates were collected on days 1, 2, 3, and 5 to quantify secreted levels of IFNγ (A), IL-17a (B), IL-6 (C), IL-2 (D), and IL-10 (E) by ELISA. Data ± SEM represent readings from one mouse of 2-3 mice per group per experiment, n= at least 4 experiments.
We next asked if the CD5-CK2 signaling pathway also promotes TCR signaling-initiated proliferation and cell-cycle entry in CD8+ T-cells. We found that the extent of CD8+ T-cell proliferation following anti-CD3 stimulated recapitulated the observations made with CD4+ T-cells (Fig. 4I). The frequencies of CD8+ T-cells in cell cycle from CD5WT, CD5KO or CD5ΔCK2BD mice were not significantly different from each other (Fig. 4J). This result suggests that differences in proliferation probably reflect variances in susceptibility to AICD. Overall our results indicate that the CD5-CK2 signaling pathway promotes cell cycle progression in CD4+ T-cells following TCR activation.
The CD5-CK2 signaling pathway regulates T-cell conversion to non-responsiveness
CD5WT mice immunized with 450 μg MOG35-55 peptide developed less severe EAE than mice immunized with a lower dose of 150 μg peptide, a characteristic similar to high-dose tolerance (Fig. 2A and Table II). However in both CD5KO and CD5ΔCK2BD mice, the severity of EAE directly correlated with antigen dose. This suggested that the CD5-CK2 signaling pathway may be involved in regulating T-cell response to antigen re-exposure. To directly test this, we performed an in vitro recall assay where CD4+ T-cells were activated with anti-CD3 or cognate peptide, rested, and then restimulated with anti-CD3 or MOG35-55, respectively. We measured proliferation and cell cycle entry. We observed CD4+ T-cells from CD5WT, CD5KO and CD5ΔCK2BD mice proliferated equally when restimulated with 0.1 μg/ml anti-CD3 (Fig. 5A). In contrast, at 1 μg/ml anti-CD3, CD5KO and CD5ΔCK2BD T-cells proliferated more vigorously than CD5WT T-cells. We next wanted to determine if this property of CD5 was active if T-cells were stimulated with cognate peptide. MOG35-55 peptide-activated and rested CD4+ T-cells from 2D2.CD5WT, 2D2.CD5KO or 2D2.CD5ΔCK2BD mice were restimulated with 1 and 10 μg/ml MOG35-55 peptide, and proliferation was measured. The proliferation of CD4+ T-cells from 2D2.CD5ΔCK2BD mice was several folds greater than CD4+ T-cells from either 2D2.CD5WT and 2D2.CD5KO mice at both MOG35-55 concentrations (Fig. 5B). We then evaluated cell cycle entry using the EdU incorporation assay. We observed that fewer CD4+ T-cells from CD5WT mice stimulated with anti-CD3 or MOG35-55 peptide entered cell cycle than similarly stimulated CD4+ T-cells from CD5KO or CD5ΔCK2BD mice (Fig. 5C, 5D). Consistently, CD5ΔCK2BD CD4+ T-cells responded more vigorously to restimulation than CD5KO CD4+ T-cells. Just as CD4+ T-cells, CD8+ T-cells from CD5KO mice or CD5ΔCK2BD mice responded more vigorously when restimulated than CD5WT CD8+ T-cells (Fig. 5E).
FIGURE 5.

CD5-CK2 signaling pathway-deficient T-cells are resistant to development of anergy. Purified CD4+ T-cells were stimulated for 24 h with anti-CD3 or MOG35-55 peptide, rested for 72 h, and then re-stimulated for 48 h with anti-CD3 or MOG35-55 peptide. A and B, Frequency of CD4+ T-cells incorporating [3H]thymidine following 48 h re-stimulation with varying concentrations of anti-CD3 (A) or MOG35-55 peptide (B). Data ± SEM represent the average of 2 mice per group per experiment, n= 3 experiments. C and D, EdU incorporation by CD4+ T-cells restimulated with anti-CD3(C) or MOG35-55 peptide (D). Cells were pulse labeled with EdU after 48 h re-stimulation. E, EdU incorporation in CD8+ T-cells 48 h after re-stimulation with 1 μg/ml anti-CD3. F and G, EdU incorporation in co-cultures of F, CFSE-labeled CD5WT and unlabeled CD5ΔCK2BD CD4+ T-cells restimulated with anti-CD3 mAb or G, CFSE-labeled 2D2.CD5WT and unlabeled 2D2.ΔCK2BD CD4+T-cells restimulated with MOG35-55 for the indicated times. H, Effect of in vitro restimulation on in vivo primed T-cells. CD5WT mice and CD5ΔCK2BD mice were immunized with 150 μg MOG35-55 peptide in CFA, s.c. Seven days later CD4+ T-cells from spleens from both groups of mice were co-cultured in the presence of MOG35-55 peptide and proliferation was assessed by EdU incorporation at the indicated time points. Data ± SEM represent the average of 2-3 mice per group per experiment, n= at least 3 experiments. Bars above plots denote regions of statistical significance. *p<0.05, **p<0.01, ***p<0.001 with Student t test.
Considering WT-CD5 expressing T-cells and ΔCK2BD-CD5 expressing T-cells produce different levels of cytokines, we evaluated their proliferation in co-cultures following restimulation with anti-CD3 or MOG35-55 peptide. At all-time points of measurement, we observed significantly greater proportions of ΔCK2BD-CD5 expressing T-cells in cell-cycle compared to WT-CD5 expressing T-cells (Fig 5F, 5G). The CFSE dilution assay showed that CD5-CK2 signaling deficient T-cells went through one or more generations of cell division than WT T-cells (Supplemental Fig 3A). The decreased proliferation in WT-CD5 expressing T-cells was not associated with enhanced cell death (Supplemental Fig 3B).
We next evaluated if the CD5-CK2 signaling pathway also regulated re-exposure to antigen when the initial priming was performed in vivo. CD5WT or CD5ΔCK2BD mice were immunized with MOG35-55 peptide and seven days later, CD4+ T-cells from spleens and lymph nodes were isolated from each group of mice and co-cultured with MOG35-55 peptide. We again observed that a significantly greater proportion of CD5ΔCK2BD T-cells incorporated EdU than CD5WT T-cells (Fig. 5H). There was no difference in T-cell death between the two groups of mice (Supplemental Fig. 3C). Overall our results show that the CD5-CK2 signaling pathway regulates response to re-exposure to antigen.
CD5 regulates cytokine production in CD4+ T-cells
2D2.CD5KO and 2D2.CD5ΔCK2BD CD4+ T-cells cultured under non-polarizing conditions contain fewer IFNγ+ expressing T-cells than 2D2.CD5WT counterparts (Fig. 3A). Therefore we examined if the CD5-CK2 signaling pathway regulated antigen receptor-induced cytokine production. To address this question, naïve CD4+ T-cells from 2D2.CD5WT, 2D2.CD5KO and 2D2.CD5ΔCK2BD mice were stimulated with MOG35-55 peptide (10 and 100 μg/ml) and supernates collected on days 1, 2, 3 and 5 were analyzed for levels of IFNγ, IL-17a, IL-6 and IL-2 and IL-10. At each time point, the levels of IFNγ, IL-17a and IL-6 in 2D2.CD5ΔCK2BD T-cell cultures were dramatically greater than those in CD4+ T-cell cultures from 2D2.CD5WT mice (Figs. 6A, B, and C and Supplemental Figs. 3A, B, and C). Although T-cell cultures form 2D2.CD5KO mice contained higher levels of these three cytokines than 2D2.CD5WT T-cell cultures (day 2-day5), they were consistently lower than 2D2.CD5ΔCK2BD T-cell cultures. The levels of IL-2 and IL-10, cytokines associated with regulatory T-cell function followed different kinetics. Although IL-2 levels in T-cell cultures from 2D2.CD5WT and 2D2.CD5KO increased rapidly, the cytokine persisted longer than when the T-cells were obtained from 2D2.CD5WT mice (Fig. 6D and Supplemental Fig. 3D). In contrast, detectable levels of IL 2 in T-cell cultures from 2D2.CD5ΔCK2BD did not increase beyond day 1 of stimulation. The level of this cytokine had contracted to basal levels by day 3 in 2D2.CD5KO and 2D2.CD5ΔCK2BD T-cells cultures and by day 5 in 2D2.CD5WT T-cell cultures. This contraction is probably associated with T-cell exhaustion. IL-10 levels rose and contracted in 2D2.CD5ΔCK2BD T-cell cultures (Fig. 6E and Supplemental Fig. 3E). In contrast, IL-10 levels continuously increased in T-cell cultures from 2D2.CD5WT and 2D2.CD5KO mice (Fig. 6E). A striking finding is that although MOG35-55 peptide-stimulated unpolarized and Th17 polarized 2D2.CD5ΔCK2BD T-cell cultures contain fewer Th1 and Th17 cells, respectively (Fig 3A), the levels of IFNγ and IL-17a produced were the greatest. This would suggest that the CD5-CK2 signaling pathway is needed for the expansion of these subsets rather than production of these cytokines.
We also determined the cytokine levels in cultures of anti-CD3 stimulated T-cells. Cytokine levels in CD4+ T-cell cultures stimulated with anti-CD3 in the presence of irradiated APC reflected the general trend of MOG35-55-stimulated cultures. However, the main difference was that the CD5KO T-cell cultures contained greater levels of all cytokines than that in the T-cell cultures from CD5WT or CD5ΔCK2BD mice (Supplemental Fig. 4).
CD5 signaling differentially regulates Th1 vs. Th17 disease
Th1 and Th17 T-cells are the major effector T-cell populations in MS and EAE that cross-regulate each other (39). Furthermore, we recently showed that MS broadly stratifies into a Th1 or Th17 predominant disease (31). We therefore wanted to determine if loss of CD5-CK2 signaling pathway differentially affects the outcome of EAE disease induced by encephalitogenic Th1 cells vs. Th17 cells. To address this question, encephalitogenic Th1 or Th17 cells generated from MOG35-55 peptide-immunized CD5WT, CD5KO or CD5ΔCK2BD mice were adoptively transferred into CD5WT mice and development of EAE was monitored. For Th1 disease we found that encephalitogenic CD5ΔCK2BD Th1 cells induced the most severe EAE mean maximum disease score based on accumulative score and mortality (Fig. 7A and Table IV). Although EAE induced by encephalitogenic CD5KO Th1 cells was more severe that that induced by CD5WT Th1 cells, the difference was not statistically significant. Unlike Th1 disease, encephalitogenic Th17 cells from CD5ΔCK2BD mice induced slightly less severe EAE than CD5WT Th17 cells; however, the difference was not significant (Fig. 7B and Table IV). Th17 cells from CD5KO mice induced the least severe disease.
FIGURE 7.
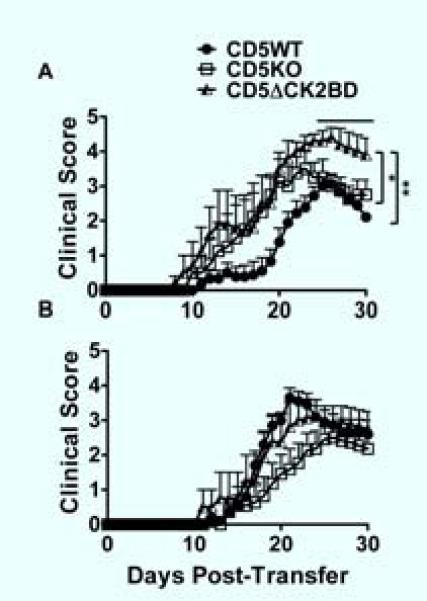
The CD5-CK2 signaling pathway differentially affects Th1 and Th17 EAE. Th1 and Th17 EAE were induced by passive transfer of encephalitogenic CD5WT, CD5KO, or CD5ΔCK2BD T-cells. Splenocytes from immunized donor mice of each CD5 genotype were depleted of CD8+ and CD25+ cells and re-stimulated under Th1 (A) and Th17 (B) polarizing conditions with MOG35-55 peptide for 72 h. Cells were transferred to sublethally irradiated CD5WT mice to induce disease. Data±SEM are from two separate experiments, 6-11 mice per group. Bar above plot denote region of statistical significance. *p<0.05, **p<0.01 with Student t test.
Table IV.
Analysis of disease parameters for EAE induced by passive transfer of Th1- or Th17-polarized encephalitogenic T-cellsfrom CD5WT, CD5KO, and CD5ΔCK2BD into CD5WTmice
| Cell Source (CD8CD25- Splenocytes) |
Day of Onset |
Max Score | Time to Peak |
Accumulative Score |
Incidence | Mortality | |
|---|---|---|---|---|---|---|---|
| Th1 | CD5WT | 19.9±3.3 | 3.4±0.8 | 23.3±3.0 | 30.7±9.8 | 9/9 | 0/9 |
| CD5KO | 16.0±4.5 | 3.6±1.1 | 20.0±2.2a | 48.0±23.1 | 6/6 | 0/6 | |
| CD5ΔCK2BD | 15.2±4.9a | 4.4±0.7a | 23.8±1.9 | 48.9±10.5a | 6/6 | 2/6 | |
| Th17 | CD5WT | 16.8±1.5 | 3.8±0.9 | 20.6±1.9 | 37.5±8.3 | 10/10 | 1/10 |
| CD5KO | 21.5±3.7b | 2.6±1.2b | 22.3±3.6 | 19.9±7.7b | 11/11 | 1/11 | |
| CD5ΔCK2BD | 18.5±4.2 | 3.3±1.4 | 20.1±3.3 | 30.4±13.3 | 8/8 | 1/8 |
Data are presented as mean ± SEM (n= 6-11 mice).
p<0.05; Disease induced with CD5ΔCK2BD Th1 cells vs. disease induced with CD5WT Th1 cells
p<0.05; Disease induced with CD5KO Th17 cells vs. disease induced with CD5WT Th17 cells
Discussion
The development of the CD5KO mouse more than 15 years ago led to a major revision in our understanding of CD5 biology (1). That study reversed the existing dogma that CD5 was a costimulator to that of an attenuator in T-cell activation. Using the CD5KO mouse we determined that CD5 had an important role in promoting survival of activated T-cells (28). This discovery resolved the puzzle as to why loss of negative regulation in the CD5KO mice did not lead to the development of spontaneous autoimmunity. We subsequently demonstrated that the prosurvival activity required the CK2 binding domain located within the distal portion of CD5 cytoplasmic tail (27). In this study we developed a knock-in mouse with a 12-nucleotide deletion in exon 10 of CD5 gene encoding the codons for the four amino acids (S458-S461) necessary for CK2 to interact with CD5. This mouse gave us the opportunity to examine the consequence of loss of the CD5-dependent CK2 signaling pathway in a context that maintains physiological regulation of CD5 expression. We found that the expression of CD5 on all T and B-lymphoid populations in this CD5-dependent CK2 signaling deficient mouse was identical to that in the CD5WT mouse. This observation suggests that the CD5-CK2 signaling pathway does not alter TCR or BCR signaling events that directly affect CD5 expression (11, 14, 40, 41). In fact we found that Ca2+ mobilization and upregulation of CD25 or CD69 following TCR stimulation was unaltered in both CD4+ and CD8+ T-cells from the CD5ΔCK2BD mouse. Both of these proximal parameters of antigen receptor signaling are tyrosine kinase dependent and were elevated in T-cells from the CD5KO mouse as previously reported (1). Although, the loss of CD5-CK2 signaling did not dramatically alter mature T-cell populations in the C57BL/6 mouse, the absolute numbers of CD4+CD8+ DP cells were significantly lower than those seen in both the CD5WT and CD5KO counterparts. Currently we do not understand the underlying mechanism of this phenotype and we are developing appropriate animal models to address this question.
We previously reported that EAE in CD5KO mice was significantly less severe and delayed in onset compared to that in CD5WT mice (28). Remarkably, we found that EAE in CD5ΔCK2BD knock-in mice essentially recapitulated the CD5KO mice in severity and onset. This is an intriguing observation because the CD5KO mouse unlike CD5ΔCK2BD mouse lacks the CD5-ITIM mediated regulation in addition to the CD5-CK2 signaling pathway. CD5ΔCK2BD mice and CD5WT mice have similar numbers of nTregs. Furthermore, nTregs from CD5ΔCK2BD mice are less efficient than that from CD5WTmice (unpublished observation). Thus it is unlikely that nTregs in CD5ΔCK2BD mice are responsible for the lower severity of EAE than CD5WT mice. With increase in MOG35-55 peptide immunization dose CD5WT mice developed less severe EAE, whereas CD5ΔCK2BD and CD5KO mice developed disease earlier in onset and with greater severity. The outcome of this study slightly differs from our previous findings that showed EAE in CD5KO mice reconstituted with a CD5 transgene expressing CD5 with the same microdeletion was significantly less severe in both severity and incidence than that in CD5KO mice (27). In our previous study, the CD5ΔCK2BD transgene was expressed at lower levels than the endogenous CD5 in CD5WT mice and was under the control of the CD2 promoter, with no dynamic regulation of expression by TCR signals. These significant distinctions could account for the differences between these studies. Nevertheless, the overall findings show that the CD5-signaling pathway is an important regulator for setting threshold for response to immunization and represents a critical signaling pathway in T-cells.
We determined that the CD5-CK2 signaling pathway was necessary for the differentiation of naïve CD4+ T-cells to Th17 or Th2 cells stimulated with cognate peptide under polarizing conditions. Similarly, the CNS of CD5-CK2 signaling deficient mice with EAE also contained fewer IL-17 expressing Th Cells but not Th1 than that is CD5WT mice. A recent report showed that CD5 is an important co-stimulator for Th17 differentiation; however the mechanism was not resolved (38). Another recent study made the elegant finding that CK2 is recruited to JAK1 and JAK2 and participates in the activation of STAT3 and STAT5 (37). STAT3 is required for Th17 differentiation and STAT5 is required for Th2 differentiation (42, 43). Additional studies still need to be performed to elucidate the precise CD5-CK2 signaling pathway that promotes Th2 and Th17 differentiation. However, one mechanism for promoting Th17 generation might be by regulating STAT1 activation (44). In fact we find that the loss of CD5-CK2 signaling pathway leads to hyperactivation of STAT1. We also observed enhanced activation of STAT3 in CD5-CK2 signaling deficient Th17 cells. This enhanced STAT3 activation may be a consequence of gain of STAT1 activity, which has been recently observed in humans (45). Although STAT3 phosphorylation in response to STAT3 activating cytokines such as IL-6 and IL-21 was augmented, the study showed that the enhanced STAT1 activity inhibited Th17 differentiation. This crossregulation by STAT1 on STAT3 induced genes may be one explanation for our finding that in the absence of CD5-CK2 signaling pathway Th17 differentiation was inhibited but greater levels of both IL-17A and IL-6 was produced by activated CD5ΔCK2BD T-cells. We also found that the differentiation of naïve CD4+ T-cells to Th1 cells stimulated with cognate peptide under non-polarizing conditions was diminished if the CD5-CK2 signaling pathway was ablated. This observation is consistent with decreased IFNγ-stimulated STAT1 activation when CK2 activity is inhibited (37). However, under polarizing conditions, the absence of CD5-CK2 signaling led to increased Th1 differentiation. This finding perhaps explains the enhanced passive EAE observed when encephalitogenic Th1 cells are generated from CD5ΔCK2BD donor mice. Overall our results now support a growing paradigm for a Th differentiation that involves cross-talk of cytokine-dependent JAK-STAT signaling and the CD5-CK2 activation pathway.
CK2 is an important participant in various stages of cell division, including cell-cycle entry (26). Therefore, our finding that CD4+ and CD8+ T-cells hypoproliferate in response to TCR-stimulation with anti-CD3 or cognate peptide was predictable. Since CK2 is also an important regulator of cell survival, and we showed in this and a previous study that ablation of CD5-dependented CK2 activation leads to enhanced AICD, it was important to determine if the decreased proliferation merely reflects enhanced death (27). Using the thymidine analog EdU we found that T-cells from the CD5ΔCK2BD mouse exhibited diminished cell-cycle entry, demonstrating that the hypoproliferation was independent of cell death.
The in vitro recall response following a short rest after primary stimulation is an effective assay for determining threshold for development of anergy in T-cells. We found that CD5-CK2 signaling-deficient T-cells proliferated more efficiently, with greater proportion entering into cell cycle than CD5WT T-cells. This in vitro result suggests that the CD5-CK2 signaling pathway plays an essential role in setting threshold for the development of anergy and thus recapitulates the enhanced severity of EAE with increasing dose of MOG35-55 peptide in CD5KO mice and CD5ΔCK2BD mice.
This study has revealed that the CD5-CK2 signaling pathway has a unique role in promoting differentiation of Th2 and Th17 cells and in regulating the threshold for development of anergy. We suggest that manipulation of the CD5-CK2 signaling pathway might be useful for the improvement of immune responses to vaccines, or conversely for attenuating dysregulated T-cells in autoimmunity.
Supplementary Material
Acknowledgements
We thank Simer Preet Singh, Tethia Mbana, and PingAr Yang for their technical assistance, the UAB Comprehensive Arthritis, Musculoskeletal and Autoimmunity Center Analytic and Preparative Cytometry Facility (P30 AR48311), the UAB Center for AIDS Research Flow Cytometry Core (P30 AI027767), Epitope Recognition Immunoreagent Core (ERIC) (P30 AR 48311), the UAB Comprehensive Cancer Center Transgenic Animal Shared Facility (P30 AR48311, P30 CA-13148), and the UAB Animal Resources Program (G20 RR025858 and G20 RR022807-01).
Footnotes
Supported by National Institutes of Health Grants T32 AR07450 and 5K12GM088010 to CS, NS064261 to PD, AI1076562 to CR, and from the National Multiple Sclerosis Society RG3891 to CR
Abbreviations used in this paper: 7-AAD, 7-Aminoactinomycin; AICD, activation-induced cell death; BAC, bacterial artificial chromosome; BMDC, bone marrow dendritic cells; DLN, draining lymph node; DN, double negative; DP, double positive; EAE, experimental autoimmune encephalomyelitis; EdU, 5-ethynyl-2′-deoxyuridine; MOG, myelin oligodendrocyte glycoprotein; SP, single positive.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Tarakhovsky A, Kanner SB, Hombach J, Ledbetter JA, Muller W, Killeen N, Rajewsky K. A role for CD5 in TCR-mediated signal transduction and thymocyte selection. Science. 1995;269:535–537. doi: 10.1126/science.7542801. [DOI] [PubMed] [Google Scholar]
- 2.Bikah G, Carey J, Ciallella JR, Tarakhovsky A, Bondada S. CD5-mediated negative regulation of antigen receptor-induced growth signals in B-1 B cells. Science. 1996;274:1906–1909. doi: 10.1126/science.274.5294.1906. [DOI] [PubMed] [Google Scholar]
- 3.Perez-Villar JJ, Whitney GS, Bowen MA, Hewgill DH, Aruffo AA, Kanner SB. CD5 negatively regulates the T-cell antigen receptor signal transduction pathway: involvement of SH2-containing phosphotyrosine phosphatase SHP-1. Mol. Cell Biol. 1999;19:2903–2912. doi: 10.1128/mcb.19.4.2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berland R, Wortis HH. Origins and functions of B-1 cells with notes on the role of CD5. Annu. Rev. Immunol. 2002;20:253–300. doi: 10.1146/annurev.immunol.20.100301.064833. [DOI] [PubMed] [Google Scholar]
- 5.Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28:639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 6.Rodamilans B, Munoz IG, Bragado-Nilsson E, Sarrias MR, Padilla O, Blanco FJ, Lozano F, Montoya G. Crystal structure of the third extracellular domain of CD5 reveals the fold of a group B scavenger cysteine-rich receptor domain. J. Biol. Chem. 2007282:12669–12677. doi: 10.1074/jbc.M611699200. [DOI] [PubMed] [Google Scholar]
- 7.Resnick D, Pearson A, Krieger M. The SRCR superfamily: a family reminiscent of the Ig superfamily. Trends Biochem. Sci. 1994;19:5–8. doi: 10.1016/0968-0004(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 8.Garza-Garcia A, Esposito D, Rieping W, Harris R, Briggs C, Brown MH, Driscoll PC. Three-dimensional solution structure and conformational plasticity of the N-terminal scavenger receptor cysteine-rich domain of human CD5. J. Mol. Biol. 2008;378:129–144. doi: 10.1016/j.jmb.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Soldevila G, Raman C, Lozano F. The immunomodulatory properties of the CD5 lymphocyte receptor in health and disease. Curr. Opin. Immunol. 2011;23:310–318. doi: 10.1016/j.coi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown MH, Lacey E. A ligand for CD5 is CD5. J. Immunol. 2010;185:6068–6074. doi: 10.4049/jimmunol.0903823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azzam HS, Grinberg A, Lui K, Shen H, Shores EW, Love PE. CD5 expression is developmentally regulated by T cell receptor (TCR) signals and TCR avidity. J. Exp. Med. 1998;188:2301–2311. doi: 10.1084/jem.188.12.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kassiotis G, Zamoyska R, Stockinger B. Involvement of avidity for major histocompatibility complex in homeostasis of naive and memory T cells. J. Exp. Med. 2003;197:1007–1016. doi: 10.1084/jem.20021812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berland R, Fiering S, Wortis HH. A conserved enhancer element differentially regulates developmental expression of CD5 in B and T cells. J. Immunol. 2010;185:7537–7543. doi: 10.4049/jimmunol.1002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azzam HS, DeJarnette JB, Huang K, Emmons R, Park CS, Sommers CL, El-Khoury D, Shores EW, Love PE. Fine tuning of TCR signaling by CD5. J. Immunol. 2001;166:5464–5472. doi: 10.4049/jimmunol.166.9.5464. [DOI] [PubMed] [Google Scholar]
- 15.Smith K, Seddon B, Purbhoo MA, Zamoyska R, Fisher AG, Merkenschlager M. Sensory adaptation in naive peripheral CD4 T cells. J. Exp. Med. 2001;194:1253–1261. doi: 10.1084/jem.194.9.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ling KW, van Hamburg JP, de Bruijn MJ, Kurek D, Dingjan GM, Hendriks RW. GATA3 controls the expression of CD5 and the T cell receptor during CD4 T cell lineage development. Eur. J. Immunol. 2007;37:1043–1052. doi: 10.1002/eji.200636485. [DOI] [PubMed] [Google Scholar]
- 17.Gary-Gouy H, Harriague J, Dalloul A, Donnadieu E, Bismuth G. CD5-negative regulation of B cell receptor signaling pathways originates from tyrosine residue Y429 outside an immunoreceptor tyrosine-based inhibitory motif. J. Immunol. 2002;168:232–239. doi: 10.4049/jimmunol.168.1.232. [DOI] [PubMed] [Google Scholar]
- 18.Stamou P, de Jersey J, Carmignac D, Mamalaki C, Kioussis D, Stockinger B. Chronic exposure to low levels of antigen in the periphery causes reversible functional impairment correlating with changes in CD5 levels in monoclonal CD8 T cells. J. Immunol. 2003;171:1278–1284. doi: 10.4049/jimmunol.171.3.1278. [DOI] [PubMed] [Google Scholar]
- 19.Ryan KR, McCue D, Anderton SM. Fas-mediated death and sensory adaptation limit the pathogenic potential of autoreactive T cells after strong antigenic stimulation. J. Leukoc. Biol. 2005;78:43–50. doi: 10.1189/jlb.0205059. [DOI] [PubMed] [Google Scholar]
- 20.Tabbekh M, Franciszkiewicz K, Haouas H, Lecluse Y, Benihoud K, Raman C, Mami-Chouaib F. Rescue of tumor-infiltrating lymphocytes from activation-induced cell death enhances the antitumor CTL response in CD5-deficient mice. J. Immunol. 2011;187:102–109. doi: 10.4049/jimmunol.1004145. [DOI] [PubMed] [Google Scholar]
- 21.Calvo J, Vilda JM, Places L, Simarro M, Padilla O, Andreu D, Campbell KS, Aussel C, Lozano F. Human CD5 signaling and constitutive phosphorylation of C-terminal serine residues by casein kinase II. J. Immunol. 1998;161:6022–6029. [PubMed] [Google Scholar]
- 22.Raman C, Kuo A, Deshane J, Litchfield DW, Kimberly RP. Regulation of casein kinase 2 by direct interaction with cell surface receptor CD5. J. Biol. Chem. 1998;273:19183–19189. doi: 10.1074/jbc.273.30.19183. [DOI] [PubMed] [Google Scholar]
- 23.Raman C, Kimberly RP. Differential CD5-dependent regulation of CD5-associated CK2 activity in mature and immature T cells: implication on TCR/CD3-mediated activation. J. Immunol. 1998;161:5817–5820. [PubMed] [Google Scholar]
- 24.Montenarh M. Cellular regulators of protein kinase CK2. Cell Tissue Res. 2010;342:139–146. doi: 10.1007/s00441-010-1068-3. [DOI] [PubMed] [Google Scholar]
- 25.Filhol O, Cochet C. Protein kinase CK2 in health and disease: Cellular functions of protein kinase CK2: a dynamic affair. Cell Mol. Life Sci. 2009;66:1830–1839. doi: 10.1007/s00018-009-9151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: From birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol. Life Sci. 2009;66:1817–1829. doi: 10.1007/s00018-009-9150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Axtell RC, Xu L, Barnum SR, Raman C. CD5-CK2 binding/activation-deficient mice are resistant to experimental autoimmune encephalomyelitis: protection is associated with diminished populations of IL-17-expressing T cells in the central nervous system. J. Immunol. 2006;177:8542–8549. doi: 10.4049/jimmunol.177.12.8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Axtell RC, Webb MS, Barnum SR, Raman C. Cutting edge: critical role for CD5 in experimental autoimmune encephalomyelitis: inhibition of engagement reverses disease in mice. J. Immunol. 2004;173:2928–2932. doi: 10.4049/jimmunol.173.5.2928. [DOI] [PubMed] [Google Scholar]
- 29.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- 31.Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos JG, Mair R, Christakos A, Kolkowitz I, Katz L, Killestein J, Polman CH, de Waal Malefyt R, Steinman L, Raman C. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nature Med. 2010;16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tarakhovsky A, Muller W, Rajewsky K. Lymphocyte populations and immune responses in CD5-deficient mice. Eur. J. Immunol. 1994;24:1678–1684. doi: 10.1002/eji.1830240733. [DOI] [PubMed] [Google Scholar]
- 33.Kersh GJ, Hedrick SM. Role of TCR specificity in CD4 versus CD8 lineage commitment. J. Immunol. 1995;154:1057–1068. [PubMed] [Google Scholar]
- 34.Ordonez-Rueda D, Lozano F, Sarukhan A, Raman C, Garcia-Zepeda EA, Soldevila G. Increased numbers of thymic and peripheral CD4+ CD25+Foxp3+ cells in the absence of CD5 signaling. Eur. J. Immunol. 2009;39:2233–2247. doi: 10.1002/eji.200839053. [DOI] [PubMed] [Google Scholar]
- 35.Dasu T, Qualls JE, Tuna H, Raman C, Cohen DA, Bondada S. CD5 plays an inhibitory role in the suppressive function of murine CD4(+) CD25(+) T(reg) cells. Immunol. Lett. 2008;119:103–113. doi: 10.1016/j.imlet.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brossard C, Semichon M, Trautmann A, Bismuth G. CD5 inhibits signaling at the immunological synapse without impairing its formation. J. Immunol. 2003;170:4623–4629. doi: 10.4049/jimmunol.170.9.4623. [DOI] [PubMed] [Google Scholar]
- 37.Zheng Y, Qin H, Frank SJ, Deng L, Litchfield DW, Tefferi A, Pardanani A, Lin FT, Li J, Sha B, Benveniste EN. A CK2-dependent mechanism for activation of the JAK-STAT signaling pathway. Blood. 2011;118:156–166. doi: 10.1182/blood-2010-01-266320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Wit J, Souwer Y, van Beelen AJ, de Groot R, Muller FJ, Klaasse Bos H, Jorritsma T, Kapsenberg ML, de Jong EC, van Ham SM. CD5 costimulates for stable human Th17 development by promoting IL-23R expression and sustained STAT3 activation. Blood. 2011;118:6107–6114. doi: 10.1182/blood-2011-05-352682. [DOI] [PubMed] [Google Scholar]
- 39.Jager A, Kuchroo VK. Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation. Scand. J. Immunol. 2010;72:173–184. doi: 10.1111/j.1365-3083.2010.02432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berland R, Wortis HH. An NFAT-dependent enhancer is necessary for anti-IgM-mediated induction of murine CD5 expression in primary splenic B cells. J. Immunol. 1998;161:277–285. [PubMed] [Google Scholar]
- 41.Yang Y, Contag CH, Felsher D, Shachaf CM, Cao Y, Herzenberg LA, Herzenberg LA, Tung JW. The E47 transcription factor negatively regulates CD5 expression during thymocyte development. Proc. Natl. Acad. Sci. U S A. 2004;101:3898–3902. doi: 10.1073/pnas.0308764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Z, Laurence A, O’Shea JJ. Signal transduction pathways and transcriptional regulation in the control of Th17 differentiation. Sem. Immunol. 2007;19:400–408. doi: 10.1016/j.smim.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paul WE. What determines Th2 differentiation, in vitro and in vivo? Immunol. Cell Biol. 2010;88:236–239. doi: 10.1038/icb.2010.2. [DOI] [PubMed] [Google Scholar]
- 44.Villarino AV, Gallo E, Abbas AK. STAT1-activating cytokines limit Th17 responses through both T-bet-dependent and -independent mechanisms. J. Immunol. 2010;185:6461–6471. doi: 10.4049/jimmunol.1001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, Flatot J, Migaud M, Chrabieh M, Kochetkov T, Bolze A, Borghesi A, Toulon A, Hiller J, Eyerich S, Eyerich K, Gulacsy V, Chernyshova L, Chernyshov V, Bondarenko A, Grimaldo RM, Blancas-Galicia L, Beas IM, Roesler J, Magdorf K, Engelhard D, Thumerelle C, Burgel PR, Hoernes M, Drexel B, Seger R, Kusuma T, Jansson AF, Sawalle-Belohradsky J, Belohradsky B, Jouanguy E, Bustamante J, Bue M, Karin N, Wildbaum G, Bodemer C, Lortholary O, Fischer A, Blanche S, Al-Muhsen S, Reichenbach J, Kobayashi M, Rosales FE, Lozano CT, Kilic SS, Oleastro M, Etzioni A, Traidl-Hoffmann C, Renner ED, Abel L, Picard C, Marodi L, Boisson-Dupuis S, Puel A, Casanova JL. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 2011;208:1635–1648. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.