

Abstract
Growing evidence exists for a new role for apoptotic cell recognition and clearance in immune homeostasis. Apoptotic cells at all stages, irrespective of membrane integrity, elicit a signature set of signaling events in responding phagocytes, both professional and non-professional. These signaling events are initiated by receptor-mediated recognition of apoptotic determinants, independently of species, cell type, or apoptotic stimulus. We propose that the ability of phagocytes to respond to apoptotic targets with a characteristic set of signaling events comprises a second distinct dimension of innate immunity, as opposed to the traditional innate discrimination of self vs. non-self. We further propose that a loss or abnormality of the signaling events elicited by apoptotic cells, as distinct from the actual clearance of those cells, may predispose to autoimmunity.
Keywords: Innate immunity, apoptosis, autoimmunity, inflammation, phagocytosis, signal transduction
Introduction and overview
Cells are continually dying throughout the body. Such death occurs in one of two predominant forms: physiologic (apoptosis) or pathologic (necrosis). The very large number of dying cells, even in health, requires that cell corpses, whether apoptotic or necrotic, be recognized and cleared in a rapid, efficient manner. Professional phagocytes, such as macrophages and dendritic cells, are primarily charged with the duty of clearance, but all cells, including fibroblasts and epithelial cells, are capable of dead cell clearance. Importantly, the consequences of these two forms of cell death for surrounding tissues are drastically different. Apoptotic cell death is a conserved, cell-autonomous process that ensures specifically targeted removal of dying cells in an orderly and non-inflammatory fashion. In contrast, necrotic cell death, which also triggers phagocytosis, is marked by rapid and disorganized cellular swelling and rupture, and is typically seen in association with inflammation and tissue injury.
In this review, we will focus primarily on the signaling events induced in healthy surrounding cells by their interaction with apoptotic vs. necrotic targets. As far as possible, we will distinguish between signaling events initiated by the specific recognition of apoptotic vs. necrotic targets and signaling events initiated by the common machinery of phagocytosis. Our data support the view that dead cells are not merely cleared by phagocytes, but instead elicit a potent affirmative response that critically modulates the survival, proliferation, and inflammatory activity of interacting cells. We propose that this response to dead cells comprises an important and under-appreciated dimension of innate immunity. This added dimension depends upon the discrimination of apoptotic and necrotic cells from viable cells, as opposed to the discrimination of “self” from “non-self”, typically considered the crucial focus of immunity. Appreciation of these two independent dimensions of innate immune recognition has important implications for the pathogenesis of autoimmunity, in particular the popular “delayed clearance model”. As we will discuss, the impaired clearance of apoptotic targets has two important consequences, whose roles in the development of autoimmunity have not been properly distinguished: persistence of apoptotic corpses, and loss of apoptotic target-initiated affirmative signaling.
Discrimination of apoptotic from necrotic corpses
The differential response by surrounding tissues to apoptotic vs. necrotic targets led to the hypothesis that properties unique to the dying cell determine the mode and outcome of clearance [1]. Moreover, the fact that clearance of apoptotic targets occurs before rupture of the cell membrane focused attention on the presumptive expression of apoptotic cell surface determinants for phagocytosis, or so-called “eat me” signals [2]. Identification of these surface determinants, which may include alterations in lipid and carbohydrate composition as well as the appearance of oxidation products or other modified molecules, remains an area of ongoing investigation [1]. Complementary studies have implicated several phagocyte-specific molecules as putative receptors for apoptotic corpses [1].
It is critical to note that the analysis of these putative receptors has, in many cases, been incomplete. Most functional tests have evaluated apoptotic target “uptake”, without discriminating between actual recognition and engulfment. Indeed, in those cases for which the functional roles of particular gene products were assessed, the actions of those molecules (for example, the Class A scavenger receptor, the ABC-type transporter ABC1, and the Mer tyrosine kinase) were found to be limited to the phagocytic process, and to play no role in specific recognition [3–5]. Genetic studies of cell death in Caenorhabditis elegans further illuminate this issue. Engulfment in C. elegans involves the products of at least seven genes, which act within redundant converging pathways for the clearance of dead cells [6,7]. The mammalian orthologues of these gene products (such as Rac1, DOCK180 and ELMO) have also been implicated in the clearance of dead cells [6–8]. Importantly, recent work has shown that the engulfment pathways encoded by these genes in C. elegans are not specific for cells undergoing apoptotic cell death [9]. Rather, these genes are required for the clearance of both apoptotic and necrotic corpses.
To date, then, the phagocyte receptors that mediate the recognition of apoptotic vs. necrotic cells remain elusive. Since apoptotic and necrotic targets share a common pathway for phagocytosis, it would seem that the differential response to these two targets must lie at the level of target recognition. In keeping with this notion, we have found that discrimination between apoptotic and necrotic corpses occurs at the level of target recognition, or binding, and is independent of engulfment [10]. As shown by quantitative and objective fluorescence assays, the recognition by macrophages of each type of dead cell occurs via a saturable, receptor-mediated process [10]. Notably, these two receptor-mediated processes are distinct and non-competing [10].
Recognition-dependent anti-inflammatory responses by apoptotic cells
The uniquely defining property of an apoptotic corpse is its ability to be removed without inflammation. Our experiments have shown that the distinct modes of recognition for apoptotic vs. necrotic targets by engulfing macrophages are linked to their opposing effects on the inflammatory response [10]. The anti-inflammatory activity of apoptotic cells has been documented by several groups, and is readily observed at the level of inhibition of secretion of inflammatory cytokines from engulfing macrophages [11,12]. In contrast, necrotic cells, which are recognized by a distinct mechanism, tend to enhance pro-inflammatory macrophage responses (although alone they are usually not sufficient to trigger those responses) [10]. Typically, necrosis is induced by brief heating; but other methods, including freeze-thaw lysis, yield similar results. Strikingly, the inhibitory effect of apoptotic targets is dominant to the stimulatory effect of necrotic targets [10,13], demonstrating unequivocally that the acquisition of a cell-associated anti-inflammatory activity represents a gain-of-function by the apoptotic cell. Moreover, this inhibitory effect persists at all stages of apoptotic cell death, irrespective of cell membrane integrity [10,13,14].
In a series of studies, we have made the following observations on the anti-inflammatory response to apoptotic targets by macrophages [10,14]. First, recognition of apoptotic targets inhibited transcription of several cytokine genes, including interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) [14]. Apoptotic inhibition occurred independently of new protein synthesis, and, therefore, represented an immediate-early response [14]. Second, transcriptional reporters, such as a luciferase construct driven by the authentic IL-8 promoter or a simplified NF-κB-dependent promoter, reliably recapitulated the inhibitory effect of apoptotic targets [14]. Third, the inhibitory effect of apoptotic targets, as assessed by NF-κB-dependent transcriptional activity 5 h after exposure to apoptotic targets, was a direct consequence of recognition [14]. An autocrine/paracrine contribution by secreted transforming growth factor-β (TGF-β) was excluded both by blocking antibodies and by pharmacological inhibition of TGF-β type I receptor kinase activity [14]. These findings do not exclude a secondary role for TGF-β, or other secreted factors, which may act at later times to amplify and disseminate the modulatory response [11,12]. Finally, since NF-κB-dependent transcriptional activity was unaffected by the cytoskeletal inhibitor cytochalasin D, which effectively blocked engulfment, modulation was dependent on apoptotic cell recognition and independent of engulfment [10,14].
Proximal signaling events elicited by recognition of apoptotic vs. necrotic corpses
These observations prompted us to examine the proximal signaling events that are induced in primary macrophage cultures (elicited peritoneal and bone marrow-derived) by the recognition and/or phagocytosis of apoptotic vs. necrotic targets [13,15]. We hypothesized that signaling events for which apoptotic and necrotic targets elicit opposite responses must be triggered by distinct receptor-mediated recognition, while signaling events for which apoptotic and necrotic targets elicit similar responses are triggered by the shared machinery of phagocytosis. We also examined the response to cell-sized latex beads, as a non-receptor-mediated trigger of phagocytosis. We focused on early signaling events, within 15 min of exposure, that could impact the survival or proliferative potential of responding macrophages, particularly within the phosphatidylinositol 3-kinase (PI3K)/Akt axis and the family of mitogen-activated protein kinase (MAPK) modules. Our results are summarized in Figure 1.
Figure 1.

Discrete signaling events elicited within phagocytes by the recognition vs. engulfment of dead cells. The clearance of dead cells by phagocytes, both professional and non-professional, is associated with a number of early signaling events. These signaling events can be broken up into: (A) those mediated by the recognition of apoptotic vs. necrotic corpses, and (B) those mediated by their engulfment via a common phagocytic machinery. (A) Receptor-mediated discrimination of apoptotic vs. necrotic corpses elicits directionally opposite responses in pro-inflammatory transcription (denoted here as NFκB-dependent transcriptional activity) and the activity of the signaling kinases ERK1/2, JNK1/2 and p38. Apoptotic cells, at all stages, irrespective of the integrity of the plasma membrane, initiate identical signaling events. (B) Signaling events linked to the engulfment of apoptotic and necrotic cells are directionally similar and lead to the activation of Akt and its downstream targets, GSK3β and BAD. Latex particles, which are taken up in a receptor-independent manner, elicit no recognition-dependent signaling events, but, like dead cells, activate Akt via their engulfment.
With respect to the PI3K/Akt axis, a major regulator of cell survival, we hypothesized that the phagocytosis of dead cells (apoptotic or necrotic) might confer a survival advantage on professional phagocytic cells such as macrophages [15]. For example, in situations in which a relative deficiency of survival factors results in apoptotic death, it would be crucial that phagocytic cells remain viable to facilitate apoptotic cell clearance. In accord with this hypothesis, apoptotic and necrotic targets both promoted the survival of macrophages induced to undergo apoptosis by withdrawal of serum and survival factors [15]. Apoptotic and necrotic targets activated Akt in a dose-dependent manner. The effect of dead cells on survival seemed to depend on phagocytosis, as prior exposure to colchicine, a microtubular inhibitor, blocked the activation of Akt by apoptotic and necrotic targets [15]. In support of the notion that activation of Akt is dependent on phagocytosis, rather than specific recognition of apoptotic and necrotic cells, exposure to latex beads, a neutral phagocytic stimulus, also activated Akt [15].
In marked contrast, the effects of apoptotic vs. necrotic targets on MAPK signaling appeared to depend on recognition, as these two types of targets induced opposite signaling events [13,15]. Exposure to apoptotic cells strongly inhibited both basal and macrophage-colony stimulated factor (M-CSF)-induced phosphorylation of ERK1/2 [13,15]. Inhibition of ERK1/2 by apoptotic targets occurred to an equivalent extent whether apoptotic targets were added before or after M-CSF stimulation [15]. Notably, while contact with macrophages was required for apoptotic targets to exert their inhibitory effect, they were potently inhibitory even when present at far fewer that one target per macrophage, suggesting that phagocytosis was not required [13]. This is consistent with our observations of brief, serial binding of targets to macrophages. In accord with their inhibition of ERK1/2, apoptotic targets inhibited M-CSF-stimulated proliferation by up to 100% [15]. Opposite to the effects of apoptotic cells, exposure to necrotic cells stimulated proliferation and activated ERK1/2 in a dose-dependent manner [15]. Latex beads, a neutral phagocytic stimulus, had no effect on proliferation or ERK1/2 phosphorylation [13,15].
We also examined the effects of apoptotic and necrotic cells on two additional MAPK modules, Jun N-terminal kinases 1 and 2 (JNK1/2) and p38 [13]. In contrast to their inhibitory effect on ERK1/2 activity, apoptotic cells induced dose-dependent phosphoryl-ation of both JNK1/2 and p38. The concentration dependency of the JNK1/2 and p38 responses to apoptotic cells was similar to that of the ERK1/2 response. Although the JNK and p38 modules are activated in response to a wide variety of stresses and inflammatory stimuli, exposure to necrotic cells had no detectable effect on these two pathways, either alone or administered prior to M-CSF stimulation. Latex beads, like necrotic cells, had no effect on the levels of active JNK1/2 and p38. The divergent responses triggered in the three major MAPK modules by apoptotic and necrotic cells emphasizes the distinct processes of recognition and proximal signaling that are engaged by these different types of targets [10,13,15].
Ubiquity of the responses to apoptotic recognition
As the ability to ingest apoptotic cells is not restricted to professional phagocytes, we asked whether non-professional phagocytic cells can also discriminate apoptotic and necrotic targets and respond to apoptotic modulation in an anti-inflammatory manner similar to that by macrophages. For both mouse fibroblasts and human epithelial cells, as for macrophages, the specific recognition of apoptotic targets inhibited pro-inflammatory transcription [16,17]. We have observed similar specific apoptotic modulation in a wide variety of other cell types [17]. Indeed, we have observed no exceptions to date to the ubiquity of this cellular response to apoptotic recognition. Even non-phagocytic B- and T-lymphocytes exhibit this response to the recognition of apoptotic cells [17].
We have examined the full repertoire of responses to apoptotic vs. necrotic recognition in several of these cell types. To date, we have found that the differential response of apoptotic vs. necrotic targets on MAPK modules is conserved in mouse fibroblasts and mouse epithelial cells [16, 17]. Taken together, these results reinforce the view that the process of apoptotic recognition, linked to the modulation of such vital cell processes as inflammation and proliferation, is widely conserved.
Two-dimensionality of innate immune discrimination
Our observations prompt a fundamental reconsideration of the classical view of immune discrimination. The innate immune system is commonly viewed simply as a preprogrammed host response with predetermined specificity to non-self (especially microbial) invaders. In contrast to this essentially one-dimensional view of innate responsiveness, along a self vs. other (non-self) axis, our data suggest that the recognition of apoptotic determinants provides a second, distinct dimension of innate immune discrimination. These two independent criteria of innate recognition are essentially antagonistic with regard to functional inflammatory outcome. Importantly, the specific modulatory activity of apoptotic cells is neither species-specific nor influenced by self/non-self discrimination. The response of professional and non-professional phagocytes to apoptotic targets appears to be conserved, regardless of whether the targets derive from the same or different species or represent a homotypic or heterotypic interaction.
Figure 2 is a graphical representation of immune responsiveness as a function of these two independent criteria of innate immune recognition. As depicted, immune responsiveness is an integrated function of (at least) these two inputs. Within a population of responding cells, integration of these two independent criteria of responsiveness results in a continuum of potential responses within a two-dimensional field. Indeed, our cell culture experiments demonstrate the graded nature of responsiveness [10,13,14,17]. It remains to be determined whether individual cells can express such a continuum of responses or instead exist in alternative and discrete states. We hypothesize that the integration of responsiveness along these two axes corresponds to specific functional states.
Figure 2.
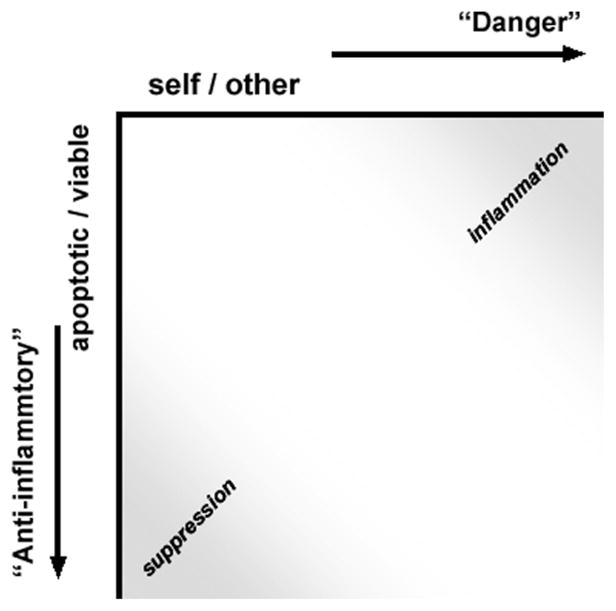
Two distinct criteria of innate immune recognition. Innate immune responsiveness is an integrated function of two independent criteria of immune recognition. One criterion is the well-appreciated discrimination along a self vs. other (non-self) axis. The second reflects the discrimination between viable and dead (particularly apoptotic) cells. These two distinct criteria of recognition are represented here along orthogonal axes. Whereas recognition of non-self determinants engages a signal transduction pathway linked to inflammatory outcomes, apoptotic recognition triggers distinct signaling events that result in antagonistic, anti-inflammatory outcomes. The placement of necrotic cells in this schema is less obvious. At least with regard to inflammatory responses, necrotic cells are more like pathogenic invaders than apoptotic cells, although their ability to elicit inflammatory responses tends to be modest at best (10,13).
Dual consequences of impaired clearance of apoptotic cells
A prevalent and popular model for the pathogenesis of autoimmunity is the delayed clearance model [18–20]. According to this model, autoimmunity can ensue if delayed or reduced clearance of apoptotic cells leads to their persistence in tissues past the point of cell membrane rupture. The model presumes that such late apoptotic cells will behave like necrotic cells, because of their release of potentially inflammatory intracellular contents, and that subsequent uptake and presentation of apoptotic antigens in the context of inflammatory signals will result in autoimmunity. Several studies in mice, in which genetic deficiencies of gene products involved in the engulfment of apoptotic targets (for example, C1q and the MER receptor tyrosine kinase) gave rise both to delayed clearance of apoptotic cells and to the development of systemic autoimmunity [19,20], lend support to this model.
Our data showing that recognition of apoptotic targets elicits an affirmative set of signaling events in responding phagocytes [10,13–17] suggest an alternative interpretation for the association between impaired clearance of apoptotic cells and the development of systemic autoimmunity (Figure 3). Indeed, most discussions of this association have neglected the fact that impaired clearance of apoptotic targets has two important consequences, whose roles in the development of autoimmunity have not been properly distinguished: (i) the persistence of the apoptotic corpses, and (ii) the loss of apoptotic target-initiated affirmative signaling [13]. We hypothesize that it is the second consequence—namely, the diminution or absence of apoptotic cell-dependent proximal signaling events in professional phagocytes—that plays the larger role in the development of autoimmunity [13].
Figure 3.

Exposure to apoptotic cells induces multiple signaling events that contribute to immune homeostasis. (A) The interaction of phagocytes with apoptotic cells results in a number of signaling events that are dependent upon both receptor-mediated recognition and engulfment of the apoptotic target (summarized in Figure 1). Among the signaling events induced by apoptotic cells, we speculate that some play a critical role in maintaining self-tolerance and preventing autoimmunity. (B) According to the model we suggest, loss of self-tolerance, with resultant autoimmunity, may occur through one of two fundamental mechanisms. First, impaired clearance of apoptotic cells, as may occur through targeted deletion of relevant genes, results in a decreased interaction of apoptotic cells with phagocytes, and therefore a decreased amount of tolerogenic signals within the phagocyte. The role of antigenic persistence is less clear, since apoptotic cells, at all stages, irrespective of membrane integrity, elicit identical signaling events. Alternatively, inherited or acquired abnormalities within the tolerogenic signaling pathways that are induced by apoptotic cells may constitute a predisposing background for the development of autoimmunity.
Importantly, there are at least three strong predictions of these two models that allow their discrimination. In comparing the models, we took advantage of the fact that necrotic and apoptotic targets have opposing effects on both pro-inflammatory transcription and the activity of the ERK, JNK, and p38 MAPK modules [10,13,15]. First, the delayed clearance model predicts that late apoptotic cells, which have lost membrane integrity, should behave like necrotic cells, whereas we predicted that signaling events triggered by late apoptotic cells should resemble those of early apoptotic cells. In fact, as discussed above, late apoptotic cells were indistinguishable from early apoptotic cells in all of their effects on macrophages. Late apoptotic cells inhibited basal and M-CSF-induced ERK1/2 activity, they activated JNK1/2 and p38, and they inhibited pro-inflammatory transcription [10,13,14]. Second, the delayed clearance model predicts that the signaling events induced by necrotic cells should be dominant over those induced by apoptotic cells. This prediction arises from the following considerations. Since apoptotic cells are generated continuously in vivo, delayed clearance of apoptotic cells will lead to the co-existence of both early and late apoptotic cells. Immunity will result only if the inflammatory signals in response to the presumed inflammatory intracellular contents of late apoptotic cells can override the anti-inflammatory effects of early apoptotic cells. Since the intracellular contents of necrotic cells should be at least as inflammatory as those of late apoptotic cells, we pitted apoptotic cells against necrotic cells. Again, in sharp contrast to the prediction of the delayed clearance model, the signaling events induced by apoptotic cells were strongly dominant over those induced by necrotic cells [10,13]. Of particular note, late apoptotic cells were just as dominant as early apoptotic cells in terms of inhibiting ERK1/2 activity and stimulating JNK1/2 and p38 [13]. Finally, in direct opposition to the delayed clearance model, which predicts that the leaked cellular contents of late apoptotic cells should be pro-inflammatory, soluble material recovered from these cells had no inflammatory effect [10,13]. Thus, in striking contrast to the predictions of the delayed clearance model, apoptotic cells appear to be functionally equivalent throughout their existence, irrespective of membrane integrity.
Abnormalities of apoptotic cell-dependent signaling in phagocytes from multiple murine models of autoimmunity
Taken together, our results argue strongly against the delayed clearance model, as currently formulated. Indeed, independent data demonstrate that the persistence of apoptotic corpses is not in and of itself sufficient to produce autoimmunity. Targeted deletion of CD14 or the mannose binding lectin led to the in vivo accumulation of apoptotic cells in the absence of autoimmunity [21,22].
The questions then are, how do apoptotic cells contribute to adaptive immune responsiveness normally, and how do aberrations associated with their clearance lead to autoimmunity? We hypothesize that, just as apoptotic cells affirmatively modulate innate immune responsiveness in cells that recognize them, so too do they specifically suppress the ability of antigen-presenting cells to stimulate antigen-specific adaptive immune responses.
According to the model we propose, loss of self-tolerance with resultant autoimmunity may occur through acquired or genetic abnormalities that interfere with the affirmative set of signaling events induced in phagocytes by the recognition and/or engulfment of apoptotic targets. As shown in Figure 3, abnormalities predisposing to autoimmunity can occur at multiple steps proximal to the generation of necessary tolerogenic signals, affecting not only the recognition and/or engulfment of apoptotic corpses (as in the targeted deletion of C1q and MER [19, 20]), but also any of the proximal signaling events or cascades elicited by specific recognition of apoptotic corpses [10,13–17].
Consistent with our hypothesis, macrophages from mice of all the major inbred murine models of spontaneous autoimmunity, including multiple strains that develop systemic lupus erythematosus (SLE), as well as the autoimmune diabetes-prone NOD strain, have an identical apoptotic cell-dependent abnormality in the expression of multiple cytokines [23,24]. Affected SLE-prone strains include MRL/+, MRL/lpr, NZB, NZW, NZB/W F1, BXSB, and LG/J. No similar defect in cytokine expression can be found in macrophages from 16 nonautoimmune strains, including three strains that develop type II (non-autoimmune) diabetes mellitus [23,24]. Importantly, in the absence of apoptotic cells, cytokine expression by these autoimmune-prone strains is completely comparable to that by nonautoimmune mice [23,24]. Furthermore, abberant cytokine expression is not the sole apoptotic cell-dependent abnormality observed in macrophages from auto-immune-prone mice. Macrophages from NOD and the same SLE strains also have a reversible defect in the activity of the cytoplasmic G-protein Rho, a key regulator of the cytoskeleton, resulting in profound abnormalities of adhesion and cytoskeletal organization [25,26]. Once again, no similar abnormalities were observed in macrophages from multiple non-autoimmune control strains. Also, in the absence of apoptotic cells, Rho activity, adhesion, and cytoskeletal organization were comparable between auto-immune and nonautoimmune mice [25,26]. These results provide strong support for the notion that abnormalities in the affirmative signaling events induced by apoptotic cells may be causally related to the development of autoimmunity.
Conclusions
Our studies have revealed that apoptotic cells affirmatively elicit specific signaling responses and functional outcomes in all cells with which they interact. Most significantly, apoptotic cells potently modulate the proliferation, survival, and inflammatory activity of responding cells. Our work leads us to suggest that apoptotic cells play an important role in normal immune homeostasis, and that aberrations in the recognition and signaling responses to apoptotic cells (as opposed to apoptotic cell engulfment) contribute to immune pathology. Recent work demonstrating the opposing effects of apoptotic and necrotic cells on immune homeostasis [27] also supports the view that the disparate effects of these two types of dead cells on immune function arise through signaling events initiated upon their receptor-dependent discrimination [10]. We suggest that acquired and/or genetic abnormalities that affect the signaling pathways elicited by dead cell discrimination play a major role in the development of immune pathology and, in particular, autoimmunity [13,23–26].
References
- 1.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: Clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 2.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation: Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Platt N, Suzuki H, Kurihara Y, Kodama T, Gordon S. Role for the class A macrophage scavenger receptor in the phagocytosis of apoptotic thymocytes in vitro. Proc Natl Acad Sci USA. 1996;93:12456–12460. doi: 10.1073/pnas.93.22.12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marguet D, Luciani MF, Moynault A, Williamson P, Chimini G. Engulfment of apoptotic cells involves the redistribution of membrane phosphatidylserine on phagocyte and prey. Nat Cell Biol. 1999;1:454–456. doi: 10.1038/15690. [DOI] [PubMed] [Google Scholar]
- 5.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 6.Kinchen JM, Cabello J, Klingele D, Wong K, Feichtinger R, Schnabel H, Schnabel R, Hengartner MO. Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C.elegans. Nature. 2005;434:93–99. doi: 10.1038/nature03263. [DOI] [PubMed] [Google Scholar]
- 7.Reddien PW, Horvitz HR. CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat Cell Biol. 2000;2:131–136. doi: 10.1038/35004000. [DOI] [PubMed] [Google Scholar]
- 8.Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC, Macara IG, Madhani H, Fink GR, Ravichandran KS. Unconventional Rac-GEF activity is mediated through the Dock180–ELMO complex. Nat Cell Biol. 2002;4:574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- 9.Chung S, Gumienny TL, Hengartner MO, Driscoll M. A common set of engulfment genes mediates removal of both apoptotic and necrotic cell corpses in C. elegans. Nat Cell Biol. 2000;2:931–937. doi: 10.1038/35046585. [DOI] [PubMed] [Google Scholar]
- 10.Cocco RE, Ucker DS. Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol Biol Cell. 2001;12:919–930. doi: 10.1091/mbc.12.4.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 13.Patel VA, Longacre A, Hsiao K, Fan H, Meng F, Mitchell JE, Rauch J, Ucker DS, Levine JS. Apoptotic cells, at all stages of the death process, trigger characteristic signaling events that are divergent from and dominant over those triggered by necrotic cells: Implications for the delayed clearance model of autoimmunity. J Biol Chem. 2006;281:4663–4670. doi: 10.1074/jbc.M508342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cvetanovic M, Ucker DS. Innate immune discrimination of apoptotic cells: Repression of pro-inflammatory macrophage transcription is coupled directly to specific recognition. J Immunol. 2004;172:880–889. doi: 10.4049/jimmunol.172.2.880. [DOI] [PubMed] [Google Scholar]
- 15.Reddy SM, Hsiao KH, Abernethy VE, Fan H, Longacre A, Lieberthal W, Rauch J, Koh JS, Levine JS. Phagocytosis of apoptotic cells by macrophages induces novel signaling events leading to cytokine-independent survival and inhibition of proliferation: Activation of Akt and inhibition of extracellular signal-regulated kinases 1 and 2. J Immunol. 2002;169:702–713. doi: 10.4049/jimmunol.169.2.702. [DOI] [PubMed] [Google Scholar]
- 16.Mitchell JE, Cvetanovic M, Tibrewal N, Patel V, Colamonici OR, Li MO, Flavell RA, Levine JS, Birge RB, Ucker DS. The presumptive phosphatidylserine receptor is dispensable for innate anti-inflammatory recognition and clearance of apoptotic cells. J Biol Chem. 2006;281:5718–5725. doi: 10.1074/jbc.M509775200. [DOI] [PubMed] [Google Scholar]
- 17.Cvetanovic M, Mitchell JE, Patel V, Avner BS, Su Y, van der Saag PT, Witte PL, Fiore S, Levine JS, Ucker DS. Specific recognition of apoptotic cells reveals a ubiquitous and unconventional innate immunity. J Biol Chem. 2006;281:20055–20067. doi: 10.1074/jbc.M603920200. [DOI] [PubMed] [Google Scholar]
- 18.Gaipl US, Voll RE, Sheriff A, Franz S, Kalden JR, Herrmann M. Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun Rev. 2005;4:189–194. doi: 10.1016/j.autrev.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell DA, Pickering MC, Warren J, Fossati-Jimack L, Cortes-Hernandez J, Cook HT, Botto M, Walport MJ. C1q deficiency and autoimmunity: The effects of genetic background on disease expression. J Immunol. 2002;168:2538–2543. doi: 10.4049/jimmunol.168.5.2538. [DOI] [PubMed] [Google Scholar]
- 20.Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devitt A, Parker KG, Ogden CA, Oldreive C, Clay MF, Melville LA, Bellamy CO, Lacy-Hulbert A, Gangloff SC, Goyert SM, Gregory CD. Persistence of apoptotic cells without autoimmune disease or inflammation in CD14−/− mice (2004) J Cell Biol. 2004;167:1161–1170. doi: 10.1083/jcb.200410057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stuart LM, Takahashi K, Shi L, Savill J, Ezekowitz RA. Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol. 2005;174:3220–3226. doi: 10.4049/jimmunol.174.6.3220. [DOI] [PubMed] [Google Scholar]
- 23.Koh JS, Wang Z, Levine JS. Cytokine dysregulation induced by apoptotic cells is a shared characteristic of murine lupus. J Immunol. 2000;165:4190–4201. doi: 10.4049/jimmunol.165.8.4190. [DOI] [PubMed] [Google Scholar]
- 24.Fan H, Longacre A, Meng F, Patel V, Hsiao K, Koh JS, Levine JS. Cytokine dysregulation induced by apoptotic cells is a shared characteristic of macrophages from NOD and SLE-prone mice. J Immunol. 2004;172:4834–4843. doi: 10.4049/jimmunol.172.8.4834. [DOI] [PubMed] [Google Scholar]
- 25.Longacre A, Koh JS, Hsiao K-H, Gilligan H, Fan H, Patel VA, Levine JS. Macrophages from lupus-prone MRL mice are characterized by abnormalities in Rho activity, cytoskeletal organization, and adhesiveness to extracellular matrix proteins. J Leukoc Biol. 2004;76:971–984. doi: 10.1189/jlb.0604346. [DOI] [PubMed] [Google Scholar]
- 26.Fan H, Patel VA, Longacre A, Levine JS. Abnormal regulation of the cytoskeletal regulator Rho typifies macrophages of the major murine models of spontaneous autoimmunity. J Leuko Biol. 2006;79:155–165. doi: 10.1189/jlb.0705408. [DOI] [PubMed] [Google Scholar]
- 27.Albert ML. Death-defying immunity: Do apoptotic cells influence antigen processing and presentation? Nat Rev Immunol. 2004;4:223–231. doi: 10.1038/nri11308. [DOI] [PubMed] [Google Scholar]