

Abstract
The ability of cells to pack, use and duplicate DNA remains one of the most fascinating questions in biology. To understand DNA organization and dynamics, it is important to consider the physical and topological constraints acting on it. In the eukaryotic cell nucleus, DNA is organized by proteins acting as spools on which DNA can be wrapped. These proteins can subsequently interact and form a structure called the chromatin fibre. Using a simple geometric model, we propose a general method for computing topological properties (twist, writhe and linking number) of the DNA embedded in those fibres. The relevance of the method is reviewed through the analysis of magnetic tweezers single molecule experiments that revealed unexpected properties of the chromatin fibre. Possible biological implications of these results are discussed.
Keywords: chromatin, topology, DNA
1. DNA supercoiling and transcription
According to the ‘central dogma’ of molecular biology, the DNA double helix codes for the sequence of all proteins in the cell. However, in eukaryotic cells, the coding sequences can vary from 90 per cent (in yeast) to less than 1.5 per cent of the DNA sequence (in higher eukaryotes, such as humans). A role of non-coding DNA in the assembling of the hierarchical architecture of the genome has been, therefore, proposed. This architecture must, at a time, compact DNA in the nucleus and let it be accessible when requested by the cell functioning. Besides the geometrical puzzling that these questions imply, the architecture of DNA and its dynamical modifications have to deal with the mechanical response of DNA that behaves as a rather rigid screw.
In particular, gene transcription imposes strong mechanical constraints. The crucial step of protein synthesis, the transcription elongation, is performed by a dedicated enzyme, the RNA-polymerase (RNAP). RNAP opens locally the double helix, reads it and makes a copy of the coding DNA template onto a RNA stretch. The transcription activation requires the formation of a large protein complex, consisting of up to 60 different proteins. Owing to the large dimension of the transcription complex, it is very unlikely that the RNAP rotates around DNA during the elongation process [1]: the DNA has, therefore, to rotate inside the RNAP. This process leads to topological and mechanical constraints upstream and downstream of the transcription site. Indeed, as the elongation complex progresses along the genomic sequence, the DNA double helix in front of it becomes overwound (positively supercoiled), whereas the DNA behind it becomes underwound (negatively supercoiled). This is the so-called twin-supercoiled-domain (TSD) model, first introduced by Liu & Wang [2] and extensively acknowledged since (for a review, see Lavelle [3]). In this paper, we will deal with the problem of building the appropriate theoretical frame where mechanical and topological constraints acting on DNA can be accounted for, in the perspective of studying the implications of these constraints for different biological processes.
2. DNA topology and White–Fuller theorem
Topology of DNA is a long-standing problem, since Vinograd et al. [4] introduced in 1965 the idea that the conformation of DNA both in eukaryotes and prokaryotes is related to topological quantities. They showed that circular DNA chromosomes isolated from small viruses may be in a highly compact and wrapped conformation. A proper characterization of the different conformations, or ‘topoisomers’ (as those displayed in figure 1) was then introduced soon after by Fuller with the mathematical definition of the three relevant topological quantities, namely the twist Tw, the writhe Wr and the linking number Lk [6,7].
Figure 1.
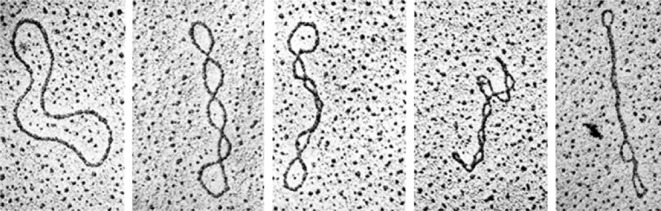
Supercoiling: different topological states (topoisomers) of circular DNA molecules. Reproduced with permission from Kornberg [5].
Since then, a rather large amount of work has been done to study the relationship between twist and writhe for closed and open curves, especially in relation to torsionally stressed DNA including in the presence of binding proteins [8–12]. We largely refer to these works in the following derivations.
When one extremity of a ribbon is rotated around the local tangent to its axis, the ribbon modifies its conformation by splitting up the imposed rotation into two complementary contributions: its twist and its writhe.
2.1. Twist
The ribbon twist is equal to the excess of torsion induced around its own axis. If r(s) defines the ribbon axis trajectory, with s the arc-length along the DNA path, then we can define the tangent, normal and binormal vectors, respectively, by
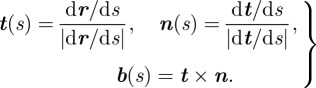 |
2.1 |
The normal vector then leads to the definition of an intrinsic local reference frame following the ribbon axis, called the Frenet reference frame, whose normal coincides with the principal normal vector to the curve. The twist of a ribbon can then be decomposed into the sum of the twist of the Frenet frame plus the twist of the ribbon relative to the Frenet frame [8]. The twist of a straight DNA is simply given by the excess number of turns of the double helix with respect to its relaxed state. For a bent DNA, the calculation is more delicate. We will explicitly express this term for a special case of interest in §5.
2.2. Writhe
The writhe of a closed ribbon of length L depends instead on the curve followed by the ribbon axis in the three-dimensional space. It measures the coiling of the axis, and its computation is quite complicated. It is generally evaluated through a double integral according to a method based on Gauss theorem, leading to the formula
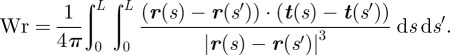 |
2.2 |
The Gauss integral can be efficiently integrated numerically. Nevertheless, an alternative method has been introduced by Fuller [7], which has the advantage of a direct geometrical interpretation and of a more straightforward generalization to open curves [11].
Consider again the unit vector tangent to the closed ribbon, t(s). The tangent vector vertex, T(s), lies on the unit sphere. As s varies from 0 to L, it describes a closed curve on this sphere, called tangent indicatrix [9].
Fuller's first theorem states that the writhe of the curve r(s) can be calculated from the signed area 𝒜 enclosed by the tangent indicatrix T(s), namely Wr = 𝒜/2π − 1 (mod 2). Under a few hypotheses, Fuller's second theorem [7] permits one to get rid of this congruence when one calculates the writhe difference between two closed curves, r1(s) and r2(s). The hypotheses of the theorem impose that one of the curves can be obtained from the other by a continuous deformation in space such that (i) at any point along the curve and at any moment during the transformation, the tangent vector should assume opposite direction with respect to any other intermediate state, (ii) the curve is non-self-intersecting all along the transformation, and (iii) the tangent to the curve varies continuously. Provided these conditions, it is possible to show that the difference in writhe between the two curves corresponds to the area 𝒮 swept out by the unique shortest geodesic arc from the running point T1(s) to the running point T2(s) on the unit sphere, divided by 2π, i.e. 𝒮/2π. This allows one, in particular, to calculate the writhe of a DNA chain with respect to a referring state, that could be a straight DNA, if the deformation satisfies the previous hypotheses.
2.3. Linking number
For a filament whose ends are oriented in fixed directions, if one of them is rotated around its tangent, then the sum of the twist and the writhe variations will always correspond to the number of turns applied. This property is formalized by the famous White–Fuller theorem
| 2.3 |
where Lk is the filament linking number. For a closed ribbon, or a ribbon with clamped ends, the linking number is, therefore, a topological invariant.
3. Magnetic tweezers: wrapping DNA
The effect of mechanical constraints on the topology of a single DNA molecule can be nowadays experimentally explored, thanks to magnetic tweezers manipulation. This is a single-molecule technique that permits controlled application of force and torsion on a biomolecule of interest. In a typical set-up, one extremity of a single DNA molecule is attached to a glass surface, the other extremity to a micrometre-sized paramagnetic bead. Permanent magnets induce a magnetic moment in the magnetic bead, which experiences a force owing to the magnetic field gradient of the magnets. Moreover, the magnetic bead can be rotated to a controlled number of turns by rotating the magnets (figure 2).
Figure 2.
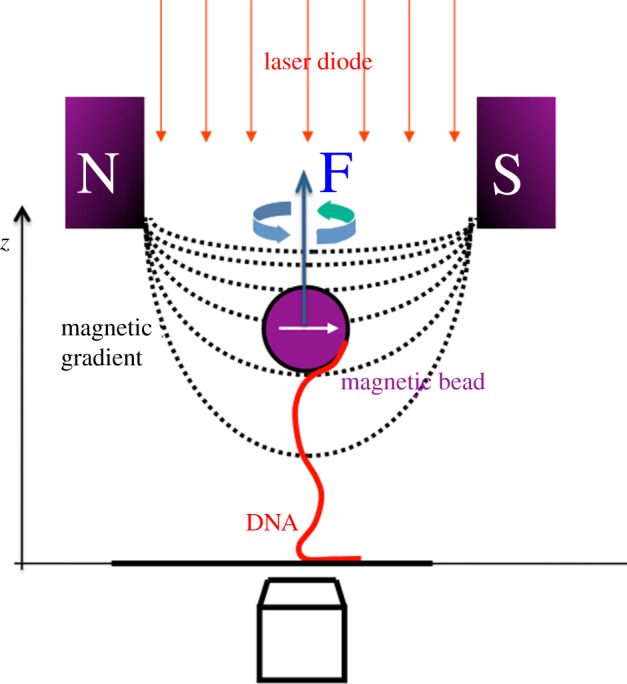
Schematic of the magnetic tweezers set-up. See the main text for details.
The effect of torsion on a DNA molecule is not very different from what we can experience in everyday life by twisting a rope. At high pulling force, the application of a torque twists the molecule on its axis. If the pulling force is now released, while maintaining constant the bead rotation, the twist of the molecule becomes unstable and the molecule tends to writhe on itself, forming typical super-coiled structures called plectonemes. A plectoneme is a loop of helices twisted together as we are used to experience with electric cables.
The instability that converts twist to writhe is the result of a competition between bending and twisting energies, the latter becoming too large for large torsion. The transition to plectonemes arises at a critical torque [13], and plectoneme formation absorbs a significant portion of the mechanical stress exerted on the DNA. Since in plectonemes, the DNA wraps on itself, their formation shortens the molecule, and is therefore detectable by measuring, at constant force, the height of the magnetic bead as a function of the number of turns n applied, i.e. of the linking number variation ΔLk = n. The rotation response of DNA has been studied in detail [13–18] and we now dispose of good fitting functions that may be adjusted to the rotation–extension curves very precisely.
Magnetic tweezers have also given spectacular results on the mechanics of DNA [19–21], revealing unexpected structural changes, such as the existence of over-stretched S-DNA [22] or over-twisted P-DNA [23], and allowing the study of important conformational transitions such as the formation of cruciforms [24]. While the biological relevance of cruciforms is known, the existence in vivo of the two unexpected altered DNA forms has still to be demonstrated. However, it seems plausible that they can appear, at least locally, owing to DNA–protein interaction. The second field where optical and magnetic tweezers have given very interesting results is precisely the study on the interaction of DNA with enzymes. These studies include the action of topoisomerases [25,26], of RNAP during transcription initiation [27,28], or of remodelling enzymes [29]. For a more exhaustive review on tweezers set-up and applications, see Lavelle et al. [30].
4. DNA in the eukaryotic cell: chromatin and chromatin fibre
Dealing with metres of DNA in a 10 nm nucleus is, evidently, an overwhelming problem. In the nucleus of eukaryotic cells, the genetic material is organized into a complex DNA–protein assembly, called chromatin. The nucleosome (figure 3) is the fundamental unit of chromatin. It is formed by a spool of proteins (histones) on which 147 DNA base pairs (bp) are wrapped in about 1.65 turns. Nucleosomes are more or less regularly spaced on the genome, with a repeat length Nr of the order of 200 bp (but rather variable depending on organisms, tissues, genomic regions and approximately in the range 155 to 240 bp). Nucleosome core particles (NCPs) form then a bead on a string array, with short stretches of bare DNA—the linker-DNA—connecting adjacent nucleosomes.
Figure 3.
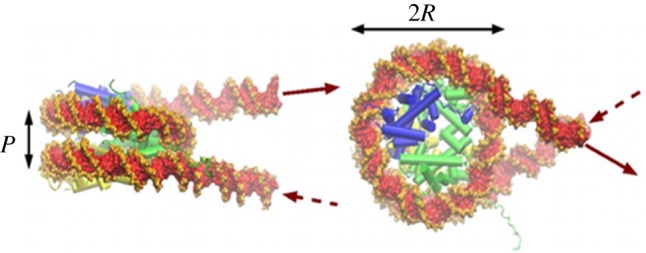
Structure of the nucleosome. In green, the histone tetramer (H3-H4)2, in blue and yellow, the two histone dimers (H2A-H2B). The figure has been obtained starting from the crystallographic structure [31], to which were added two short entering and exiting DNA segments (courtesy of Richard Lavery).
The higher levels of organization remain largely speculative. However, it is generally accepted that attractive interactions between nucleosomes [32] fold this array into a fibre about 30 nm in diameter (figure 4).
Figure 4.
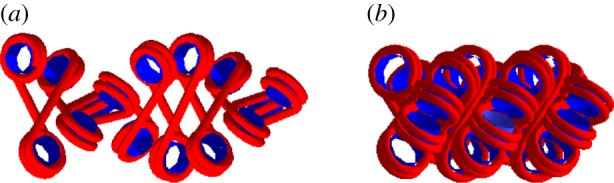
One of the many possible models of chromatin fibre, in a more condensed (a) and a less condensed (b) configuration. The regular spacing between nucleosomes allows for the formation of a compact structure, probably stabilized by stacking nucleosome–nucleosome interactions.
Although fibres have been observed in vitro, their existence in vivo remains an open—and highly debated—question [33,34] since most attempts to visualize these fibres in vivo have failed so far, except for some rare images obtained in nuclei which are transcriptionally largely inactive (chicken erythrocytes and echinoderm sperm) [35,36].
Since the entry–exit DNA should be phased with respect to the protein core, the precise orientation of a nucleosome with respect to the previous one is largely determined by the linker-DNA length and by the torsional constraint applied to the fibre. The shortness of the linker DNA is indeed hardly compatible with its bending, so that one can reasonably assume the linkers as straight (except, of course, in the case of additional protein binding). This assumption led Woodcock et al. [37] to propose a simple, geometrical model for the chromatin structure at the 30 nm scale, called the two-angle model, which describes the fibre by the following three parameters: the entry–exit angle α, the rotational angle β and the linker length (figure 5).
Figure 5.
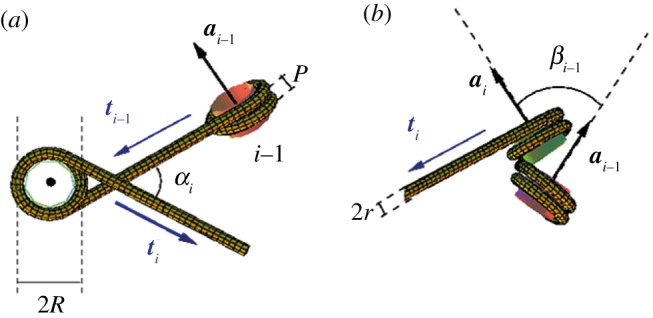
Schematic of the DNA winding pattern along two neighbouring nucleosomes in the two-angle model. (a) View down the NCP axis ai. (b) View down the linker direction ti−1. The angle α is the dihedral angle (ti−1, ai, ti) and β the dihedral angle (ai−1, ti−1, ai) standing for the twist (modulo 2π) of the DNA linker.
We believe that the physical properties of the chromatin fibre may govern the structural changes necessary for the functioning and dynamics of chromatin. Since the chromatin fibre is, in vivo, organized in loops with fixed end, the linking number is generally a conserved quantity. We therefore proposed a method to calculate the twist, writhe and linking number of the embedded DNA [38], based on the work of Fuller [6], for any conformation of the fibre obtained using the canonical two-angle model of Woodcock et al. [37]. The possibility to calculate these topological quantities provides an important element for modelling the dynamic behaviour of fibres. Following this idea, we proposed a possible mechanism for the condensation of chromatin during mitosis [39].
5. Topology into the fibre
5.1. Reference crystallographic relaxed state
The number nNCP of bp in contact with the protein core, and therefore the angle α, may vary as a consequence of a partial unwrapping of the DNA. A reference value for α is obtained from the X-ray crystallographic data, giving a superhelix path with a radius R = 4.18 nm and pitch P = 2.39 nm [40]. The crystallographic superhelix corresponds thus to 1.65 turns, this covering nNCP0 = 126 bp and, consequently, the reference angle α0 amounts to α0 = 1.65 × 2π − 3π = 0.3π or 0.94 rad (54°).
The angle β depends on the degree of overtwist of the linker, but also on the linker length itself and depends on α in a non-trivial way: the position of the DNA grooves at the exiting point of the previous NCP introduces indeed a phase that has to be taken into account. It is useful to introduce a reference value β0, corresponding to the crystallographic NCP structure with relaxed (untwisted) linkers, which amounts to β0 = 2π(Nr − nNCP0)/h0, with h0 the number of bp per helical turn in a straight and relaxed DNA in the classical B form.
5.2. Twist in the fibre
In the two-angle model, the repeat is composed of two parts: a superhelical DNA with solenoidal shape in the NCP, followed by a straight linker DNA, eventually twisted. The left-handed solenoid described by the DNA axis is represented by the parametric curve r(σ) = (−R cos(2πσ/P), R sin(2πσ/P), σ), with σ the arc-length along the NCP axis, related to the arc-length along the solenoidal curve, s, by the relation ds = |dr/dσ| dσ.
Since the twist is an extensive quantity, its calculation for DNA in a fibre can be done at the level of a single nucleosome as a function of angles α and β following the derivation of White & Bauer [8].
The overall twist per nucleosome tw can, therefore, be expressed as the sum of two twist contributions: (i) from the linker-DNA, twlinker = (Nr − nNCP)/h and (ii) from the NCP, twNCP = nNCP/h′ − τ(3π + α)/2π, where we indicate with h (respectively, h′) the number of bp in a linker (respectively, NCP) DNA period. The last term in twNCP is the twist contribution owing to the bending of the DNA axis. The tortuosity τ corresponds to the absolute value of the total torsion T obtained by integrating the local torsion τL = −2πP/(4π2R2 + P2) over one helical turn of the DNA axis path in the NCP [8] and finally amounts to 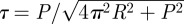 .
.
It is now possible to introduce an explicit twist dependence on α and β, by calculating tw and β as a function of the number of wrapped bp nNCP, then recombining the two expressions. Moreover, in general biological applications it is usual to refer to a relative DNA twist, i.e. to calculate the difference between the total twist tw and a reference twist tw0, which corresponds to the twist of a straight and relaxed B-DNA of the same length. After subtraction of this term, the final expression obtained is
| 5.1 |
Here 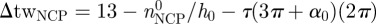 is the difference in twist between the wrapped DNA and a corresponding straight, relaxed DNA stretch of the same length, as measured at α = α0.
is the difference in twist between the wrapped DNA and a corresponding straight, relaxed DNA stretch of the same length, as measured at α = α0.
5.3. Writhe in the fibre
The writhe is known to be non-extensive and, as shown by Starostin [11], the total writhe of a curve is given by the sum of the writhes of its parts plus the surface of the spherical polygon composed by the set of geodesics closing each part.
However, assuming the chromatin fibre as a regular structure composed of identical repetitive nucleosomes, it is possible to calculate its writhe from the writhe of the individual units, up to a boundary term. In the case of a chromatin fibre, the DNA indicatrix T(s) can indeed be naturally divided into N parts, namely the N nucleosomes, in correspondence to ti = t(si), the direction of the (straight) linker i. Denote by Ti the vertex point on the unit sphere that corresponds to ti. The total writhe of the DNA in the fibre is then given, accordingly to Starostin [11] and Barbi et al. [38], by the following addition rule:
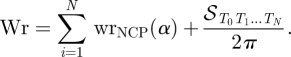 |
5.2 |
Here, wrNCP(α) is the writhe of the ith nucleosome whose indicatrix had been closed by a geodesic, while 𝒮T0T1 … TN/2π is the area enclosed in the spherical polygon of vertices T0, T1, … ,TN, and represents the writhe of the ‘carrying’ structure formed by the sequence of linkers, or linker skeleton.
This second term 𝒮T0T1
… TN in equation (5.2) is the non-extensive contribution, given by the signed area of the spherical polygon connecting all points Ti that correspond to the linker directions. Nevertheless, it should be very useful, at this point, to ‘remove’ the non-extensivity by an appropriate partition of this area. Once chosen an arbitrary point C on the unit sphere, the area 𝒮T0T1
… TN can also be written as 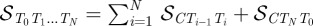 . This partition allows a natural decomposition of the writhe into single nucleosome contributions: up to a single boundary term 𝒮CTNT0/2π, the writhe can be expressed as the sum
. This partition allows a natural decomposition of the writhe into single nucleosome contributions: up to a single boundary term 𝒮CTNT0/2π, the writhe can be expressed as the sum 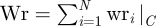 , where we define the writhe wri | C of nucleosome i
with respect to point
C as
, where we define the writhe wri | C of nucleosome i
with respect to point
C as
| 5.3 |
Moreover, there exists a special point, C = F, defined by the director of the fibre axis, for which 𝒮FTi−1Ti = 𝒮(α, β) is the same for all the nucleosomes (figure 6). For this particular choice, we can define an effective writhe per nucleosome, wr(α, β), such that
| 5.4 |
Figure 6.
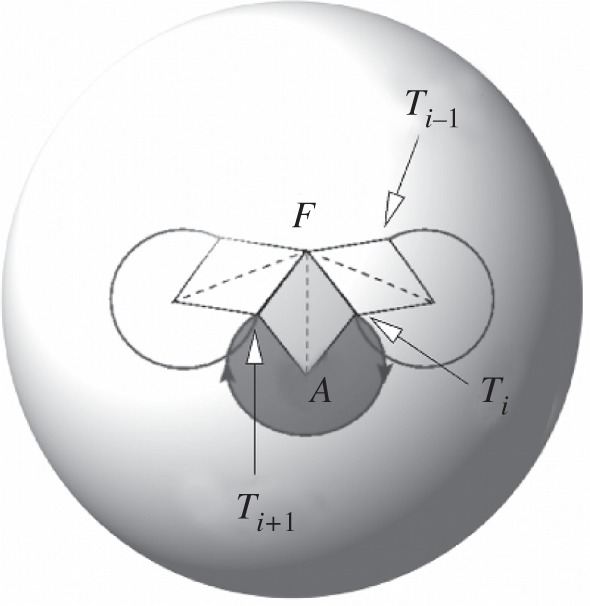
A schematic of the writhe calculation for a nucleosome in a regular fibre. Scales and length have been arbitrarily chosen for the sake of clarity. The curve represents the tangent indicatrix of DNA in the fibre. Point Ti represents the vertex of the ith linker tangent vector. The circular part corresponds to the indicatrix of the DNA wrapping around the nucleosome. The central point is the direction of the fibre axis. The surface 𝒮(α, β) of equation (5.4) corresponds to the spherical triangle FTiTi+1.
5.4. Linking number per nucleosome
The particular choice of the reference point F leads to an expression for the writhe which is not only additive, but also provides identical contributions for each nucleosome, thus supplying for a consistent definition of a writhe per nucleosome wr(α, β), independent of the fibre length N. Summing up the twist and writhe contributions, we have finally the following expression for the DNA linking number excess in the fibre ΔLk:
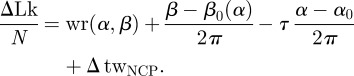 |
5.5 |
5.5. Possible extensions to irregular fibres
Up to now, we have considered perfectly regular fibres. In this case, the fibre is always straight, its writhe is zero, and the DNA linking number calculated here can be associated with the fibre twist: a change in α and β will indeed modify the fibre architecture and consequently induce a rotation of one fibre end with respect to the other.
However, it has been pointed out that consecutive nucleosomes may have significantly altered topology owing to alternative nucleosome spacing [41] and topological models accounting for alternating linker DNA crossings [42] as well as for irregular spacing between nucleosomes [43] have been proposed. In principle, alternate nucleosomal spacing or alternate linker DNA crossing1 can be accounted for by our model, through a redefinition of the repetition unit in the fibre: this would imply a decomposition of the fibre into di-nucleosomes, the calculation of the linking number contribution per di-nucleosome with respect to the local fibre axis, and a redefinition of the fibre backbone based on the new partition. The calculation of the di-nucleosome linking number may be quite cumbersome but it can be obtained, and the principle of calculation remains the same.
On the other hand, linker DNA folding heterogeneity can be also addressed. An extension of the previous calculation can be made to include fibres for which the structural parameters α and β change from nucleosome to nucleosome in a periodic way along the fibre. In that case, some additional terms should be included in the DNA linking number expression, essentially owing to the different orientation of the local fibre axis with respect to a reference straight axis. Such additional terms can therefore be associated with the bending of the fibre axis, i.e. with the fibre writhe, and the total DNA linking number can therefore be rewritten as the sum of fibre twist and writhe [38].
6. Wrapping chromatin fibres
Micromanipulation by magnetic tweezers of chromatin fibres has been realized by the team of Jean-Louis Viovy [44–46]. Chromatin fibres of tens of nucleosomes, reconstituted in vitro and inserted between two fragments of naked DNA are manipulated—stretched and twisted.
The rotation–extension response of the nucleosome assembly showed rather unexpected results. Unlike what was observed for DNA, the fibres can absorb a very high number of turns, with little or no change in length (high torsional resilience). Intuitively, these results imply that nucleosomes must be able to store a portion of the torsion induced in the fibre. Calculating the topological parameters of the fibre is an essential step in order to interpret these results. We have therefore proposed a statistical physics model, including the existence of structural transitions of the nucleosome between three different states, characterized by different topological contributions to the fibre linking number [45]. These three states, previously identified in minicircles studies [47,48], are indeed characterized by a different pathway for the entering and exiting linker DNAs: the negatively crossed state corresponds to the standard crystallographic structure; in the positively crossed state, the two linker DNAs cross instead in the opposite, positive way; and in the open state, DNA is partially unwrapped from the NCP and linker DNAs do not cross any more (figure 7). The corresponding Lk contributions per nucleosome can be calculated as previously described and give, respectively, Lkneg = −1.4, Lkopen = −0.7, Lkpos = −0.4. The free energies for the three conformations can be estimated experimentally. The stability of the different states eventually depends on the applied torque: the application of a positive (respectively, negative) torque to the fibre will favour the states with the most positive (respectively, negative) linking number per nucleosome. The resulting dynamic equilibrium between the different conformations acts, therefore, as a topological buffer, absorbing the additional torsion, thanks to the rearrangement of the internal nucleosome structure [45].
Figure 7.
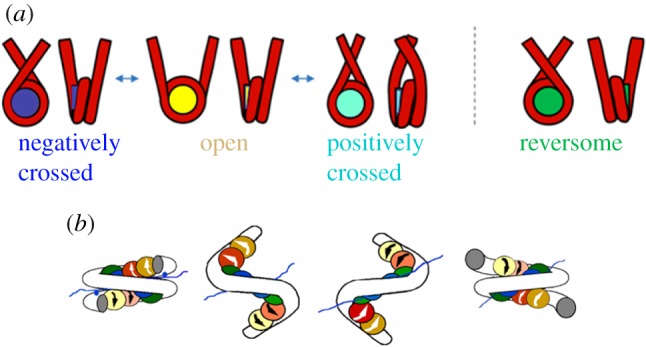
(a) The four states of the nucleosome discussed in the main text. (b) An image of the chiral transition of the nucleosome from the standard (crystallographic) conformation into the reversome state. Here, histones are represented by the coloured spheres and DNA by the white tube.
7. A new nucleosome state: the reversome
Even more unexpected, chromatin fibres display a strong hysteretic behaviour when submitted to extensive positive torsion [46]. Again, this effect can be explained by a structural rearrangement of individual nucleosomes, but implying this time a much larger deformation, with a Lk variation of about two turns per nucleosome. The altered form should be metastable in order to explain the presence of hysteresis, and has to be stabilized by the application of a large positive torque. If magnetic tweezers measurements allowed one to deduce the value of Lk for this altered form of the nucleosome, its precise structure was completely unknown. Relying on the analysis of the nucleosome normal modes to identify its flexible parts, we have obtained a coarse-grained model of the nucleosome, that we have studied by means of Brownian dynamics simulations. In the simulations, an inversion of chirality of the nucleosome is induced by the application of an external torque (and provided that linker DNAs are in the open configuration) [46,49]. The resulting structure is a right-handed nucleosome, that we named reversome, for reversed nucleosome (figure 7). Calculations of the linking number contribution for this mirror-inversed nucleosome (with a slightly different α) give Lkrev ≃ 0.85, in agreement with the experimental estimation of a linking number difference of about two turns. More recently, we used the same experimental and theoretical methods to study fibres with and without an additional protein known to compact the fibre, the linker histone H5, and showed that linker histone incorporation maintains chromatin fibre plasticity [44].
8. Perspectives: the ‘reversome wave’
Single fibre micromanipulation experiments suggest the existence of an inverted structure of the nucleosome, the reversome, stabilized by a significantly high torsional stress. This plasticity may play a role in vivo, if a torque constrains the fibre. As we have discussed, this is indeed the case in the elongation phase of transcription, when DNA screws through the RNAP, turning on its axis. We then wanted to address the broader question of whether and how transcription may take place in a condensed fibre, characterized by a regular array of nucleosomes stacked on each other (see again figure 4). In these conditions, steric hindrance prevents nucleosomes to move and change conformation. However, the first nucleosome, at the border between the condensed fibre and its decondensed part where transcription is in progress, is free to move. It will therefore pass to its reversome state under the effect of the high torque exerted by the incoming polymerase. After the transition, the torque on the fibre is partially relaxed, since about two turns are absorbed in the transition. However, it starts to increase again with the progression of the RNAP and eventually reaches the critical torque required to induce a new transition. The following nucleosome then passes to the reversome state and so on, with a domino effect, that forms a ‘reversome front’ progressing along the fibre (figure 8). This mechanism acts as a ‘topological buffer’ to absorb excess of positive supercoiling [50]. We have estimated the speed of this reversome front, which results to be about 10 times larger than the speed of the RNAP, thus indicating that it spreads rapidly far from the transcribing region. This model mainly applies to intense transcription events, where local loss of histones during passage of the polymerase is observed [3,51]. In that case, in the vicinity of the polymerase, elongation factors [3,52] help to destabilize nucleosomes (and reversomes, which are less stable), while complete reversome transition could happen farther downstream.
Figure 8.
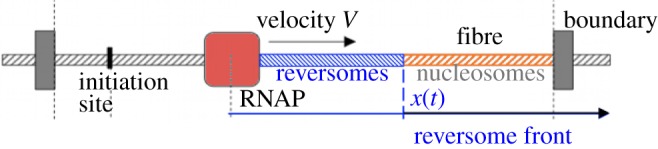
Schematic of the progress of a reversome front (blue) along a compact fibre of nucleosomes, induced by the advancing RNAP (red square). The two ends of the fibre are supposed to be clamped in a loop, as frequently observed in cells.
Although this scenario remains for the moment only a speculative suggestion, recent experimental results [53] showed a wave of destabilization of nucleosomes, which grows very rapidly along the fibre, about 10 times faster than the motion of the RNAP. These observations can be interpreted in the frame of our model [54], and possibly lead to a first verification of our hypothesis.
In conclusion, if DNA behaves as an electric cable, the nucleosome to reversome transition makes the chromatin fibre similar to a telephone cord (figure 9). An increasing linking number can not only be stored in the cord twist and writhe, but also in a third form of distortion: some of the telephone cord spires can be inverted, thus modifying locally the internal architecture of the cord, and relaxing the torsional stress elsewhere. Dealing with topological features of DNA represents an essential and a rich tool in order to understand and model biological processes, either in vitro or in vivo, helping in deciphering the underlying physical and mechanical constraints that can suggest new scenarios for complex phenomena.
Figure 9.
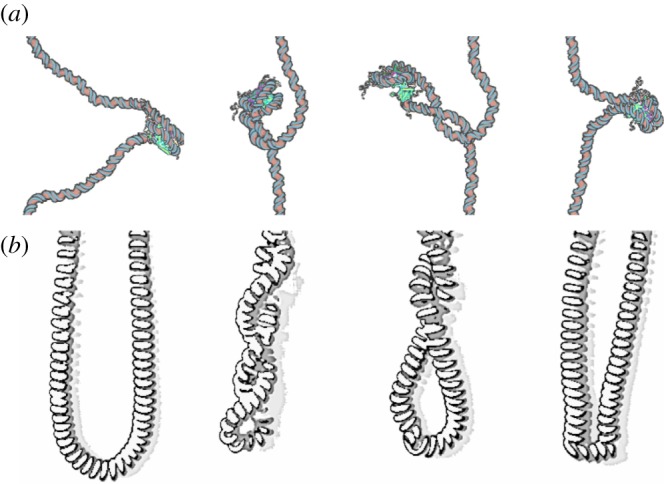
Analogy between (a) the reversome transition and (b) the local inversion of spires in a telephone cord. The nucleosome to reversome transition is obtained by means of Brownian dynamics simulations on a coarse-grained model [55].
Footnotes
A linker DNA bending can be included in the model by adding one more local angle [44].
References
- 1.Cook P. R. 1999. The organization of replication and transcription. Science 284, 1790–1795 10.1126/science.284.5421.1790 (doi:10.1126/science.284.5421.1790) [DOI] [PubMed] [Google Scholar]
- 2.Liu L. F., Wang J. C. 1987. Supercoiling of the DNA template during transcription. Proc. Natl Acad. Sci. USA 84, 7024–7027 10.1073/pnas.84.20.7024 (doi:10.1073/pnas.84.20.7024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavelle C. 2007. Transcription elongation through a chromatin template. Biochimie 89, 516–527 10.1016/j.biochi.2006.09.019 (doi:10.1016/j.biochi.2006.09.019) [DOI] [PubMed] [Google Scholar]
- 4.Vinograd J., Lebowitz J., Radloff R., Watson R., Laipis P. 1965. The twisted circular form of polyoma viral DNA. Proc. Natl Acad. Sci. USA 53, 1104–1111 10.1073/pnas.53.5.1104 (doi:10.1073/pnas.53.5.1104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kornberg A. 1980. DNA replication, p. 29. San Francisco, CA: W. H. Freeman. [Google Scholar]
- 6.Fuller F. B. 1971. The writhing number of a space curve. Proc. Natl Acad. Sci. USA 68, 815–819 10.1073/pnas.68.4.815 (doi:10.1073/pnas.68.4.815) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuller F. B. 1978. Decomposition of the linking number of a closed ribbon, a problem from molecular biology. Proc. Natl Acad. Sci. USA 75, 3557–3561 10.1073/pnas.75.8.3557 (doi:10.1073/pnas.75.8.3557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White J. H., Bauer W. R. 1982. Calculation of the twist and writhe for representative models of DNA. J. Mol. Biol. 189, 329–341 10.1016/0022-2836(86)90513-9 (doi:10.1016/0022-2836(86)90513-9) [DOI] [PubMed] [Google Scholar]
- 9.Maggs A. C. 2001. Writhing geometry at finite temperature: random walks and geometric phases for stiff polymers. J. Chem. Phys. 114, 5888–5896 10.1063/1.1353545 (doi:10.1063/1.1353545) [DOI] [Google Scholar]
- 10.Rossetto V., Maggs A. C. 2003. Writhing geometry of open DNA. J. Chem. Phys. 21, 9864–9874 10.1063/1.1569905 (doi:10.1063/1.1569905) [DOI] [Google Scholar]
- 11.Starostin E. L. 2005. On the writhe of non-closed curves. In Physical and numerical models in knot theory including applications to the life sciences, vol. 36 (eds Calvo J., Millett K., Rawdon E., Stasiak A.), pp. 525–545 Singapore: World Scientific; 10.1142/9789812703460_0026 (doi:10.1142/9789812703460_0026) [DOI] [Google Scholar]
- 12.van der Heijden G. H. M., Peletier M. A., Planque R. 2007. On end rotation for open rods undergoing large deformations. Q. Appl. Math. 65, 385–402 [Google Scholar]
- 13.Moroz J. D., Nelson P. 1997. Torsional directed walks, entropic elasticity, and DNA twist stiffness. Proc. Natl Acad. Sci. USA 94, 14 418–14 422 10.1073/pnas.94.26.14418 (doi:10.1073/pnas.94.26.14418) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marko J. F., Siggia E. D. 1995. Statistical mechanics of supercoiled DNA. Phys. Rev. E 52, 2912–2938 10.1103/PhysRevE.52.2912 (doi:10.1103/PhysRevE.52.2912) [DOI] [PubMed] [Google Scholar]
- 15.Bouchiat C., Mezard M. 1998. Elasticity theory of a supercoiled DNA molecule. Phys. Rev. Lett. 80, 1556–1559 10.1103/PhysRevLett.80.1556 (doi:10.1103/PhysRevLett.80.1556) [DOI] [Google Scholar]
- 16.Neukirch S., van der Heijden G. H. M., Thompson J. M. T. 2002. Writhing instabilities of twisted rods: from infinite to finite length. J. Mech. Phys. Solids 50, 1175–1191 10.1016/S0022-5096(01)00130-2 (doi:10.1016/S0022-5096(01)00130-2) [DOI] [Google Scholar]
- 17.Neukirch S. 2004. Extracting DNA twist rigidity from experimental supercoiling data. Phys. Rev. Lett. 93, 198107. 10.1103/PhysRevLett.93.198107 (doi:10.1103/PhysRevLett.93.198107) [DOI] [PubMed] [Google Scholar]
- 18.Clauvelin N., Audoly B., Neukirch S. 2009. Elasticity and electrostatics of plectonemic DNA. Biophys. J. 96, 3716–3723 10.1016/j.bpj.2009.02.032 (doi:10.1016/j.bpj.2009.02.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith S., Finzi L., Bustamante C. 1992. Direct mechanical measurements of the elasticity of single DNA molecules by using magnetic beads. Science 258, 1122–1126 10.1126/science.1439819 (doi:10.1126/science.1439819) [DOI] [PubMed] [Google Scholar]
- 20.Bustamante C., Marko J. F., Siggia E. D., Smith S. 1994. Entropic elasticity of lambda-phage DNA. Science 265, 1599–1600 10.1126/science.8079175 (doi:10.1126/science.8079175) [DOI] [PubMed] [Google Scholar]
- 21.Strick T. R., Allemand J.-F., Bensimon D., Bensimon A., Croquette V. 1996. The elasticity of a single supercoiled DNA molecule. Science 271, 1835–1837 10.1126/science.271.5257.1835 (doi:10.1126/science.271.5257.1835) [DOI] [PubMed] [Google Scholar]
- 22.Cluzel P., Lebrun A., Heller C., Lavery R., Viovy J.-L., Chatenay D., Caron F. 1996. DNA: an extensible molecule. Science 271, 792–794 10.1126/science.271.5250.792 (doi:10.1126/science.271.5250.792) [DOI] [PubMed] [Google Scholar]
- 23.Allemand J. F., Bensimon D., Lavery R., Croquette V. 1998. Stretched and overwound DNA forms a Pauling-like structure with exposed bases. Proc. Natl Acad. Sci. USA 95, 14 152–14 157 10.1073/pnas.95.24.14152 (doi:10.1073/pnas.95.24.14152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramreddy T., Sachidanandam R., Strick T. R. 2011. Real-time detection of cruciform extrusion by single-molecule DNA nanomanipulation. Nucleic Acids Res. 39, 4275–4283 10.1093/nar/gkr008 (doi:10.1093/nar/gkr008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dekker N. H., Rybenkov V. V., Duguet M., Crisona N. J., Cozzarelli N. R., Bensimon D., Croquette V. 2002. The mechanism of type IA topoisomerases. Proc. Natl Acad. Sci. USA 99, 12 126–12 131 10.1073/pnas.132378799 (doi:10.1073/pnas.132378799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charvin G., Bensimon D., Croquette V. 2003. Single-molecule study of DNA unlinking by eukaryotic and prokaryotic type-II topoisomerases. Proc. Natl Acad. Sci. USA 100, 9820–9825 10.1073/pnas.1631550100 (doi:10.1073/pnas.1631550100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Revyakin A., Ebright R. H., Strick T. R. 2004. Promoter unwinding and promoter clearance by RNA polymerase: detection by single-molecule DNA nanomanipulation. Proc. Natl Acad. Sci. USA 101, 4776–4780 10.1073/pnas.0307241101 (doi:10.1073/pnas.0307241101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Revyakin A., Liu R. C., Ebright H., Strick T. R. 2006. Abortive initiation and productive initiation by RNA polymerase involve DNA scrunching. Science 314, 1139–1143 10.1126/science.1131398 (doi:10.1126/science.1131398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavelle C., Praly E., Bensimon D., Le Cam E., Croquette V. 2011. Nucleosome remodelling machines and other molecular motors observed at the single molecule level. FEBS J. 278, 3596–3607 10.1111/j.1742-4658.2011.08280.x (doi:10.1111/j.1742-4658.2011.08280.x) [DOI] [PubMed] [Google Scholar]
- 30.Lavelle C., Victor J.-M., Zlatanova J. 2010. Chromatin fiber dynamics under tension and torsion. Int. J. Mol. Sci. 11, 1557–1579 10.3390/ijms11041557 (doi:10.3390/ijms11041557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davey C. A., Sargent D. F., Luger K., Maeder A. W., Richmond T. J. 2002. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 319, 1097–1113 10.1016/S0022-2836(02)00386-8 (doi:10.1016/S0022-2836(02)00386-8) [DOI] [PubMed] [Google Scholar]
- 32.Scheffer M. P., Eltsov M., Bednar J., Frangakis A. S. In press Nucleosomes stacked with aligned dyad axes are found in native compact chromatin in vitro. J. Struct. Biol. 10.1016/j.jsb.2011.11.020 (doi:10.1016/j.jsb.2011.11.020) [DOI] [PubMed] [Google Scholar]
- 33.Maeshima K., Hihara S., Eltsov M. 2010. Chromatin structure: does the 30-nm fibre exist in vivo? Curr. Opin. Cell Biol. 22, 291–297 10.1016/j.ceb.2010.03.001 (doi:10.1016/j.ceb.2010.03.001) [DOI] [PubMed] [Google Scholar]
- 34.Fussner E., Ching R. W., Bazett-Jones D. P. 2011. Living without 30 nm chromatin fibers. Trends Biochem. Sci. 36, 1–6 10.1016/j.tibs.2010.09.002 (doi:10.1016/j.tibs.2010.09.002) [DOI] [PubMed] [Google Scholar]
- 35.Scheffer M. P., Eltsov M., Frangakis A. S. 2011. Evidence for short-range helical order in the 30-nm chromatin fibers of erythrocyte nuclei. Proc. Natl Acad. Sci. USA 108, 16 992–16 997 10.1073/pnas.1108268108 (doi:10.1073/pnas.1108268108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horowitz R. A., Agard D. A., Sedat J. W., Woodcock C. L. 1994. The three-dimensional architecture of chromatin in situ: electron tomography reveals fibers composed of a continuously variable zig-zag nucleosomal ribbon. J. Cell Biol. 125, 1–10 10.1083/jcb.125.1.1 (doi:10.1083/jcb.125.1.1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woodcock C. L., Grigoryev S. A., Horowitz R. A., Whitaker N. A. 1993. Chromatin folding model that incorporates linker variability generates fibers resembling the native structures. Proc. Natl Acad. Sci. USA 90, 9021–9025 10.1073/pnas.90.19.9021 (doi:10.1073/pnas.90.19.9021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbi M., Mozziconacci J., Victor J. M. 2005. How the chromatin fiber deals with topological constraints. Phys. Rev. E 71, 031 910. 10.1103/PhysRevE.71.031910 (doi:10.1103/PhysRevE.71.031910) [DOI] [PubMed] [Google Scholar]
- 39.Mozziconacci J., Lavelle C., Barbi M., Lesne A., Victor J. M. 2006. A physical model for the condensation and decondensation of eukaryotic chromosomes. FEBS Lett. 580, 368–372 10.1016/j.febslet.2005.12.053 (doi:10.1016/j.febslet.2005.12.053) [DOI] [PubMed] [Google Scholar]
- 40.Luger K., Maeder A. W., Richmond R. K., Sargent D. F., Richmond T. J. 1997. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–259 10.1038/38444 (doi:10.1038/38444) [DOI] [PubMed] [Google Scholar]
- 41.Stein A. 1980. DNA wrapping in nucleosomes. The linking number problem re-examinated. Nucleic Acids Res. 8, 4803–4820 10.1093/nar/8.20.4803 (doi:10.1093/nar/8.20.4803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Worcel A., Strogatz S., Riley D. 1981. Structure of chromatin and the linking number of DNA. Proc. Natl Acad. Sci. USA 78, 1461–1465 10.1073/pnas.78.3.1461 (doi:10.1073/pnas.78.3.1461) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grigoriev S. A., Ioffe L. B. 1981. The dependence of the linking number of a circular minichromosome upon the shape and the orientation of its nucleosomes. FEBS Lett. 130, 43–46 10.1016/0014-5793(81)80661-8 (doi:10.1016/0014-5793(81)80661-8) [DOI] [PubMed] [Google Scholar]
- 44.Recouvreux P., Lavelle C., Barbi M., Conde e Silva N., Le Cam E., Victor J.-M., Viovy J.-L. 2011. Linker histones incorporation mantains chromatin fiber plasticity. Biophys. J. 100, 2726–2735 10.1016/j.bpj.2011.03.064 (doi:10.1016/j.bpj.2011.03.064) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bancaud A., et al. 2006. Structural plasticity of single chromatin fibers revealed by torsional manipulation. Nat. Struct. Mol. Biol. 13, 444–450 10.1038/nsmb1087 (doi:10.1038/nsmb1087) [DOI] [PubMed] [Google Scholar]
- 46.Bancaud A., et al. 2007. Nucleosome chiral transition under positive torsional stress in single chromatin fibers. Mol. Cell 27, 135–147 10.1016/j.molcel.2007.05.037 (doi:10.1016/j.molcel.2007.05.037) [DOI] [PubMed] [Google Scholar]
- 47.De Lucia F., Alilat M., Sivolob A., Prunell A. 1999. Nucleosome dynamics. III. histone tail-dependent fluctuation of nucleosomes between open and closed DNA conformations. Implications for chromatin dynamics and the linking number paradox. J. Mol. Biol. 285, 1101–1119 10.1006/jmbi.1998.2382 (doi:10.1006/jmbi.1998.2382) [DOI] [PubMed] [Google Scholar]
- 48.Sivolob A., Lavelle C., Prunell A. 2003. Sequence-dependent nucleosome structural and dynamic polymorphism. Potential involvement of histone H2B N-terminal tail proximal domain. J. Mol. Biol. 326, 49–63 10.1016/S0022-2836(02)01372-4 (doi:10.1016/S0022-2836(02)01372-4) [DOI] [PubMed] [Google Scholar]
- 49.Lavelle C., Recouvreux P., Wong H., Bancaud A., Viovy J.-L., Prunell A., Victor J.-M. 2009. Right-handed nucleosome: myth or reality? Cell 139, 1216–1217 10.1016/j.cell.2009.12.014 (doi:10.1016/j.cell.2009.12.014) [DOI] [PubMed] [Google Scholar]
- 50.Bécavin C., Barbi M., Victor J.-M., Lesne A. 2010. Transcription within condensed chromatin: steric hindrance facilitates elongation. Biophys. J. 98, 824–833 10.1016/j.bpj.2009.10.054 (doi:10.1016/j.bpj.2009.10.054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kulaeva O. I., Studitsky V. M. 2010. Mechanism of histone survival during transcription by RNA polymerase II. Transcription 1, 85–88 10.4161/trns.1.2.12519 (doi:10.4161/trns.1.2.12519) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Workman J. L. 2006. Nucleosome displacement in transcription. Genes Dev. 20, 2009–2017 10.1101/gad.1435706 (doi:10.1101/gad.1435706) [DOI] [PubMed] [Google Scholar]
- 53.Petesch S. J., Lis J. T. 2008. Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell 134, 74–84 10.1016/j.cell.2008.05.029 (doi:10.1016/j.cell.2008.05.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zlatanova J., Victor J. M. 2009. How are nucleosomes disrupted during transcription elongation? HFSP J. 3, 373–378 10.2976/1.3249971 (doi:10.2976/1.3249971) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lavelle C., Bancaud A., Recouvreux P., Barbi M., Victor J.-M., Viovy J.-L. 2011. Chromatin topological transitions. Prog. Theor. Phys. Suppl. 191, 30–39 [Google Scholar]