

Abstract
Purpose
Cancer associated stromal fibroblasts (CAFs) undergo transcriptional and phenotypic changes that contribute to tumor progression, but the mechanisms responsible for these changes are not well understood. Aberrant DNA methylation is an important cause of transcriptional alterations in cancer cells but it is not known how important DNA methylation alterations are to CAF behavior.
Experimental Design
We used Affymetrix exon arrays to compare genes induced by the DNA methylation inhibitor 5-aza-dC in cultured pancreatic cancer associated fibroblasts, pancreatic control fibroblasts and pancreatic cancer cell lines.
Results
We found that pancreatic CAFs and control pancreatic fibroblasts were less responsive to 5-aza-dC-mediated gene reactivation than pancreatic cancer cells (mean+/−SD of genes induced ≥5-fold was 9±10 genes in 10 pancreatic CAF cultures, 17±14 genes in 3 control pancreatic fibroblast cultures, and 134±85 genes in 4 pancreatic cancer cell lines). We examined differentially expressed genes between CAFs and control fibroblasts for candidate methylated genes and identified the disintegrin and metalloprotease, ADAM12 as hypomethylated and overexpressed in pancreatic CAF lines and overexpressed in fibroblasts adjacent to primary pancreatic adenocarcinomas.
Conclusions
Compared to pancreatic cancer cells, few genes are reactivated by DNMT1 inhibition in pancreatic CAFs suggesting these cells do not harbor many functionally important alterations in DNA methylation. CAFs may also not be very responsive to therapeutic targeting with DNA methylation inhibitors.
Introduction
Epigenetic changes in gene expression are a fundamental feature of cancer cells [1]. One of the best characterized epigenetic alterations is DNA methylation. DNA methylation patterns are heritable and are maintained after cell division mainly by the DNA methyltransferase Dnmt1. Aberrant DNA hypermethylation often occurs at CpG islands in gene promoters and is associated with a closed chromatin state and gene repression. Considerable evidence indicates that aberrant hypermethylation and gene silencing of tumor suppressors and other regulatory genes contributes to the development of pancreatic and other cancers [2]. Aberrant hypomethylation of normally methylated and silenced genes has also been described in pancreatic and other cancers as a cause of aberrant gene overexpression [3], [4].
The mechanisms responsible for these aberrant methylation patterns in cancers are not well understood, but suspected mechanisms include overexpression of DNMT1, accumulation of DNA methylation alterations with age [5], [6] and environmental influences [7], altered activity of the de novo methylating enzymes DNMT3a and DNMT3b [8], [9], and possibly from altered cellular microenvironment such as from chronic inflammation [3], [10].
Non-neoplastic stromal cells within the tumor microenvironment also undergo transcriptional and other phenotypic changes relative to normal cells that are thought to contribute to tumor progression. Pancreatic cancer is a deadly disease [11] and is well known for its extensive desmoplastic stromal response comprising an average 75% of the cells in the tumor mass [12]. It is suspected that the stromal component contributes to the resistance of pancreatic cancers to drug therapies [13], and several studies implicate differences in stromal behavior with patient outcome in pancreatic and other cancers [14], [15], [16], [17], [18]. Cancer associated fibroblasts (CAFs), the predominant cell type in the stromal microenvironment, adopt an activated phenotype during tumorigenesis, undergoing morphologic, functional, and gene expression changes relative to normal fibroblasts [19]. These activated CAFs are characterized by α-smooth-muscle actin (α-SMA) expression [20], enhanced contractile and secretory ability, and increased synthesis of collagens, extracellular matrix proteins [21] and growth factors including epithelial growth factor, platelet-derived growth factors (PDGF), basic fibroblast growth factor, hepatocyte growth factor, and transforming growth factor beta (TGF-β) [22]. Through these and other signals, CAFs interact intimately with tumor cells to promote their growth, and therefore CAFs are of considerable interest as a therapeutic target. Indeed, recent strategies to target CAFs include the use of the kinase inhibitor, imatinib to block stromal PDGF receptors [23], antibodies to block vascular endothelial growth factor (VEGF) derived from both cancer cells and CAFs to inhibit tumor angiogenesis [24] and the use of smoothened (Smo) inhibitors to block tumor-stromal Hedgehog signaling and deplete stromal fibroblasts that overexpress the hedgehog (Hh) receptor Smo [25], [26], [27], [28]. That initial trials of smoothened inhibitors in pancreatic cancers did not show evidence of benefit highlights the need for basic studies investigating tumor stromal interactions. Another potential benefit of targeting cancer stroma is to improve the accessibility of therapeutics to pancreatic cancer cells. For example, the drug nab-paclitaxel (Abraxane), a nanoparticle formulation of paclitaxel, binds to Sparc, a stromal matrix protein often highly expressed in pancreatic cancer stroma that mediates tumor stromal interactions, thereby potentially improving drug delivery to the tumor [29], [30]. Clinical trials of abraxane in pancreatic cancer show promise and in animal models there is evidence to suggest that the delivery of gemcitabine to the tumor is improved with abraxane, and with hedgehog inhibitors [25]. Efficacy of the Gemcitabine/abraxane drug combination still awaits the results of phase 3 clinical trials.
The influence of CAFs on cancer growth has been demonstrated in numerous studies [31], [32] but the molecular mechanisms underlying the CAF phenotype are not well understood. Although a major influence of CAFs is suspected to be the tumor microenvironment including influences from the cancer cells themselves, CAFs retain their tumor-promoting properties in vitro after prolonged cell culture [33], suggesting that hereditary mechanisms are responsible. Although genetic alterations in CAFs have been described, more recent studies investigating the possibility that CAFs undergo clonal genetic alterations similar to cancer cells have found no evidence for such alterations [33], [34], [35], [36], [37], [38]. In the absence of widespread genetic mutations, it is reasonable to suspect that epigenetic mechanisms such as DNA methylation, which are mitotically heritable, are responsible for the stable gene expression changes in CAFs. Indeed, several genes implicated in tumor-stromal interactions and induced in CAFs by coculture with cancer cells, such as SPARC [29], [30], COX-2 and matrix-metalloproteinases (MMPs) [39] are regulated by DNA methylation. Furthermore, CAFs are subjected to the same influences in the tumor microenvironment which are thought to contribute to methylation changes in cancer cells, such as chronic inflammation [34].
The heterogeneity of CAFs from different tumors and even within the same tumor [14], [30] also suggests that CAFs have multiple sources which could contribute to epigenetic differences. In addition to activation of resident fibroblasts in the tumor microenvironment, some CAFs are bone-marrow derived mesenchymal precursor cells [40], [41] and perhaps epithelial or endothelial cells undergoing mesenchymal transition [21], [42]. Because these differentiation and transdifferentiation processes are regulated by epigenetic mechanisms [43], CAFs derived from different sources have different epigenetic and transcriptional profiles compared to their resident tissue counterparts.
Only a few studies have begun to explore epigenetic mechanisms for these transcriptional changes in CAFs. One of the first studies to provide evidence of DNA methylation differences between normal and tumor associated stromal cells took a genome-wide approach using methylation specific digital karyotyping (MSDK) to profile epithelial cells, myoepithelial cells and stromal fibroblasts during breast tumor progression [35]. Another approach combining laser capture microdissection with methylation specific PCR (MSP) of candidate genes identified frequent promoter methylation of GSTP1 and RARB2 in prostate tumor-associated stromal cells relative to normal prostate stromal cells [44]. A third approach using methylation-sensitive SNP array analysis (MSNP) demonstrated focal gains in methylation and global hypomethylation in gastric cancer-associated myofibroblasts [36].
One useful strategy for identifying methylation events is to perform a gene reactivation screen using DNA methylation inhibitors such as 5-aza-2′-deoxycytidine (5-aza-dC), which selectively targets the DNMT1 enzyme for depletion. This method has the advantage of identifying the DNA methylation alterations that regulate transcription and therefore more likely to be functionally important. Although this approach has successfully identified methylation events in cancer cells from multiple tumor types [45] to our knowledge this approach has not been used to identify genes regulated by methylation in CAFs. To determine whether DNA methylation are similarly important regulators of gene expression changes in CAFs as for cancer cell lines, we performed a genome-wide reexpression analysis of human pancreatic cancer-associated fibroblasts and pancreatic cancer cell lines using 5-aza-dC.
Materials and Methods
Culture of Cell lines
Primary cultures of stromal fibroblasts, designated cancer associated fibroblasts (CAFs) CAF9, CAF11, CAF12, CAF13, CAF14, CAF15, CAF16, CAF18, CAF19, CAF20, CAF21, CAF22, CAF25, and CAF35 were established from surgically-resected pancreatic cancer tissue from 13 patients (6 males mean ± standard deviation (SD) age of 58±7 years) with clinically sporadic pancreatic ductal adenocarcinoma. The cancers were all moderate to poorly differentiated with a mean tumor size of 3.5 cm. These primary CAF cultures were established as previously described [27] as were two hTERT-immortalized control fibroblasts (SC2 and SC3, established from surgically resected non-neoplastic pancreas tissue from 2 females (mean age, 63 years) [27]. The immortalized human pancreatic Nestin-expressing (HPNE) cell line was generously provided by Dr. Michel Ouellette (University of Nebraska Medical Center) [46]. Four pancreatic cancer cell lines, including A32-1, Panc2.8, Panc3.014 and Panc215 established from primary pancreatic ductal adenocarcinomas at our institution and described previously [47] were also used in this study. All cell lines were maintained in DMEM (4.5 mg/mL glucose; Invitrogen) containing 10% FBS under standard conditions as described previously [48]. All studies were performed with approval from the Johns Hopkins Committee for Clinical Investigation.
5-aza-dC treatment and RNA extraction
Cells were plated at 2×105 cells per T75 flask and treated the following day with 1 µmol/L 5-aza-2′-deoxycytidine (5-aza-dC, Sigma) [49] for 4 days during the exponential growth phase, with a change of media and drug every 24 hours. On day 5 when cells were near-confluent, they were washed with cold PBS and total RNA isolated using a Qiagen kit (Qiagen) or the mirVana miRNA kit (Ambion), according to manufacturer's instructions as described previously [50].
Exon array and selection of genes for validation
The Affymetrix GeneChip Human Exon 1.0ST Array platform was used to analyze gene expression patterns in untreated and 5-aza-dC treated fibroblasts and cancer cell lines, as previously described [27]. We are in compliance with the Minimum Information about a Microarray Experiment guidelines and have submitted our microarray data set to the Gene Expression Omnibus repository (GEO Accession #GSE20911).
Quantitative RT-PCR (qRT-PCR)
1 µg total RNA was reverse transcribed according to the manufacturer's instructions (Invitrogen). The resulting cDNA was amplified on an ABI 7300 Real-Time PCR thermocycler using SYBR GREEN PCR Master Mix and recommended PCR conditions (Applied Biosystems). The housekeeping gene GAPDH was used for normalization. Primer specificity was confirmed by melting curve analysis. Results are expressed as normalized expression values relative to the indicated cell line ( = 2−ΔΔCt). All PCR reactions were performed in triplicate. Primers sequences are as follows: Stratifin RTsense: 5′-TCTGATCCAGAAGGCCAAG-3′; Stratifin RTantisense: 5′-GTTTCGCTCTTCGCAGGAG-3′; TKTL1 RTsense: 5′-AGCTCCGGCCACCCTACATCATG-3′; TKTL1 RTantisense: 5′-TGCCACATCCACAAACGACAGTCT-3′; ADAM12 RTsense: 5′-AAATGAAGGTCTCATTGCCAG-3′; ADAM12 RTantisense: 5′-AGAATTACCCGTGTAATTTCGAG-3′; GAPDH RTsense: 5′-CAACAGCCTCAAGATCATCAG-3′; GAPDH RTantisense: 5′-ACTGTGGTCATGAGTCCTTC-3′.
Bisulfite Sequencing
The methylation status of the 5′ CpG islands of candidate genes was determined by bisulfite sequencing. Bisulfite modification and PCR was performed as previously described [51]. Bisulfite Modified Sequencing (BMS) primers were as follows: TKTL1 BMS forward: 5′-TGTGTAGAGAAAGAAGATTTTGTATT-3′, TKTL1 BMS reverse: 5′-CCCTTTAAAATCTAAAAACCCACTC-3′, and internal sequencing primer TKTL1 BMS-Seq: 5′-GATTGTAGGAGAGAAGATGAG-3′; ADAM12 BMS forward: 5′-TTTAGTTTTAGTTTGAAAAGTTGGA-3′, ADAM12 BMS reverse: 5′-CTAAACTCTTCTAACCTTTCAT-3′, and internal sequencing primer ADAM12 BMS-Seq: 5′-TTCTAACACAAACCAACCTTAACC-3′. Purified products were sequenced at the Johns Hopkins Core Sequencing Facility.
Western Blot Analysis of DNMT1 Expression
Western blot analysis was performed using lysates from untreated or 5-aza-dC-treated HPNE or CAF12 cells. A Bradford assay was used to estimate protein concentration, using bovine serum albumin (BSA, Invitrogen) as a standard. Equal amounts of protein (40 µg per lane) were separated by 4–12% gradient SDS gel electrophoresis (Invitrogen), transferred to nitrocellulose membranes, and incubated in blocking solution (5% milk in TBS with 0.1% Tween 20) for 30 minutes. Membranes were incubated for 60 minutes with an anti-DNMT1 polyclonal antibody (generously provided by Dr. Bill Nelson, JHU) at 1∶200 dilution or a monoclonal antibody against GAPDH (Cell Signaling, Danvers, MA) at 1∶2000 dilution. Secondary HRP-linked antibodies (Santa Cruz Biotechnology, and Amersham Biosciences) were applied at 1∶2000 dilution and proteins detected using an ECL kit (Amersham Biosciences).
Pancreatic Adenocarcinoma Tissue Microarrays and Adam12 Immunohistochemistry
The expression of Adam12 protein was examined utilizing immunohistochemical (IHC) labeling of formalin-fixed, paraffin-embedded tissue microarrays (TMAs) using a DAKO Autostainer (DAKO, Carpinteria, CA). Four TMAs containing a total of 72 different surgically resected pancreatic ductal adenocarcinomas and corresponding normal pancreas tissues were constructed as previously described [29]. Adam12 IHC staining was performed as previously described [29] using a rabbit anti-human Adam12 antibody (A2601, Sigma) at a 1∶100 dilution and a 60 minute incubation time. Labeling was performed using the Envision-sPlus Detection Kit (DAKO).
Statistical Analysis
Descriptive statistical values and plots were generated using Microsoft Excel package or the Partek® Genomics Suite™ v6.3 beta. The Robust Multichip Average (RMA) method was used to normalize the raw intensity measurements of all probe sets. Gene expression values were then obtained using the one-step Tukey's biweight method. Two-way ANOVA was done to identify significant expression changes between untreated and 5-aza-dC-treated fibroblasts or cancer cell lines, or between CAFs and control fibroblasts. Differences were considered significant at P<0.05, and values reported are means ± SD.
Results
Global gene expression analysis of human pancreatic fibroblasts and cancer cell lines treated with 5-aza-dC
We established primary pancreatic CAF cultures and control fibroblast cultures as previously described [38]. Using global gene expression profiling, we previously identified gene expression differences in pancreatic CAFs relative to control fibroblasts [27]. Hierarchical clustering of the gene expression profiles of untreated CAF and control fibroblasts is provided in Figure S1. Suspecting that these changes in gene expression were in part mediated by changes in DNA methylation, we compared the gene expression profiles of CAF and control fibroblast cultures before and after treatment with 5-aza-dC using Affymetrix Exon Arrays (ST 1.0) (CAF11, CAF12, CAF13, CAF15, CAF16, CAF18, CAF19, CAF22, CAF25, and CAF35 for CAFs; HPNE, SC2, and SC3 for control fibroblast cells, respectively).
We first confirmed 5-aza-dC depletion of the Dnmt1 enzyme by Western blot. 5-aza-dC treatment of HPNE and CAF12 cells resulted in depletion of Dnmt1 protein at 1 µM concentrations but not GAPDH control protein (Figure 1). To further examine the effect of 5-aza-dC concentrations, we compared the number of genes induced by 1 µM and 10 µM of 5-aza-dC in two additional CAFs, CAF19 and CAF35, and found no major difference in the number of genes induced between these drug concentrations (data not shown). We therefore used 1 µM concentrations of 5-aza-dC to treat each CAF. We treated CAF cells for 4 days as this was sufficient duration for cell proliferation. Longer treatments (for 5–7 days) often resulted in CAFs growing to confluence (data not shown). Consistent with this, we performed proliferation assays on CAF19 at different 5-aza-dC concentrations (0 µM, 1 µM, 5 µM, 10 µM, and 20 µM) and found that growth was not significantly affected at the 1 µM concentration we employed (Figure S2).
Figure 1. Effect of 5-aza-dC treatment on DNMT1 protein levels by Western blot analysis of pancreatic CAF and control fibroblast cultures.
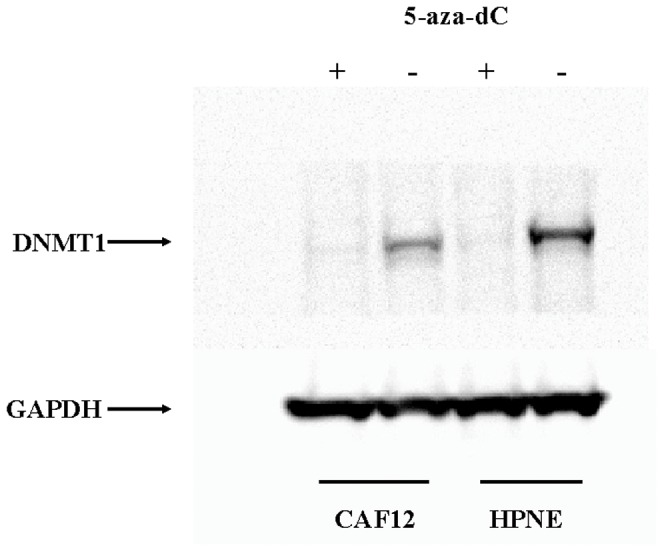
DNMT1 protein is depleted (relative to GAPDH) in 5-aza-dC treated HPNE and CAF12 cells.
After comparing the expression profiles of each CAF before and after 5-aza-dC treatment, we first identified genes that were upregulated by an overall fold-change average of ≥2.0 in all ten 5-aza-dC-treated CAFs relative to untreated CAFs. We chose a 2-fold cut-off as more modest differences in RNA were considered more likely to be related to experimental variation. Since we were interested in identifying genes silenced in CAFs whose expression was induced by 5-aza-dC, we then excluded the subset of genes that were expressed in untreated CAFs. This criterion identified 42 candidate genes (Table 1).
Table 1. Genes upregulated by an overall fold change of ≥2.0 in ten 5-aza-dC-treated pancreatic CAFs relative to untreated CAFs.
| Gene symbol | Gene Assignment | P value | Average Fold Change | Upregulated in 1 or more control fibroblasts |
| DAZL | NM_001351//deleted in azoospermia-like | 0.016 | 11.4 | Yes |
| SPANXB1 | NM_032461//SPANX family, member B1 | 0.296 | 8.9 | Yes |
| GTSF1 | NM_144594//gametocyte specific factor 1 | 0.047 | 6.4 | Yes |
| MT1G | NM_005950//metallothionein 1G | 0.022 | 4.5 | Yes |
| MAGEB2 | NM_002364//melanoma antigen family B, 2 | 0.055 | 4.3 | Yes |
| ASB5 | NM_080874//ankyrin repeat and SOCS box-containing 5 | 0.048 | 3.3 | Yes |
| HAPLN1 | NM_001884//hyaluronan and proteoglycan link protein 1 | 0.15 | 3.3 | Yes |
| MYH3 | NM_002470//myosin, heavy chain 3, skeletal muscle, embryonic | 0.062 | 3.1 | No |
| IL18 | NM_001562//interleukin 18 (interferon-gamma-inducing factor) | 0.064 | 3.1 | Yes |
| HIST1H1T | NM_005323//histone cluster 1, H1t | 0.071 | 3 | Yes |
| TKTL1 | NM_012253//transketolase-like 1 | 0.034 | 2.9 | Yes |
| KRT81 | NM_002281//keratin 81 | 0.03 | 2.8 | Yes |
| MT1H | NM_005951//metallothionein 1H | 0.067 | 2.8 | Yes |
| TRIM55 | NM_033058//tripartite motif-containing 55 | 0.119 | 2.7 | Yes |
| LAPTM5 | NM_006762//lysosomal protein transmembrane 5 | 0.011 | 2.7 | Yes |
| H2AFB1 | NM_001017990//H2A histone family, member B1 | 0.107 | 2.6 | Yes |
| NXPH2 | NM_007226//neurexophilin 2 | 0.006 | 2.6 | No |
| LCE2D | NM_178430//late cornified envelope 2D | 0.366 | 2.5 | Yes |
| ANXA3 | NM_005139//annexin A3 | 0.005 | 2.5 | Yes |
| OBP2B | NM_014581//odorant binding protein 2B | 0.542 | 2.4 | Yes |
| SPANXE | NM_145665//SPANX family, member E | 0.142 | 2.4 | Yes |
| UPK1B | NM_006952//uroplakin 1B | 0.037 | 2.4 | Yes |
| PRY | NM_004676//PTPN13-like, Y-linked | 0.581 | 2.4 | Yes |
| MAGEA4 | NM_001011548//melanoma antigen family A, 4 | 0.042 | 2.4 | Yes |
| SUSD2 | NM_019601//sushi domain containing 2 | 0.047 | 2.3 | Yes |
| MYL7 | NM_021223//myosin, light chain 7, regulatory | 0.04 | 2.3 | No |
| SFN | NM_006142//stratifin | 0.06 | 2.2 | Yes |
| IL20RB | NM_144717//interleukin 20 receptor beta | 0.205 | 2.2 | Yes |
| AQP1 | NM_198098//aquaporin 1 (Colton blood group) | 0.024 | 2.2 | Yes |
| MAEL | NM_032858//maelstrom homolog (Drosophila) | 0.052 | 2.2 | Yes |
| IFI27 | NM_001130080//interferon, alpha-inducible protein 27 | 0.014 | 2.2 | Yes |
| ACTC1 | NM_005159//actin, alpha, cardiac muscle 1 | 0.022 | 2.2 | No |
| TRIML2 | NM_173553//tripartite motif family-like 2 | 0.042 | 2.2 | No |
| VAMP8 | NM_003761//vesicle-associated membrane protein 8 (endobrevin) | 0.016 | 2.1 | Yes |
| TMEM92 | NM_153229//transmembrane protein 92 | 0.049 | 2.1 | Yes |
| KRT14 | NM_000526//keratin 14 | 0.11 | 2.1 | Yes |
| KRTAP13-4 | NM_181600//keratin associated protein 13-4 | 0.22 | 2.1 | No |
| ANKRD1 | NM_014391//ankyrin repeat domain 1 (cardiac muscle) | 0.197 | 2.1 | Yes |
| TYROBP | NM_003332//TYRO protein tyrosine kinase binding protein | 0.067 | 2.1 | Yes |
| TUBA4A | NM_006000//tubulin, alpha 4a | 0.1 | 2 | Yes |
| KRT17 | NM_000422//keratin 17 | 0.051 | 2 | No |
| LYPD1 | NM_144586//LY6/PLAUR domain containing 1 | 0.001 | 2 | Yes |
To further confirm the 5-aza-dC mediated gene induction we performed qRT-PCR on the gene Stratifin (14-3-3 sigma) and Transketolase-like protein 1 (TKTL1) because their expression is regulated by methylation in other cell types and the exon array analysis identified their expression as induced by 5-aza-dC [52] [53]. Consistent with the exon array result, stratifin (SFN) mRNA levels were low or undetectable in all untreated fibroblasts (CAFs and control fibroblasts), and increased expression of SFN mRNA was detected in the majority of the 5-aza-dC treated fibroblasts (Fig. 2 A and B). We observed near complete methylation of the SFN promoter region in all CAFs by bisulfite sequencing (data not shown). Similarly, TKTL1 mRNA levels were undetectable in almost all untreated fibroblasts except SC2 cells, and increased expression of TKTL1 was detected in the majority of the 5-aza-dC treated fibroblasts (Fig. 2 C). Consistent with this, we found evidence of methylation of 5′ CpGs of TKTL1 in fibroblasts by MSP (data not shown). Bisulfite sequencing of 18 CpG sites upstream of the TKTL1 transcription start site (Fig. 2 D) revealed that all or most CpG sites were fully methylated in the non-expressing fibroblast lines, HPNE and SC3 and in CAF19 and CAF25 (Fig. 2 E). In contrast, the TKTL1-expressing line, SC2, lacked methylation of many of these sites, supporting a role for DNA methylation in the regulation of TKTL1 expression. We also identified the upregulation of DAZL and ANXA3 (data not shown), genes previously reported to be induced by 5-aza-dC [36], NDN, reported to be imprinted [54], and MT1G, reported to be methylated in other cancers [55].
Figure 2. Effect of 5-aza-dC treatment on SFN and TKTL1 mRNA expression in 5-aza-dC treated pancreatic CAFs and control fibroblasts, and bisulfite sequencing analysis of TKTL1.

(A) Affymetrix exon array analysis of SFN mRNA expression in five pancreatic CAFs before (green) and after (red) 5-aza-dC treatment. (B) and (C) Quantitative RT-PCR analysis of SFN and TKTL1 mRNA expression relative to GAPDH mRNA in pancreatic fibroblast cultures before (blue bars) and after (red bars) 5-aza-dC treatment. Each assay was performed in triplicate. Data are means of three independent experiments; bars are SD values. (D) Top: TKTL1 gene structure and distribution of CpG dinucleotides. Short vertical bars represent CpG sites. Arrow points to transcriptional start site. Below: Bisulfite genomic sequencing analysis in pancreatic CAFs and control fibroblasts. Open circles represent unmethylated CpG sites, solid black circles methylated CpG sites, and hatched circles partially methylated CpG sites. (E) Bisulfite sequencing chromatograms of the TKTL1 promoter in a pancreatic CAF (CAF19) and control fibroblast lines (HPNE and SC2). Arrows point to cytosine residues.
We examined RNA profiles to determine if there were any differential responses to 5-aza-dc in CAFs compared to control fibroblasts. Thirty-five of the 42 genes induced in CAFs by 5-aza-dc (Table 1) were also induced in one or more of the control pancreatic fibroblast lines indicating that few if any genes are selectively and consistently upregulated by 5-aza-dC in CAFs. DAZL was the most highly induced gene in four CAFs (CAF12, CAF15, CAF16, and CAF19) and was among the top ten genes induced in two CAFs (CAF18 and CAF22) and one control fibroblast (HPNE) (data not shown). We performed hierarchical clustering of the genes induced by 5-aza-dC in CAFs and control fibroblasts to determine if there were observable differences in the overall patterns of gene response to 5-aza-dC, but there was no clustering by fibroblast class (Figure S3). Five of the seven CAFs clustered together, the other two CAFs clustering with the control fibroblasts.
Further evidence for this paucity of genes silenced by DNA methylation in CAFs comes from evaluating the genes underexpressed in CAFs. We identified genes that were underexpressed by a mean of ≥4.0-fold in all CAFs relative to pancreatic control fibroblasts (Table S2). Notably, there was no overlap between this list of 86 underexpressed genes and the genes induced by a mean of 2-fold by 5-aza-dc in CAFs (Table 1). These data indicate that few if any genes consistently underexpressed in CAFs are silenced by DNA methylation.
Identification of candidate genes for hypomethylation analysis
We have previously identified 200 genes, such as Smo, upregulated in pancreatic CAFs relative to pancreatic control fibroblasts [27]. To identify candidate hypomethylated genes in CAFs, we merged this list of genes overexpressed in pancreatic CAFs with the list of 581 genes induced by ≥3.0 fold in at least one fibroblast cell line by 5-aza-dC treatment. This criterion identified 49 candidate genes (Table S1) some of which may be regulated by methylation, and potentially hypomethylated in pancreatic CAFs relative to control fibroblasts.
5-aza-dC treatment of human pancreatic CAFs, control fibroblasts and cancer cell lines
We next compared the responses of CAFs and control fibroblasts to 5-aza-dC with the responses of pancreatic cancer cell lines. We generated an individual gene list for each fibroblast line or cell line and then counted the total number of genes induced by ≥5-fold in each line (Figure 3). We then compared the average number of genes induced by ≥5-fold by 5-aza-dC in the ten CAFs, the 3 control fibroblast lines, and the 4 pancreatic cancer cell lines. Significantly fewer genes were induced by 5-aza-dC in CAFs than in pancreatic cancer cell lines (P = 0.0009). The number of genes induced by ≥5-fold in the ten CAFs was only 9±10 genes (mean+/−standard deviation) compared to 17±14 genes induced by ≥5-fold in the three pancreatic control fibroblasts, and 123±86 genes induced by ≥5-fold in the 4 pancreatic cancer cell lines (Figure 3). There was no significant difference in the number of genes induced by ≥5.0 fold in CAFs and in pancreatic control fibroblasts. To ensure that we had selected a reasonable fold-change cutoff for comparison, we also compared the number of genes induced by ≥3-fold. Similar differences were also observed: The number of genes induced ≥3-fold by 5-aza-dC was 83±47 genes induced in control fibroblasts and 48±42 genes induced in CAFs and 430±271 genes in the pancreatic cancer cell lines (P = 0.0006 for CAFs relative to cancer cell lines). There were no significant differences related to patient age or gender in the number of genes induced by 5-aza-dC.
Figure 3. Number of genes induced by 5-aza-dC treatment in individual pancreatic CAFs, control fibroblasts and cell lines.
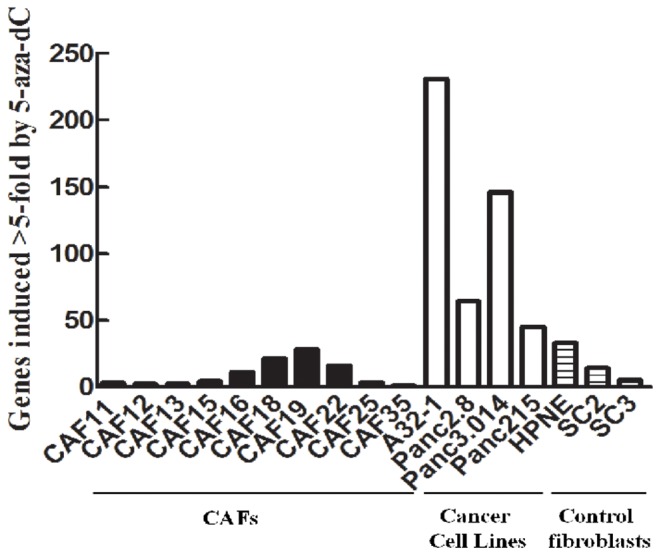
An average of 123±86 genes were induced 5-fold or more by 5-aza-dC treatment in four pancreatic cancer cell lines, 9±10 genes in ten pancreatic CAFs (P = 0.0009) and 17±14 genes in three control pancreatic fibroblast lines.
Differential methylation of the candidate hypomethylated gene ADAM12
Our analysis of genes selectively upregulated by 5-aza-dC in CAFs yielded no genes known to be regulated by DNA methylation that influence stromal fibroblast behavior. We also examined our lists of genes differentially expressed in CAFs relative to control pancreatic fibroblasts for candidates that influence tumor/stromal interactions. One candidate overexpressed in CAFs was ADAM12. ADAM12 (a disintegrin and metalloprotease 12), a regulator of cell-cell and cell-matrix interactions, is overexpressed in cancer-associated fibroblasts and is implicated in tumor progression [56]. We first confirmed upregulation of ADAM12 mRNA in CAFs relative to control fibroblasts using quantitative RT-PCR. Consistent with our exon array result (Fig. 4a), ADAM12 mRNA was highly overexpressed in the pancreatic control fibroblast derived from a pancreatitis specimen (SC3) and nine CAFs relative to the control fibroblasts HPNE and SC2 (Fig. 4b).
Figure 4. Analysis of ADAM12 mRNA expression in pancreatic CAFs and control fibroblasts.

(A) Affymetrix exon array analysis of ADAM12 mRNA expression in pancreatic CAFs and control fibroblasts. (B) Quantitative RT-PCR analysis of ADAM12 mRNA expression in pancreatic CAFs and control fibroblasts after normalization to GAPDH levels. Each assay was performed in triplicate. Data are means of three independent experiments; bars are SD values.
To determine if ADAM12 was differentially methylated in CAFs compared to control fibroblasts, we performed bisulfite sequencing of the ADAM12 gene promoter in 9 CAFs and 3 control fibroblast lines. We amplified a 481-bp region upstream of the ADAM12 transcription start site containing 38 CpG sites (Figure 5a). Bisulfite sequencing revealed that 29 of 38 (76%) CpG sites were fully methylated in the control fibroblast line HPNE (Figure 5b). By contrast, six of nine CAFs (CAF12, CAF14, CAF15, CAF16, CAF20, and CAF22) were completely (100%) unmethylated at all CpG sites. The remaining CAFs were partial methylated at 2 to 7 of 38 CpG sites and fully unmethylated at all other CpGs (Figure 5a). The control fibroblasts SC2 and SC3 were fully methylated at CpGs near the 5′ end of the sequenced region and partially methylated at CpGs near the 3′ end of this region. They were fully unmethylated at all other CpGs. Thus, there was relative hypomethylation at multiple CpGs between the fibroblasts that expressed ADAM12 and those that did not, implicating aberrant hypomethylation of ADAM12 as a mechanism for its overexpression in pancreatic CAFs compared to control pancreatic fibroblasts.
Figure 5. Bisulfite sequencing analysis of ADAM12.

(A) Top: ADAM12 gene structure and distribution of CpG dinucleotides. Short vertical bars represent CpG sites. Arrow points to transcriptional start site. Below: Bisulfite genomic sequencing analysis in pancreatic CAFs and control fibroblasts. Open circles represent unmethylated CpG sites, solid black circles methylated CpG sites, and hatched circles partially methylated CpG sites. (B) Bisulfite sequencing chromatograms of the ADAM12 promoter in a pancreatic CAF (CAF19) and control fibroblast line (HPNE). Arrows point to cytosine residues.
To determine if Adam12 protein was overexpressed in the stroma of primary pancreatic adenocarcinomas, we performed immunohistochemistry on tissue microarrays. While Adam12 protein expression was not detected in fibroblasts surrounding the normal pancreatic duct (Figure 6b), CAFs of primary pancreatic adenocarcinomas were positive for Adam12 protein expression (Figure 6c). Adam12 expression was also observed in pancreatic epithelial cells and invasive pancreatic cancers.
Figure 6. Immunohistochemical analysis of Adam12 protein expression in tissue microarrays.

(A) Adam12 protein expression is undetectable in the granule cell layer of the brain (negative control tissue). (B) Stromal fibroblasts (arrows) surrounding normal pancreatic duct do not label Adam12. (C) Cancer associated fibroblasts (arrows) in a primary pancreatic adenocarcinoma are strongly positive for Adam12 protein; magnification, 20×.
Discussion
Using a drug-based gene reactivation approach combined with global gene expression profiling, we find that human pancreatic CAFs are significantly less sensitive than cancer cell lines to 5-aza-dC-mediated gene expression changes. While an average of 134 genes were induced 5-fold or more by 5-aza-dC treatment of four human pancreatic cancer cell lines, an average of only 10 genes were induced in seven pancreatic CAFs. We confirmed 5-aza-dC-induced depletion of Dnmt1 protein and induction of genes known to be induced by 5-aza-dC. We also observed comparable Dnmt1 protein levels by Western blot or in DNMT1 RNA levels by Exon array in pancreatic CAFs and cancer cell lines (data not shown), suggesting that the limited number of genes induced by 5-aza-dC was not due to a lack of DNMT1 expression in CAFs or a lack of Dnmt1 inhibition by 5-aza-dC. This is consistent with the recent observation that hypomethylation in gastric CAFs could not be attributed to any difference in mRNA levels of DNMT1, DNMT3a, or DNMT3b between normal and cancer-associated myofibroblasts [36]. Overall, our results indicate that pancreatic CAFs do not undergo promoter DNA hypermethylation to the same extent as pancreatic cancer cells.
Our results are consistent with one previous report that 5-aza-dC-induced approximately twice as many genes (61 genes induced ≥4-fold) in a bladder cancer cell line (T24) than in normal fibroblasts (34 genes induced ≥4-fold) [45]. Similarly, we found an average of 17 genes was induced ≥5-fold in our pancreatic control fibroblasts. The 123 genes induced by 5-aza-dC in the 4 pancreatic cancer cells lines in this study was almost identical to the number of genes induced in 4 different pancreatic cancer cell lines (n = 131) in an earlier study by our group [30], [57]. This result is consistent with the hypothesis that CAFs are not subject to many promoter hypermethylation-induced gene silencing events compared to neoplastic epithelial cells. Previously, we demonstrated that 5-aza-dC treatment of the HPV E6E7-immortalized pancreatic non-neoplastic cell line, HPDE induced the expression of 93 genes (>5-fold with the Affymetrix U133 microarray) [30], [57], which is higher than the number of genes induced in fibroblasts but somewhat less than the number induced in pancreatic cancer cell lines. We also find that fibroblasts from non-neoplastic pancreata also have significantly fewer genes induced by 5-aza-dC than pancreatic cancer cell lines. These control fibroblasts respond similarly to 5-aza-dC as CAFs respond with respect to the numbers of genes.
Our findings indicate that despite residing in the same tumor microenvironment and undergoing similar environmental influences, it is the cancer cells that acquire most of the gene silencing DNA methylation alterations. Perhaps this is not surprising as CAFs are not thought to be under the same selective pressures as cancer cells. It was particularly notable that there was no evidence of silencing of tumor suppressor genes in CAFs supporting the notion that these cells are not under clonal selection [34]. In addition to clonal selection pressures, one of the proposed mechanisms for DNA methylation events in cancer cells are the mutations occurring during clonal expansion, some of which have been reported to influence DNA methylation. For example, amplifications in PML-RAR recruit DNA methyltransferases to promoters and cause hypermethylation [58]. Because CAFs do not undergo a similar clonal expansion from oncogenic and tumor suppressor mutations, it is not expected that they would acquire mutations that contribute to altered methylation patterns. Pancreatic and other cancer cells undergo methylation-induced silencing of numerous growth regulatory genes during clonal expansion (e.g. p16, hMLH1, p14arf, SPARC, RELN, TFPI-2 and others [3], [30], [57], [59], [60], [61], [62]) and it is notable that we found no evidence that these genes are silenced by methylation in CAFs. In addition, age-related changes in DNA methylation did not appear to have had a major influence on fibroblast gene silencing. Although age-related DNA methylation are thought to be an important contributor to the methylation alterations of cancer cells such changes typically induce partial methylation of a small percentage of cells not sufficient to induce widespread gene silencing [63]. One explanation for the relative lack of methylation found in CAFs is the relative plasticity of fibroblasts as compared to epithelial cells. Studies comparing DNA methylation patterns at different states of differentiation indicate that cells undergo methylation-induced silencing of genes associated with pluripotency, development, and imprinting, suggesting that terminally differentiated cells would undergo more DNA methylation and would be more susceptible to 5-aza-dC induced reexpression of these genes [64], [65]. The plasticity of fibroblasts suggests that these cells are less differentiated than epithelial cells, consistent with the observation that they are less responsive than epithelial cells to 5-aza-dC treatment.
Our results suggest that few genes are silenced by DNA methylation in CAFs. Notably, 5-aza-dC does not reactivate the expression of all genes silenced by DNA methylation and works synergistically with other epigenetic modifying drugs such as histone deacetylase (HDAC) inhibitors, so it is possible that some genes are silenced by methylation in CAFs that are not induced by 5-aza-dC alone.
We selected ADAM12 for further analysis because it was one of the few genes that we found to be overexpressed in pancreatic CAFs [27] and known to function in tumor-stromal cell interactions. We found relative hypomethylation of the ADAM12 gene promoter in nine pancreatic CAFs expressing ADAM12 relative to the non-expressing fibroblast lines HPNE and SC2. Consistent with these findings, we observed overexpression of Adam12 protein in CAFs in primary pancreatic adenocarcinomas. Adam12 overexpression has not previously reported in pancreatic cancer stroma but is known to be overexpressed in cancer cells of multiple tumor types [66], and urinary Adam12 has been evaluated as a potential marker of bladder and urinary cancers [67].
Gene hypomethylation of overexpressed genes has been observed in pancreatic and other cancers [68] and promoter hypomethylation correlates with gene transcription [69]. Studies investigating mechanisms of demethylation have investigated whether such hypomethylation is passive rather than active [70], and recently, evidence has emerged implicating an active DNA demethylase function to DNA glycosylases [71].
The present results may hold important implications for therapeutic targeting of CAFs. Combined with previous studies indicating that fibroblasts are less susceptible than cancer cells to the cytotoxic effects of 5-aza-dC ([72] and our own observations), these data suggest that CAFs would not likely be very responsive to targeting w/DNA demethylating agents. They further suggest that using methylation markers in CAFs for diagnostic purposes may not be a viable approach, since CAFs do not undergo many DNA methylation alterations compared to cancer cells.
In conclusion, we find that treatment with the DNMT1 inhibitor 5-aza-dC induces remarkably fewer genes in pancreatic CAFs than pancreatic cancer cells. Although further studies are needed to clarify the extent of epigenetic changes that may contribute to the CAF phenotype, our observations suggest that attempts to target the stromal cells therapeutically may need to focus directly on genes mediating tumor-stromal cell interactions, rather than on targeting CAFs with DNA methylation inhibitors.
Supporting Information
Hierarchical clustering of the global gene expression profiles of pancreatic fibroblasts.
(PPT)
The effect of 5-aza-dc on the proliferation of CAF19 cells as measured by MTT.
(PPTX)
Hierarchical clustering of the genes expression induced in pancreatic fibroblasts by 2-fold or more by 5-aza-dC.
(PPTX)
Genes induced by 5-aza-dC and overexpressed in pancreatic CAFs relative to control fibroblasts.
(XLSX)
Candidate hypermethylated genes underexpressed in CAFs relative to control fibroblasts.
(XLSX)
Funding Statement
This work was supported by the National Cancer Institute grants (CA62924 and RC2148346), and the Michael Rolfe Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ting AH, McGarvey KM, Baylin SB (2006) The cancer epigenome–components and functional correlates. Genes Dev 20: 3215–3231. [DOI] [PubMed] [Google Scholar]
- 2. Esteller M (2007) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 8: 286–298. [DOI] [PubMed] [Google Scholar]
- 3. Omura N, Goggins M (2009) Epigenetics and epigenetic alterations in pancreatic cancer. Int J Clin Exp Pathol 2: 310–326 Epub 2008 Nov 2015. [PMC free article] [PubMed] [Google Scholar]
- 4. Vincent A, Omura N, Hong SM, Jaffe A, Eshleman J, et al. (2011) Genome-Wide Analysis of Promoter Methylation Associated with Gene Expression Profile in Pancreatic Adenocarcinoma. Clin Cancer Res 17: 4341–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, et al. (2010) Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 20: 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matsubayashi H, Sato N, Brune K, Blackford AL, Hruban RH, et al. (2005) Age- and disease-related methylation of multiple genes in nonneoplastic duodenum and in duodenal juice. Clin Cancer Res 11: 573–583. [PubMed] [Google Scholar]
- 7. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, et al. (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102: 10604–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jin B, Yao B, Li JL, Fields CR, Delmas AL, et al. (2009) DNMT1 and DNMT3B modulate distinct polycomb-mediated histone modifications in colon cancer. Cancer Res 69: 7412–7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones PA, Liang G (2009) Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 10: 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA (2001) Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res 61: 3573–3577. [PubMed] [Google Scholar]
- 11. Vincent A, Herman JM, Schulick R, Hruban R, Goggins M (2011) Pancreatic Cancer. Lancet 378: 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boyd ZS, Raja R, Johnson S, Eberhard DA, Lackner MR (2009) A tumor sorting protocol that enables enrichment of pancreatic adenocarcinoma cells and facilitation of genetic analyses. J Mol Diagn 11: 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, et al. (2008) Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res 68: 918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, et al. (2004) Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol 2: E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cohen SJ, Alpaugh RK, Palazzo I, Meropol NJ, Rogatko A, et al. (2008) Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas 37: 154–158. [DOI] [PubMed] [Google Scholar]
- 16. Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, et al. (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14: 518–527. [DOI] [PubMed] [Google Scholar]
- 17. Iacobuzio-Donahue CA, Argani P, Hempen PM, Jones J, Kern SE (2002) The desmoplastic response to infiltrating breast carcinoma: gene expression at the site of primary invasion and implications for comparisons between tumor types. Cancer Res 62: 5351–5357. [PubMed] [Google Scholar]
- 18. Ohuchida K, Mizumoto K, Murakami M, Qian LW, Sato N, et al. (2004) Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res 64: 3215–3222. [DOI] [PubMed] [Google Scholar]
- 19. Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6: 392–401. [DOI] [PubMed] [Google Scholar]
- 20. Fujita H, Ohuchida K, Mizumoto K, Nakata K, Yu J, et al. (2010) alpha-Smooth Muscle Actin Expressing Stroma Promotes an Aggressive Tumor Biology in Pancreatic Ductal Adenocarcinoma. Pancreas [DOI] [PubMed] [Google Scholar]
- 21. Eyden B (2008) The myofibroblast: phenotypic characterization as a prerequisite to understanding its functions in translational medicine. J Cell Mol Med 12: 22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, et al. (2004) TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303: 848–851. [DOI] [PubMed] [Google Scholar]
- 23. Pietras K, Pahler J, Bergers G, Hanahan D (2008) Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med 5: e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ellis LM, Hicklin DJ (2008) VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 8: 579–591. [DOI] [PubMed] [Google Scholar]
- 25. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, et al. (2009) Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324: 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, et al. (2009) Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A 106: 4254–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walter K, Omura N, Hong SM, Griffith M, Vincent A, et al. (2010) Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clin Cancer Res 16: 1781–1789 Epub 2010 Mar 1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, et al. (2008) A paracrine requirement for hedgehog signalling in cancer. Nature 455: 406–410. [DOI] [PubMed] [Google Scholar]
- 29. Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, et al. (2007) Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol 25: 319–325. [DOI] [PubMed] [Google Scholar]
- 30. Sato N, Fukushima N, Maehara N, Matsubayashi H, Koopmann J, et al. (2003) SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions. Oncogene 22: 5021–5030. [DOI] [PubMed] [Google Scholar]
- 31. Bhowmick NA, Neilson EG, Moses HL (2004) Stromal fibroblasts in cancer initiation and progression. Nature 432: 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, et al. (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121: 335–348. [DOI] [PubMed] [Google Scholar]
- 33. Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, et al. (2004) Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 6: 17–32. [DOI] [PubMed] [Google Scholar]
- 34. Campbell I, Polyak K, Haviv I (2009) Clonal mutations in the cancer-associated fibroblasts: the case against genetic coevolution. Cancer Res 69: 6765–6768; discussion 6769. [DOI] [PubMed] [Google Scholar]
- 35. Hu M, Yao J, Cai L, Bachman KE, van den Brule F, et al. (2005) Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet 37: 899–905 Epub 2005 Jul 2010. [DOI] [PubMed] [Google Scholar]
- 36. Jiang L, Gonda TA, Gamble MV, Salas M, Seshan V, et al. (2008) Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res 68: 9900–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, et al. (2008) No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet 40: 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walter K, Omura N, Hong SM, Griffith M, Goggins M (2008) Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer Biol Ther 7: 882–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sato N, Fukushima N, Maitra A, Iacobuzio-Donahue CA, van Heek NT, et al. (2004) Gene expression profiling identifies genes associated with invasive intraductal papillary mucinous neoplasms of the pancreas. Am J Pathol 164: 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, et al. (2004) Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res 64: 8492–8495. [DOI] [PubMed] [Google Scholar]
- 41. Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, et al. (2008) Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res 68: 4331–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R (2007) Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 67: 10123–10128. [DOI] [PubMed] [Google Scholar]
- 43. Mann J, Chu DC, Maxwell A, Oakley F, Zhu NL, et al. (2010) MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 138: 705–714, 714 e701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hanson JA, Gillespie JW, Grover A, Tangrea MA, Chuaqui RF, et al. (2006) Gene promoter methylation in prostate tumor-associated stromal cells. J Natl Cancer Inst 98: 255–261. [DOI] [PubMed] [Google Scholar]
- 45. Liang G, Gonzales FA, Jones PA, Orntoft TF, Thykjaer T (2002) Analysis of gene induction in human fibroblasts and bladder cancer cells exposed to the methylation inhibitor 5-aza-2′-deoxycytidine. Cancer Res 62: 961–966. [PubMed] [Google Scholar]
- 46. Lee KM, Nguyen C, Ulrich AB, Pour PM, Ouellette MM (2003) Immortalization with telomerase of the Nestin-positive cells of the human pancreas. Biochem Biophys Res Commun 301: 1038–1044. [DOI] [PubMed] [Google Scholar]
- 47. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, et al. (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321: 1801–1806 Epub 2008 Sep 1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu J, Li A, Hong SM, Hruban RH, Goggins M (2012) MicroRNA alterations of pancreatic intraepithelial neoplasias. Clin Cancer Res 18: 981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kelloff GJ, Sullivan DC, Baker H, Clarke LP, Nordstrom R, et al. (2007) Workshop on imaging science development for cancer prevention and preemption. Cancer Biomark 3: 1–33. [DOI] [PubMed] [Google Scholar]
- 50. Yu J, Ohuchida K, Mizumoto K, Sato N, Kayashima T, et al. (2010) MicroRNA, hsa-miR-200c, is an independent prognostic factor in pancreatic cancer and its upregulation inhibits pancreatic cancer invasion but increases cell proliferation. Mol Cancer 9: 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Omura N, Li CP, Li A, Hong SM, Walter K, et al. (2008) Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma. Cancer Biol Ther 7: 1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ferguson AT, Evron E, Umbricht CB, Pandita TK, Chan TA, et al. (2000) High frequency of hypermethylation at the 14-3-3 sigma locus leads to gene silencing in breast cancer. Proc Natl Acad Sci U S A 97: 6049–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sun W, Liu Y, Glazer CA, Shao C, Bhan S, et al. (2010) TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin Cancer Res 16: 857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lau JC, Hanel ML, Wevrick R (2004) Tissue-specific and imprinted epigenetic modifications of the human NDN gene. Nucleic Acids Res 32: 3376–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kanda M, Nomoto S, Okamura Y, Nishikawa Y, Sugimoto H, et al. (2009) Detection of metallothionein 1G as a methylated tumor suppressor gene in human hepatocellular carcinoma using a novel method of double combination array analysis. Int J Oncol 35: 477–483. [DOI] [PubMed] [Google Scholar]
- 56. Iba K, Albrechtsen R, Gilpin BJ, Loechel F, Wewer UM (1999) Cysteine-rich domain of human ADAM 12 (meltrin alpha) supports tumor cell adhesion. Am J Pathol 154: 1489–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sato N, Fukushima N, Maitra A, Matsubayashi H, Yeo CJ, et al. (2003) Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res 63: 3735–3742. [PubMed] [Google Scholar]
- 58. Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, et al. (2002) Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295: 1079–1082. [DOI] [PubMed] [Google Scholar]
- 59. Fukushima N, Sato N, Ueki T, Rosty C, Walter KM, et al. (2002) Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am J Pathol 160: 1573–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sato N, Fukushima N, Chang R, Matsubayashi H, Goggins M (2006) Differential and epigenetic gene expression profiling identifies frequent disruption of the RELN pathway in pancreatic cancers. Gastroenterology 130: 548–565. [DOI] [PubMed] [Google Scholar]
- 61. Sato N, Matsubayashi H, Abe T, Fukushima N, Goggins M (2005) Epigenetic down-regulation of CDKN1C/p57KIP2 in pancreatic ductal neoplasms identified by gene expression profiling. Clin Cancer Res 11: 4681–4688. [DOI] [PubMed] [Google Scholar]
- 62. Sato N, Parker AR, Fukushima N, Miyagi Y, Iacobuzio-Donahue CA, et al. (2005) Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene 24: 850–858. [DOI] [PubMed] [Google Scholar]
- 63. Menigatti M, Truninger K, Gebbers JO, Marbet U, Marra G, et al. (2009) Normal colorectal mucosa exhibits sex- and segment-specific susceptibility to DNA methylation at the hMLH1 and MGMT promoters. Oncogene 28: 899–909. [DOI] [PubMed] [Google Scholar]
- 64. Dahl JA, Collas P (2007) Q2ChIP, a quick and quantitative chromatin immunoprecipitation assay, unravels epigenetic dynamics of developmentally regulated genes in human carcinoma cells. Stem Cells 25: 1037–1046. [DOI] [PubMed] [Google Scholar]
- 65. Freberg CT, Dahl JA, Timoskainen S, Collas P (2007) Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Mol Biol Cell 18: 1543–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kveiborg M, Albrechtsen R, Couchman JR, Wewer UM (2008) Cellular roles of ADAM12 in health and disease. Int J Biochem Cell Biol 40: 1685–1702. [DOI] [PubMed] [Google Scholar]
- 67. Frohlich C, Albrechtsen R, Dyrskjot L, Rudkjaer L, Orntoft TF, et al. (2006) Molecular profiling of ADAM12 in human bladder cancer. Clin Cancer Res 12: 7359–7368. [DOI] [PubMed] [Google Scholar]
- 68. Sato N, Maitra A, Fukushima N, van Heek NT, Matsubayashi H, et al. (2003) Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res 63: 4158–4166. [PubMed] [Google Scholar]
- 69. Laurent L, Wong E, Li G, Huynh T, Tsirigos A, et al. (2010) Dynamic changes in the human methylome during differentiation. Genome Res 20: 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kim ST, Fields PE, Flavell RA (2007) Demethylation of a specific hypersensitive site in the Th2 locus control region. Proc Natl Acad Sci U S A 104: 17052–17057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhu JK (2009) Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet 43: 143–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bender CM, Pao MM, Jones PA (1998) Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res 58: 95–101. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Hierarchical clustering of the global gene expression profiles of pancreatic fibroblasts.
(PPT)
The effect of 5-aza-dc on the proliferation of CAF19 cells as measured by MTT.
(PPTX)
Hierarchical clustering of the genes expression induced in pancreatic fibroblasts by 2-fold or more by 5-aza-dC.
(PPTX)
Genes induced by 5-aza-dC and overexpressed in pancreatic CAFs relative to control fibroblasts.
(XLSX)
Candidate hypermethylated genes underexpressed in CAFs relative to control fibroblasts.
(XLSX)