

Abstract
The elastic restoring force of tissues must be able to operate over the very wide range of loading rates experienced by living organisms. It is surprising that even the fastest events involving animal muscle tissues do not surpass a few hundred hertz. We propose that this limit is set in part by the elastic dynamics of tethered proteins extending and relaxing under a changing load. Here we study the elastic dynamics of tethered proteins using a fast force spectrometer with sub-millisecond time resolution, combined with Brownian and Molecular Dynamics simulations. We show that the act of tethering a polypeptide to an object, an inseparable part of protein elasticity in vivo and in experimental setups, greatly reduces the attempt frequency with which the protein samples its free energy. Indeed, our data shows that a tethered polypeptide can traverse its free-energy landscape with a surprisingly low effective diffusion coefficient Deff ∼ 1,200 nm2/s. By contrast, our Molecular Dynamics simulations show that diffusion of an isolated protein under force occurs at Deff ∼ 108 nm2/s. This discrepancy is attributed to the drag force caused by the tethering object. From the physiological time scales of tissue elasticity, we calculate that tethered elastic proteins equilibrate in vivo with Deff ∼ 104–106 nm2/s which is two to four orders magnitude smaller than the values measured for untethered proteins in bulk.
Keywords: force spectroscopy, protein diffusion, viscoelasticity, single molecule
While the dynamics of proteins have been studied in detail using bulk techniques their behavior when tethered and placed under force is still poorly understood. Tissue elasticity in living organisms results from the extension and recoil of proteins that are tethered to rigid structures that move under force. For example, the giant protein titin responsible for the elasticity of muscle, is mechanically anchored to both the Z disk and the M-line of the half-sarcomere (1), responding to a mechanical perturbation by elastically changing the end-to-end length (2). Hence, it is likely that one of the limiting factors in the physiological activity of a muscle fiber is ultimately determined by the dynamics of the elastic recoil of an extended and tethered polypeptide. Hummingbirds flip their wings by up to 80 Hz, and insects up to 800 Hz (3). It is interesting here to consider that the fastest muscle-driven wing beat rates never surpass a kilohertz (3). Other types of physiological activities also appear to be capped at a similar time scale. For example, salamanders can shoot and fully extend their tongues out in less than 7 ms in order to catch flying prey (4). Throughout these physiological activities it is essential that the extending polypeptide remains elastic. If at any time the speed of the muscle motion exceeded the relaxation time of the polypeptide, the muscle will cease to be elastic, greatly affecting its ability to provide a restoring force, and to store energy. For example, a significant part of the turkey’s stride results from energy stored in the passive elasticity of muscle (5). In light of these observations, it is interesting to determine the extent to which these physiological limits can be explained in terms of the diffusional dynamics of the tethered extending polypeptides.
The elasticity of a polypeptide is typically modeled using either the worm-like-chain (WLC) (6, 7) or the freely-jointed-chain models of polymer elasticity (2), where the dynamics of equilibration is assumed to be instantaneous compared to the physiological time scales. This assumption can be justified given that bulk studies of untethered proteins using Förster resonance energy transfer (FRET) probes have conclusively measured the diffusional dynamics of single polypeptides and shown that freely diffusing proteins equilibrate with a very fast diffusion coefficient of D ∼ 107–108 nm2/s (8–11). It is therefore not surprising that some theoretical studies modeling the elastic behavior of proteins use the values of D measured from untethered protein in bulk (12, 13).
A recent attempt was made to measure the internal friction of extended proteins tethered to Atomic Force Microscope (AFM) cantilevers (14, 15). Although these experiments had a limited time resolution (> 10 ms), the authors made an effort to subtract the contributions made by the cantilever in order to get an estimate of the protein’s internal friction. These experiments estimated an internal friction at 100 pN, predicting values of D ∼ 103 nm2/s for the tethered protein, which is five orders of magnitude smaller than those measured by FRET experiments. Other authors have used force spectroscopy techniques to measure the elastic dynamics of tethered polypeptides resulting in values of D ∼ 102–104 nm2/s (16–18). As a consequence, some theoretical work of proteins under force used very low values of D ∼ 1,200 nm2/s (19, 20) to match the results obtained from force spectroscopy. Similarly, a value of approximately 104 nm2/s was needed to explain the dynamics of riboswitches tethered onto optical tweezers beads (21). Thus, the diffusional dynamics of a protein estimated from bulk techniques and from force spectroscopy techniques are very disparate.
It is surprising that these contradictory results have not been questioned before. In spite of its importance for understanding tissue elasticity, the effect of tethering on the internal dynamics of a protein is still not understood. We probe the elasticity of single extended polypeptides in the submillisecond range using a fast force-spectrometer, which provides a 20-fold improvement in time resolution, in combination with Brownian dynamics and Molecular Dynamics simulations (MD). MD simulations suggest that diffusion of an isolated protein under force occur on a very fast time scale (D ∼ 108 nm2/s), in agreement with FRET measurements. These simulations were done by applying force to a single atom in the terminal end of the protein and measuring the resulting changes in length (22). However, freely diffusing polypeptides measured in bulk are far from representing the physics of elastic proteins such as titin, which by definition must always be tethered to larger structures that move in conjunction. Here we demonstrate that the large slowing down of the diffusional dynamics of a protein as measured in AFM experiments is due to the drag force exerted by the object to which it is tethered. We therefore suggest that the slow dynamics probed in force spectroscopy is an inherent property of tethering a protein to a solid object, of widespread significance for understanding the dynamics of tissue elasticity.
Results and Discussion
The advent of force-clamp spectroscopy has made it possible to probe the dynamics of single proteins extending and relaxing under a well defined force (23). This approach now permits a direct study of the relaxation times of a polypeptide under a rapid load change. However, the published response times of the feedback in force spectrometers ranges from 5–10 ms for fast AFMs (23) and up to approximately 100 ms for optical tweezers instruments (24). Although the current instrumentation can comfortably measure the kinetics of protein unfolding and refolding, it is far too slow to probe the recoil dynamics of a stretched polypeptide under force. The main limitations have been the frequency response of the piezoelectric actuators and of the cantilevers used in the AFM instruments. Here we built a new force-clamp apparatus (Fig. 1A) that makes use of a fast piezoelectric actuator (> 300 kHz) and small Olympus cantilevers with resonant frequencies > 10 kHz. The set-up can settle the applied force within approximately 150 μs (see SI Text).
Fig. 1.

Experimental design for time resolved measurements of the recoil dynamics of an extended polypeptide. (A) Schematic diagram of the force-clamp apparatus. A single polypeptide is extended between a high-speed cantilever (Olympus biolever) and a piezoelectric actuator with a high resonant frequency (300 kHz, Physik Instrumente PL-055.30). A well-tuned PID amplifier adjusts the piezoelectric actuator so as to keep the force on the polypeptide at the set point value (Fsp). Under force clamp conditions, Fc = Fpol + FD, where Fc is the force applied by the cantilever, Fpol is the elastic force produced by the molecule and FD is the flow drag force (dashed lines) from the motion of the piezoelectric actuator. (B) Typical experimental trace of the end-to-end length corresponding to the force protocol on the lower box. The inset shows a staircase of ubiquitin unfolding events (stars) measured at 180 pN providing a strong fingerprint for a single polypeptide and measures its initial length. The extended polypeptide is then repeatedly cycled between 250 pN and 100 pN in order to collect many trajectories (7 < n < 30) until the molecule detaches. (C) Example of a recoil trajectory (blue) obtained by averaging eight consecutive recoil trajectories. The averaged force trace shows a step (250 pN down to 100 pN) with a half time of approximately 33 μs, which is more than 10 times shorter than the relaxation time constant of the polypeptide measured to be approximately 1.11 ms. After recoil, the corresponding extension (green trace) is much faster with a time constant of approximately 0.68 ms. The force traces are superimposable. (D) Potential of mean force of a 250 nm long polymer calculated at two pulling forces of 250 pN and 100 pN, using the WLC model of polymer elasticity (lower box). The figure highlights the physics of our experimental design. The polypeptide starts extended at 250 pN (1) and then it is abruptly relaxed to 100 pN (2) where it recoils down to its new minimum (3), then it is switched back to the high force (4) where it extends to the PMF minimum at 250 pN (1).
Our experimental design consists of first unfolding and extending a single ubiquitin polyprotein at a force of 180 pN, resulting in a series of step increases in length of approximately 20 nm (23). This staircase uniquely identifies the ubiquitin protein and also verifies that force is applied to a single polypeptide chain (Fig. 1B). Once a poly-ubiquitin protein is fully extended, the polypeptide was repeatedly cycled between 250 pN and 100 pN in order to measure the relaxation and extension dynamics (Fig. 1B). At these high forces, the polypeptide always remains highly extended. We averaged and merged all the repeating cycles of recoil and reextension such that we obtained a smoothed recoil (blue) and extension (green) trace (Fig. 1C; top traces). A similar procedure was used to average and merge the force steps over the same trace (Fig. 1C; bottom traces). The response time achieved by the new force clamp instrumentation is shown in Fig. 1C, where approximately 70% of the command force settles with a time constant of approximately 20 μs. The residual creep in the force settles more slowly with a time constant of 1.1 ms. The observed oscillations are due to the main resonant mode of the system, as the instrument approaches the limit of its operating bandwidth (approximately 7 kHz). Crucially, the polypeptide relaxes more slowly than the force with an exponential time course with a time constant of τ = 1.17 ms. The polypeptide reextends with a faster time course (τ = 0.68 ms), while the averaged force traces appear identical. Our protocol cycles an extended polypeptide between two well defined regions of the potential of mean force (PMF), calculated from the WLC model of polymer elasticity and the applied force (25, 26) (Fig. 1D; 1–4). The PMF slope is steeper at the higher force of 250 pN (4 to 1 in Fig. 1D), which causes a faster rate of extension when compared to the rate of recoil at 100 pN (2 to 3 in Fig. 1D). This asymmetry suggests that our recordings are reporting on the underlying PMF of the polypeptide, in the millisecond time scale, as also illustrated by our MD simulations (see below).
In Fig. 2A we fit single exponentials to recoil trajectories measured at 100 pN and extension trajectories measured at 250 pN. The polypeptides can be of very different contour lengths, Lc, due to the varying length of the polyproteins that are picked up by the AFM cantilever. Thus, we can measure the relaxation time constant, τ, as a function of the contour length of the polypeptide for both types of trajectories (Fig. 2B). The results show that the value of τ grows linearly with Lc, for both the recoil and extension trajectories, albeit with very different slopes (filled circles and triangles respectively; Fig. 2B). The linear dependency of τ vs. Lc can be explained by the scaling of the PMF with the contour length of the polypeptide, where the distance between minima at 250 pN and 100 pN (ΔL; inset of Fig. 2B), increases linearly with the contour length. These measurements clearly indicate that the extended polypeptide equilibrates at each new force by sampling its PMF.
Fig. 2.

Time constants and diffusion coefficients from extension and recoil traces of single polypeptides. (A) A series of four different recoil (upper box) and extension (lower box) trajectories measured from extended polypeptides of different contour lengths LC (as labeled; 113 nm, 263 nm, 344 nm, and 391 nm). The data were fit with a single exponential to measure the value of the relaxation time constant, τ, (dashed lines). (B) Relaxation (filled circles) and extension (filled triangles) time constants, τ, as a function of the contour length of the polypeptide Lc. The asymmetry of the potential shown in Fig. 1D, results into very different values of τ for the extension and recoil, as shown in Fig. 1B. The linear contour length dependency of τ is readily reproduced by Brownian dynamics simulations using the potential shown in Fig. 1D and the values of Deff measured from these data (open circles and triangles). The linear dependency of Deff on Lc results from that of the total travel (ΔL) on Lc (inset). (C) Histograms of values for Deff measured from the recoil (top box) and extension (bottom box) traces shown in Fig. 2A. (D and E) MD simulations of the end-to-end length as a function of time for ubiquitin maintained at a constant force of 250 pN (D; black curve); relaxation after force is quenched from 250 pN to 100 pN (D; grey curves) and averaged over five such trajectories (D; blue dots). Even if each of the time-origins of each trajectory differs, we make them coincide in the plot for clarity. (E) Normalized recoil (green curve) and extension (blue curve) averaged over five trajectories. The inset shows the PMF along the end-to-end distance at 100 pN (gray curve) and 250 pN (black curve). Both curves are shifted so that their minimum is 0. (F) Plot of the measured values of Deff (circles from recoils; triangles from extensions) as a function of the viscoelastic drag, μD, measured from each cantilever. The upper squares show the values of Dmax = kBT/μD, marking the upper limit of D measured for each cantilever. We attribute the difference between Deff and Dmax to the noninstantaneous force step of the force-clamp apparatus. The open circles show the values of Deff measured from BD simulations that include the time course of the force-step for each experiment. The inset portrays a simplified mechanical representation of the principal contributions to the forces of an extending polypeptide during these experiments. The Voigt model representing the polypeptide (kpol, μpol) is in parallel with the drag (μD) generated by the motion of the piezoelectric actuator, giving μeff = μpol + μD.
Here we examine whether simple Brownian dynamics based on the PMF predicted by the WLC model can account for the time scales of relaxation of the extended polypeptides as they adjust to a new force. The time course of the end-to-end length of a model polypeptide is calculated by solving the stochastic equation of motion:
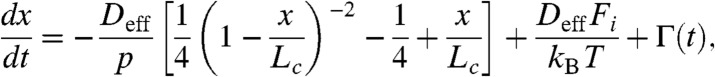 |
[1] |
where x(t) is the one-dimensional end-to-end reaction coordinate, Deff is the effective diffusion coefficient, p is the persistence length set to 0.4 nm, Fi is the applied force, kB is Boltzmann’s constant, T is the absolute temperature, and Γ(t) is white noise with 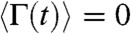 and correlation time
and correlation time 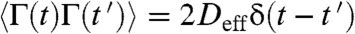 . We completed a set of simulations where the force was switched between 250 pN and 100 pN and the contour length Lc was varied from 100 nm to 500 nm. We fitted single exponentials to the simulated extensions and relaxations and measured the resulting time constants (open triangles and circles respectively; Fig. 2B). Simulations using a fixed value of Deff = 1,226 nm2/s (see below), matched the observed trends in our data, readily reproducing the difference in time constants between relaxation and extension and their linear growth with contour length (Fig. 2B). We also estimated the value of Deff directly from each relaxation trace with a simple high force approximated solution to Eq. 1 as
. We completed a set of simulations where the force was switched between 250 pN and 100 pN and the contour length Lc was varied from 100 nm to 500 nm. We fitted single exponentials to the simulated extensions and relaxations and measured the resulting time constants (open triangles and circles respectively; Fig. 2B). Simulations using a fixed value of Deff = 1,226 nm2/s (see below), matched the observed trends in our data, readily reproducing the difference in time constants between relaxation and extension and their linear growth with contour length (Fig. 2B). We also estimated the value of Deff directly from each relaxation trace with a simple high force approximated solution to Eq. 1 as
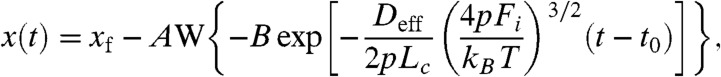 |
[2] |
where W is the Lambert W function which is defined to be the multivalued inverse of the function z = W(z)eW(z) (27), xf is the final protein length after the relaxation, A and B are fitting parameters, and t0 is the time at which the force was changed. Fits of Eq. 2 to the relaxation data shown in Fig. 2A gave a distribution of Deff values for both types of relaxation (Fig. 2C). The fitted values indicates a mean of 1,317 ± 197 nm2/s for the recoil and 1,135 ± 234 nm2/s for the extension traces, giving an average value of approximately 1,226 nm2/s, which was used in the simulations of Fig. 2B. Our observations support the view that a tethered polypeptide samples its PMF with a diffusion coefficient of Deff ∼ 103 nm2/s, which is in surprisingly close agreement with previous measurements using tethered proteins (14–18).
The measured values of Deff for tethered polypeptides are 4–5 orders of magnitude smaller than those measured from studies of polypeptides in bulk using FRET techniques (8–11). Indeed, if we use a value of Deff ∼ 108 nm2/s in our Brownian dynamics simulations, the resulting relaxation times are in the nanosecond time scale, far from those observed experimentally. Nevertheless, it could be argued that diffusion of an extended protein under force is a much slower process than the diffusion of unfolded proteins in solution as studied by FRET. To gain more insight into these discrepancies, we did all-atom MD simulations in explicit solvent of ubiquitin relaxation under force (see SI Text for details). When kept under a constant force of 250 pN, the protein is fully extended and its end-to-end distance fluctuates around an average value of L = 25.5 nm (Fig. 2D, black trace). At given moment along this trajectory, we quenched the force to a lower value of 100 pN while monitoring the relaxation of L (Fig. 2D, gray traces). These trajectories, starting from different initial configurations, were averaged to recover an average profile (Fig. 2D, blue points). It is clear that L relaxes on a nanosecond time scale, in stark contrast with the millisecond relaxation observed by our experiments, before it fluctuates around a new average value of L = 23.7 nm at 100 pN. Importantly, we checked that L does not further decay during a much longer simulation of 30 ns. Fitting these two lengths with the WLC predictions gives a contour length Lc = 28.4 nm and a persistence length p = 0.38 nm, in agreement with values usually measured experimentally (28, 29), suggesting that the full relaxation of the end-to-end distance was being reached.
We then explored the reverse scenario where the protein equilibrated at 100 pN is pulled at a constant force of 250 pN. The renormalized average of several such traces allowed direct comparison with the normalized recoil profile (Fig. 2E). The slower relaxation compared to the extension shows that AFM experiments and MD simulations are probing the same PMF. For both forces, the PMF extracted from the end-to-end distributions of equilibrium simulations further proves this point. The PMF at 250 pN is significantly stiffer than that at 100 pN, in agreement with the WLC predictions (Fig. 1D) and explains why the extension (i.e., motion along the 250 pN free energy surface) is faster than relaxation (motion along the 100 pN free-energy surface).
Independently from these extension/relaxation simulations, we estimated the diffusion coefficient along the end-to-end distance close to the minimum of each free-energy basin (see SI Text for details). The obtained values are Dpol = (5.3 ± 1.4)·108 nm2/s for L = 23.7 nm and F = 100 pN, and Dpol = (4.2 ± 1.1)·108 nm2/s for L = 25.5 nm and F = 250 pN. The obtained values are in close agreement with those measured in FRET experiments, but 5 to 6 orders of magnitude larger than those measured in our AFM experiments. Even if our method to estimate Dpol may suffer from uncertainties and other force fields may lead to some differences in the measured time scale, these factors cannot in any case explain the 6 order of magnitude difference with our experiments.
In order to understand these important discrepancies we must first consider the various contributions to the value of Deff in a protein placed under force. In the simulations, the force is directly applied to one extremity of the protein while the other is being fixed. From an experimental point of view, application of a mechanical force to a polypeptide always must involve the tethering of the polypeptide to a macroscopic object. Motion of the polypeptide is thus intimately tied to motion of the object. Any attempt of the polypeptide to move along the end-to-end length reaction coordinate of the PMF must also displace the object to which the protein is tethered. In our case, the polypeptide is tethered to an AFM cantilever and to a large gold covered surface. Thus, any change in the end-to-end length of the polypeptide must also result in drag generated between the moving surfaces (Fig. 1A). A simplified representation of these combined elements is shown in the inset of Fig. 2F, where the extended polypeptide is represented as a Voigt model with an elastic constant kpol and an internal friction of μpol. The polypeptide is placed in parallel with the drag caused by the motion of the tethered objects μD, so that friction forces are simply additive. We measured the drag force on a cantilever without attached proteins by simply touching the surface and then rapidly moving it away from the cantilever. These measurements gave a drag of 2.3 pN·ms/nm. We also measured the drag in each individual experiment after the protein detached from the cantilever and the surface started to rapidly move away (see SI Text). From these measurements we obtained an average value of 2.3 ± 0.5 pN·ms/nm. We plot the measured values of Deff as a function of μD for the individual experiments and observe that Deff has a weak dependence on the drag forces (Fig. 2F). If drag forces were the sole limiting factor affecting the motion of the polypeptide, we would expect the values of Deff to rise rapidly as drag is reduced, varying as 1/μD, which is not the case here. However, as it is apparent in Fig. 1C even in the best cases the force step applied to the polypeptide is far from approaching an ideal Heaviside step. Brownian dynamics simulations using the actual measured raw force traces readily reproduce our data. Fitting Eq. 2 to the resulting relaxation traces recovers the values of Deff measured experimentally (open circles; Fig. 2F).
Thus, it is possible to conclude that despite the considerable improvements in the time resolution of our instrumentation the surprisingly low values of Deff measured using force spectroscopy are still dominated by the drag resulting from the motion of the objects to which the polypeptide is tethered. While in our case these objects are the AFM cantilever and a gold-coated glass coverslide, such drag is an obligatory result of the elasticity of tethered polypeptides, a configuration that is widespread in biology. It is difficult to know the magnitude of the drag forces acting on the individual proteins of intact elastic tissues. However, if we consider the biological bandwidth of 800 Hz as the limit of protein elasticity, we can make some educated guesses of the biological drag coefficients and compare them with those measured in this work.
In order to estimate the drag in biological tissues, we examine the limits to the bandwidth of elastic tissues using a simple model as shown in Fig. 3. We use Brownian dynamics (with D = 1,300 nm2/s) to measure the effect of a small sinusoidal force (10 pN, peak to peak) on the extension dynamics of a prototypical tethered protein (Lc = 30 nm), as a function of the perturbation frequency, f (Fig. 3A). We analyze these traces using standard lock-in detection to measure the output signal components; the in-phase  and the out-of-phase
and the out-of-phase  amplitudes of the elastic response (triangles and circles in the lower left inset of Fig. 3B). Fitting with the corresponding real and imaginary parts of the mechanical impedance of a Kelvin-Voigt model (solid lines),
amplitudes of the elastic response (triangles and circles in the lower left inset of Fig. 3B). Fitting with the corresponding real and imaginary parts of the mechanical impedance of a Kelvin-Voigt model (solid lines), 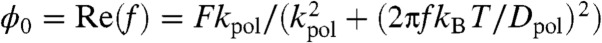 and
and  results in a perfect fit. The output power of the polypeptide oscillations calculated in dB as 10· log(Amp2(f)/Amp2(0)) is shown in Fig. 3B (triangles), where
results in a perfect fit. The output power of the polypeptide oscillations calculated in dB as 10· log(Amp2(f)/Amp2(0)) is shown in Fig. 3B (triangles), where 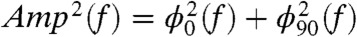 . Fitting the data with 10· log(1/(1 + (2πfkBT/kpolDpol)2)) returned values of Dpol = 1,313 nm2/s and kpol = 1.31 pN/nm (solid lines; Fig. 3B). Khatri, et al introduced an elegant expression for a frictional WLC elasticity constant kwlc = 4/Lc·(p/kBT)1/2·F3/2 (15). Substituting for the values of Lc, F, and p used in our simulations we calculate kpol = 1.31 pN/nm in good agreement with our Voigt model fits. However, a Voigt model serves only as a first approximation given that real polypeptides possess multiple relaxation times.
. Fitting the data with 10· log(1/(1 + (2πfkBT/kpolDpol)2)) returned values of Dpol = 1,313 nm2/s and kpol = 1.31 pN/nm (solid lines; Fig. 3B). Khatri, et al introduced an elegant expression for a frictional WLC elasticity constant kwlc = 4/Lc·(p/kBT)1/2·F3/2 (15). Substituting for the values of Lc, F, and p used in our simulations we calculate kpol = 1.31 pN/nm in good agreement with our Voigt model fits. However, a Voigt model serves only as a first approximation given that real polypeptides possess multiple relaxation times.
Fig. 3.

Bandwidth of an extended polypeptide. (A) Brownian dynamics computes the length of a 30 nm long polypeptide placed under a 10 pN sinusoidal load of varying frequency, f, using the measured value of Dpol = 1,300 nm2/s. The figure shows polypeptide responses at two different frequencies (25 Hz, and 250 Hz). (B) We use lock-in detection to measure the in-phase (ϕ0, triangles) and the out-of phase (ϕ90, circles) components of the elastic response of the polypeptide as a function of the load frequency, f. The solid lines (inset), correspond to the real (triangles) and imaginary (circles) parts of the mechanical impedance of a Kelvin-Voigt model (inset), fitted to the data. From ϕ0 and ϕ90 we calculate power as a function of frequency for the polypeptide (triangles). The solid line fits the Kelvin-Voigt model to the data returning values of Dpol = 1,313 nm2/s and kpol = 1.31 pN/nm. The arrow shows the -3 dB point, giving a bandwidth of 66 Hz. These measurements show that the elasticity of a polypeptide-based material is band-limited to a few hundred Hertz, in good agreement with broadly observed animal behavior.
From these considerations we can now establish the concept of elastic bandwidth. As shown in Fig. 3B, the power output follows the typical Lorentzian function of a low-pass filter. We define the corner frequency of the elastic response as being the frequency at which the power decreased by -3 dB, with respect to the power measured at zero frequency. For the simulations shown in Fig. 3A, the elastic bandwidth is 66 Hz. The half power frequency can also be straightforwardly calculated from the above considerations as;
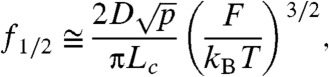 |
[3] |
where the elastic bandwidth of a polypeptide grows linearly with D. It is interesting to consider the consequences of Eq. 3. For example, simplifying the complex muscle titin protein to a polypeptide with Lc = 300 nm, p = 0.6 nm and at F = 4 pN (2) and assuming f1/2 ∼ 50 Hz (hummingbird) (3), we calculate a value of Deff ∼ 104 nm2/s. For the same titin protein operating at the limits of insect motion at f1/2 ∼ 800 Hz would give values of Deff ∼ 106 nm2/s. Our estimate of the speed limit of a tethered protein in vivo is much larger than that measured in our experimental system but still at least two orders of magnitude smaller than the values obtained by FRET and MD simulations. It is clear that the diffusional constants measured for polypeptides in solution do not apply to tethered polypeptides, and that extended polypeptides sample their PMF with values of Deff that are unique to each system and cannot be generalized.
Fig. 4 summarizes the consequences of tethering a polypeptide to an object in order to establish an elastic system. As described before, the basic PMF for a polypeptide contains an attractive potential that acts mostly at short extensions combined with the potential of the WLC which rapidly becomes dominant as the polypeptide extends (25, 26). At zero force, the PMF is downhill towards the collapsed state. Application of a constant force perturbation modifies the PMF creating an entropic barrier at relatively high extensions that can trap the collapsing polypeptide for some time, before it proceeds downhill towards the fully collapsed state (25, 26, 30). The dynamics of extended polypeptides responding to a change in the force will be determined, to a large extent, by the value of Deff. Molecules tethered to AFM cantilevers such as those used in our work will move along their PMF with values of Deff ∼ 103 nm2/s (1; Fig. 4), which readily explain our earlier observations (30, 31). It is interesting to consider that the value of Deff also affects barrier-crossing events. Indeed, transition state theory predicts that the attempt frequency for barrier-crossing events is given by Deffω0ωB/2π, where ω0 and ωB represent the curvatures of the PMF before the barrier and at the transition state (32). Thus, the kinetics of barrier crossing events that change the end-to-end length [e.g., (2); Fig. 4] will be greatly affected by tethering. An example of such barrier is the entropic barrier created by the force, which crosses over long distances along the PMF (Fig. 4) (30). In this case, the kinetics of barrier crossing will be greatly affected by drag and by the resulting Deff. By contrast, barrier crossing events that do not significantly change the end-to-end length of the molecule will be unaffected by the drag of the tethered objects. Indeed, the mechanical unfolding of ubiquitin which only has to extend by approximately 0.25 nm to cross the unfolding barrier (23) shows a very high attempt frequency (33).
Fig. 4.

Effect of tethering on the dynamics of an extending polypeptide. The act of tethering a polypeptide to an object (top inset) is an essential step in constructing an elastic system and introduces drag (μD; bottom inset) in parallel with the molecule (kpol, μpol). Polypeptide PMF’s calculated at F = 0 and F = 12 pN (25). Any motion of the polypeptide along its PMF will cause drag from the tethered objects, greatly affecting its kinetics both for simple diffusion (1) or for barrier crossing events that significantly change the end-to-end length of the molecule (2).
Our observations of the effects of tethering on the value of Deff apply to extended polypeptides where the internal friction of the molecule does not seem to play a significant role. However, under conditions where the internal friction of a polypeptide may become dominant, for example in collapsed molten globule states (34), the effects of tethering should be minimal. Furthermore, although the dynamics of a polypeptide takes place in more than one dimension, the effects of tether-drag will be mostly along the pulling coordinate. Other instruments such as optical tweezers and magnetic tweezers, which tether polypeptides to beads, will also show a substantial amount of drag giving different values of Deff for different bead sizes. Extended polypeptides probed by different instruments will show different dynamics depending of the values of Deff that result from tethering the polypeptide in each unique configuration. Thus, it is important that the drag is characterized in each instrumental configuration, and that its effects are understood and taken into account when analyzing force-spectroscopy data of extended molecules. Most importantly, the dynamics of a molecule whose kinetics is dominated by the drag of the objects to which it is tethered cannot be extrapolated to that of an untethered freely diffusing molecule in bulk. This conclusion is of wide significance for the understanding of the dynamics of numerous proteins that are tethered and under force in vivo.
Materials and Methods
Details about protein engineering and purification, experimental setup, and Brownian dynamics and MD simulations are provided in the SI Text.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Gerard Borst, for sharing his Igor application for the Lambert W function. I.P. acknowledges the Swiss National Science Foundation for a postdoctoral research grant. S.G-M. thanks the Fundación Caja Madrid and IberCaja for financial support. This work was supported by the National Institutes of Health (NIH) (NIH-HL66030 and NIH-HL61228 to J.M.F.; NIH-GM4330 to B.J.B.) and in part by National Science Foundation (NSF) through TeraGrid resources provided by National Center for Supercomputing Applications and ABE (MCA08X002 to B.J.B.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1212167109/-/DCSupplemental.
References
- 1.Linke WA. Sense and stretchability: The role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 2008;77:637–648. doi: 10.1016/j.cardiores.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 2.Li HB, et al. Reverse engineering of the giant muscle protein titin. Nature. 2002;418:998–1002. doi: 10.1038/nature00938. [DOI] [PubMed] [Google Scholar]
- 3.Greenewalt CH. The wings of insects and birds as mechanical oscillators. Proc Am Philos Soc. 1960;104:605–611. [Google Scholar]
- 4.Deban SM, Richardson JC. Cold-blooded snipers: Thermal independence of ballistic tongue projection in the salamander hydromantes platycephalus. J Exp Zool A. 2011;315A:618–630. doi: 10.1002/jez.708. [DOI] [PubMed] [Google Scholar]
- 5.Roberts TJ, Marsh RL, Weyand PG, Taylor CR. Muscular force in running turkeys: The economy of minimizing work. Science. 1997;275:1113–1115. doi: 10.1126/science.275.5303.1113. [DOI] [PubMed] [Google Scholar]
- 6.Bustamante C, Marko JF, Siggia ED, Smith S. Entropic elasticity of Lambda-Phage DNA. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 7.Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 8.Haas E, Katchalskikatzir E, Steinberg IZ. Brownian-motion of ends of oligopeptide chains in solution as estimated by energy-transfer between chain ends. Biopolymers. 1978;17:11–31. [Google Scholar]
- 9.Hagen SJ, Hofrichter J, Szabo A, Eaton WA. Diffusion-limited contact formation in unfolded cytochrome c: Estimating the maximum rate of protein folding. Proc Natl Acad Sci USA. 1996;93:11615–11617. doi: 10.1073/pnas.93.21.11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lapidus LJ, Steinbach PJ, Eaton WA, Szabo A, Hofrichter J. Effects of chain stiffness on the dynamics of loop formation in polypeptides. Appendix: Testing a 1-dimensional diffusion model for peptide dynamics. J Phys Chem B. 2002;106:11628–11640. [Google Scholar]
- 11.Moglich A, Joder K, Kiefhaber T. End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc Natl Acad Sci USA. 2006;103:12394–12399. doi: 10.1073/pnas.0604748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hummer G, Szabo A. Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc Natl Acad Sci USA. 2001;98:3658–3661. doi: 10.1073/pnas.071034098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dudko OK, Graham TGW, Best RB. Locating the barrier for folding of single molecules under an external force. Phys Rev Lett. 2011;107:208301. doi: 10.1103/PhysRevLett.107.208301. [DOI] [PubMed] [Google Scholar]
- 14.Kawakami M, Byrne K, Brockwell DJ, Radford SE, Smith DA. Viscoelastic study of the mechanical unfolding of a protein by AFM. Biophys J. 2006;91:L16–L18. doi: 10.1529/biophysj.106.085019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khatri BS, et al. Internal friction of single polypeptide chains at high stretch. Faraday Discuss. 2008;139:35–51. doi: 10.1039/b716418c. [DOI] [PubMed] [Google Scholar]
- 16.Calderon CR, Harris NC, Kiang CH, Cox DD. Analyzing single-molecule manipulation experiments. J Mol Recognit. 2009;22:356–362. doi: 10.1002/jmr.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calderon CP, Harris NC, Kiang CH, Cox DD. Quantifying multiscale noise sources in single-molecule time series. J Phys Chem B. 2009;113:138–148. doi: 10.1021/jp807908c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo SL, Li N, Lad N, Desai S, Akhremitchev BB. Distributions of parameters and features of multiple bond ruptures in force spectroscopy by atomic force microscopy. J Phys Chem C. 2010;114:8755–8765. [Google Scholar]
- 19.Minh DDL. Free-energy reconstruction from experiments performed under different biasing programs. Phys Rev E. 2006;74:061120. doi: 10.1103/PhysRevE.74.061120. [DOI] [PubMed] [Google Scholar]
- 20.Minh DDL. Multidimensional potentials of mean force from biased experiments along a single coordinate. J Phys Chem B. 2007;111:4137–4140. doi: 10.1021/jp068656n. [DOI] [PubMed] [Google Scholar]
- 21.Lin JC, Thirumalai D. Relative stability of helices determines the folding landscape of adenine riboswitch aptamers. J Am Chem Soc. 2008;130:14080–14081. doi: 10.1021/ja8063638. [DOI] [PubMed] [Google Scholar]
- 22.Sotomayor M, Schulten K. Single-molecule experiments in vitro and in silico. Science. 2007;316:1144–1148. doi: 10.1126/science.1137591. [DOI] [PubMed] [Google Scholar]
- 23.Schlierf M, Li HB, Fernandez JM. The unfolding kinetics of ubiquitin captured with single-molecule force-clamp techniques. Proc Natl Acad Sci USA. 2004;101:7299–7304. doi: 10.1073/pnas.0400033101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li PTX, Collin D, Smith SB, Bustamante C, Tinoco I. Probing the mechanical folding kinetics of TAR RNA by hopping, force-jump, and force-ramp methods. Biophys J. 2006;90:250–260. doi: 10.1529/biophysj.105.068049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berkovich R, Garcia-Manyes S, Urbakh M, Klafter J, Fernandez JM. Collapse dynamics of single proteins extended by force. Biophys J. 2010;98:2692–2701. doi: 10.1016/j.bpj.2010.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyeon CB, Morrison G, Pincus DL, Thirumalai D. Refolding dynamics of stretched biopolymers upon force quench. Proc Natl Acad Sci USA. 2009;106:20288–20293. doi: 10.1073/pnas.0905764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corless RM, Gonnet GH, Hare DEG, Jeffrey DJ, Knuth DE. Onthe Lambert W function. Adv Comput Math. 1996;5:329–359. [Google Scholar]
- 28.Li HB, et al. Multiple conformations of PEVK proteins detected by single-molecule techniques. Proc Natl Acad Sci USA. 2001;98:10682–10686. doi: 10.1073/pnas.191189098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walther KA, et al. Signatures of hydrophobic collapse in extended proteins captured with force spectroscopy. Proc Natl Acad Sci USA. 2007;104:7916–7921. doi: 10.1073/pnas.0702179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berkovich R, Garcia-Manyes S, Klafter J, Urbakh M, Fernandez JM. Hopping around an entropic barrier created by force. Biochem Biophys Res Commun. 2010;403:133–137. doi: 10.1016/j.bbrc.2010.10.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez JM, Li HB. Force-clamp spectroscopy monitors the folding trajectory of a single protein. Science. 2004;303:1674–1678. doi: 10.1126/science.1092497. [DOI] [PubMed] [Google Scholar]
- 32.Kramers HA. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica. 1940;7:284–304. [Google Scholar]
- 33.Popa I, Fernandez JM, Garcia-Manyes S. Direct quantification of the attempt frequency determining the mechanical unfolding of ubiquitin protein. J Biol Chem. 2011;286:31072–31079. doi: 10.1074/jbc.M111.264093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ptitsyn OB. Molten globule and protein folding. Adv Protein Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.