


Abstract
Prophages are phages in lysogeny that are integrated into, and replicated as part of, the host bacterial genome. These mobile elements can have tremendous impact on their bacterial hosts’ genomes and phenotypes, which may lead to strain emergence and diversification, increased virulence or antibiotic resistance. However, finding prophages in microbial genomes remains a problem with no definitive solution. The majority of existing tools rely on detecting genomic regions enriched in protein-coding genes with known phage homologs, which hinders the de novo discovery of phage regions. In this study, a weighted phage detection algorithm, PhiSpy was developed based on seven distinctive characteristics of prophages, i.e. protein length, transcription strand directionality, customized AT and GC skew, the abundance of unique phage words, phage insertion points and the similarity of phage proteins. The first five characteristics are capable of identifying prophages without any sequence similarity with known phage genes. PhiSpy locates prophages by ranking genomic regions enriched in distinctive phage traits, which leads to the successful prediction of 94% of prophages in 50 complete bacterial genomes with a 6% false-negative rate and a 0.66% false-positive rate.
INTRODUCTION
Phages, viruses that infect bacteria, have two lifestyles: lytic and lysogenic. During lysogenic growth, phages infect their host and then remain inside the microbial cell replicating with the genome. In this state, they are called prophages. These prophages will be part of the bacterial DNA in future cell divisions until appropriate environmental conditions cause them to release from their host and enter into a virulent lifestyle. The advantages of a lysogenic lifestyle for phages are numerous, including increased fecundity and increased survival within the protective bacterial environment. Integrated prophages can constitute up to 20% of a bacterial genome (1–3) and play a key role in the bacterial life cycle. Prophage integration can regulate bacterial populations, make inactive or alter the expression of some bacterial genes, and can convert non-pathogenic bacteria into pathogens and some virulent into hyper-virulent strains (4–6).
A prophage normally integrates into a genome by site-specific recombination, which is catalyzed by a family of proteins called integrases (7). These proteins recognize sequences on both the phage (attP, attachment site in the phage genome) and bacterial (attB, attachment site in the bacterial genome) genomes, and homologous recombination between these sites results in duplication of a short stretch of DNA in the continuity of the chromosome, resulting in the duplicated sites, attL and attR, flanking the inserted prophage and ready for the reverse reaction, excision of the phage from the chromosome. The att regions vary widely in total length and in the extent of the resulting duplication, which depends on the phage and its specific integration site within a bacterial genome (1,8–11). Phages often integrate into tRNA/tmRNA genes but do not exclusively use those loci as the target site for integration (12).
Identification of prophages in bacterial genomes is a difficult process. Current methodology of automated prophage identification usually relies on protein similarity searches to identify clusters of protein-encoding genes that have some similarity to known or predicted phage genes. Based on this approach, Phage_Finder (12) was one of the first automated applications for detecting prophages. Phage_Finder screens the bacterial genome with a fixed window size of 10 Kb and searches [using hidden Marokov models and BLAST (13)] for windows with at least four hits against a collection of bacteriophage proteins. These windows are then extended gene-by-gene if the annotated gene belongs to tRNAs, integrase gene, etc (12). ACLAME ProPhinder is another successful phage-finding algorithm that combines protein similarity and statistical methods (14,15). ProPhinder starts by determining phage-like coding sequences in an input bacterial genome by BLASTP similarity analysis against the ACLAME phage protein database. Then, it evaluates each phage-like genomic segment for the presence of potential prophages using statistical methods. Because these applications use homology-based approaches, they are limited to finding known prophages and it is difficult to locate those prophages that are not similar to known phages. An alternative approach for detecting prophages (DRAD) that depends on the dinucleotide relative abundance instead of sequence similarity was able to locate some of those prophages found by Prophinder and Phage_Finder as well as some novel prophages (16). No single tool is able to find all prophages in all bacterial genomes (16). This suggests that combining multiple methods or different characteristics of prophages may identify a larger set of prophages.
In this study, a bioinformatics tool (PhiSpy) was developed for identifying prophages, which focuses on the characteristics of prophages that exhibit no similarity to sequenced genomes. In particular, five distinctive similarity-agnostic characteristics were identified and their relative capabilities to define prophages were tested in the absence of homology to known phage proteins. These characteristics are protein length, transcription strand directionality, customized AT and GC skew, and the abundance of unique phage DNA sequence words. Optimized metrics were designed to quantify each of these characteristics and the random forest classification algorithm was used to predict prophages by ranking genomic regions based on those characteristics. In addition to each of these metrics, phiSpy also uses similarity-based approaches, thus enabling a complete identification of prophages in a genome. Finally, each predicted prophage region was evaluated by the identification of duplicate att sites and by phage protein similarity. PhiSpy found 94% of prophages in 50 bacterial genomes with a 6% false-negative rate and a 0.66% false-positive rate.
MATERIALS AND METHODS
Data collection
All bacterial genomes used in this analysis were retrieved from the Phage Annotation Tools and Methods server (Phantome server: http://www.phantome.org). As of March 2010, the server contained 547 complete bacterial genomes (at most 20 contigs) of which only 41 bacterial genomes (Supplemental Table S1) had 190 manually annotated prophages. All other lytic and lysogenic phage genomes were also collected from the Phantome server.
Data analysis
PhiSpy publicly available at http://phispy.sourceforge.net/ was written in python and C++. It has four steps (Supplemental Figure S1 is a flow chart of each step). Each step is described below.
Calculation of different characteristics
The first step calculates different parameters for the whole genome. The calculation of these parameters depends on a group of genes rather than a single gene. Therefore, for a complete genome, these parameters were computed using a sliding window of n genes. The average number of genes of the 190 known prophages is 39; so a window size of 40 genes was considered. The parameters are as follows:
Customized AT and GC skew. The customized AT/GC skew was calculated by modifying the cumulative skew calculation (17,18). For a group of consecutive genes, the average skew of A, C, G and T were measured using the following formula:
where n is the number of genes, Ai is the number of A nucleotide in the ith gene and so on. The customized AT and GC skews (described under ‘Results’ section) were developed and were calculated as follows:
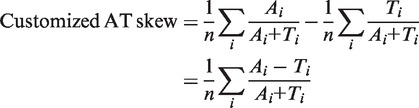 |
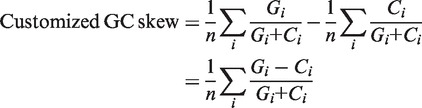 |
This customized version combines AT/GC and compensates for local deviations in the composition due to, for example, strand bias.
Difference in median protein length. The median (M) of the lengths of all the proteins in a bacterial genome was calculated. For a group of proteins in a given window, the median protein length (m) was calculated and the difference in median length was computed as (M − m).
Transcription strand orientation. For a given window size, the genes were partitioned in such a way so that all consecutive genes in a particular partition pointed in the same direction. The sum of the number of genes in the two largest partitions was taken for the window to maximize the number of consecutive genes in the same direction.
Abundance of phage words. A ‘word’ is defined as a set of 12 consecutive base pairs. Each gene was split into 12 bp long non-overlapping words (four consecutive amino acids each). A unique ‘phage word library’ was built based on the 41 bacterial genomes that have well-annotated prophages. The library was constructed as follows:
Bacterial words, B = {the ‘words’ of all bacterial genes of those 41 genomes but not including genes in prophages}
Phage words, P = {the ‘words’ of all prophage genes of those 41 genomes}
Unique ‘phage word library’ = P – B
To measure the abundance of phage ‘words’, Shannon’s index (19,20) and the frequency of the presence of phage words were calculated.
Shannon’s index was calculated by the following formula:
where pi is the frequency of those words which are present in the ‘phage word library’. The frequency of words (F) of a window was calculated by dividing the number of available phage words with the total number of words. For a given window, the abundance of phage words is F/H.
Homology. In a window of 40 genes, if there are at least 10 genes whose functional description is in phage subsystems (i.e. phage functional categories in the Phantome SEED database: http://www.phantome.org/PhageSeed/seedviewer.cgi), the window was considered a prophage window otherwise it was considered a bacterial window.
Classification algorithm
The second step of PhiSpy is to classify a window as a bacterial or a prophage window using random forests (21). A random forest is a classification algorithm that consists of multiple independent decision trees. The random forest requires a training set with multiple variables to build the forest of decision trees. In this case, there were five parameters whose values vary among distantly related genomes. If the similarities between two genomes were evolutionary significant, then they were considered as closely related genomes; otherwise, they were considered as distantly related genomes [the SEED API was used to determine relatedness (22)]. Therefore, for every group of closely related genomes, a different training set was constructed.
Training/test set. In the Phantome server, there were 547 complete bacterial genomes that had 20 contigs or fewer (as of March 2010). From these 547 bacterial genomes, 19 groups of closely related genomes were constructed, where each group has at least one genome from the set of 41 bacterial genomes with annotated prophages. These 19 groups included 114 out of 547 bacterial genomes. For each group, one genome with manually annotated prophages was used as the training set for the rest of the genomes of that group (Table 1 and Supplemental Table S2). The genomes that did not belong to any group and had no manually annotated prophages were tested using a universal generic training set (constructed in the same way described above but using all 41 bacterial genomes). The parameter ‘abundance of phage word’ was ignored in the universal generic training set (Table 2).
Table 1.
Performance analysis of phiSpy and comparison with phage_finder and prophinder
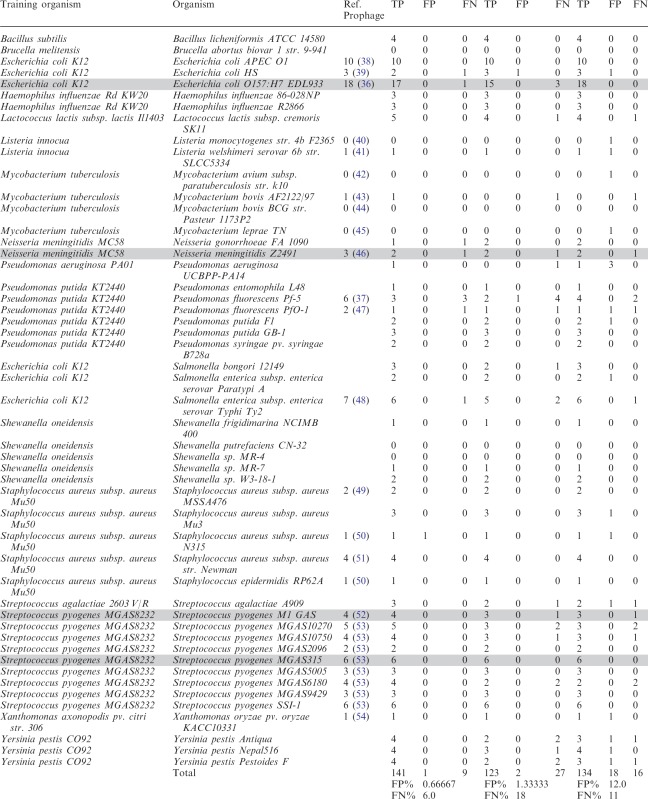 |
Table 2.
Effectiveness of different characteristics
| Closely related genomesa |
Distantly related genomesb |
|||
|---|---|---|---|---|
| GC/AT rich genome | Moderate GC% genome | GC/AT rich genome | Moderate GC% genome | |
| Transcription strand Directionality | +++ | +++ | + | + |
| Protein length | ++ | ++ | +/− | +/− |
| Customized AT skew | ++ | + | +/− | − |
| Customized GC skew | ++ | + | +/− | − |
| Abundance of phage words | ++ | ++ | − | − |
aThe effectiveness of different characteristics when the training genome and test genome are closely related.
bThe effectiveness of different characteristics when the training genome and test genome are distantly related.
The statistical software program, R (http://www.r-project.org), was used to implement the random forest (23). The random forest produces a rank for each window of the whole genome that suggests whether the window consists of bacterial or phage genes.
Processing the final rank for each gene
The third step of PhiSpy provides a prediction status—either 0 (for non prophage genes) or 1 (for prophage genes) for each gene in the genome. If the window size was n, each gene contributed to 1 to n windows. Therefore, the final rank of a particular gene was measured by taking the average rank of the window in which the gene participated. The prophage prediction status was calculated from the final rank. If the final rank was greater than half of the maximum rank of any gene in the genome, then the gene was considered as a phage gene; otherwise, it was considered as a bacterial gene.
Evaluation of the prediction
The final step is to define the att sites for the predicted prophages and the overall evaluation of the prophages. When phages integrate into their hosts’ genome, they are usually bounded by two att sites—a short repeated sequence that flanks the insertion site. To find this insertion site, for each predicted prophage region (considered an initial prediction), the following steps were followed.
Extending the predicted region up to 2000 bp on both sides.
Identifying all duplicate short DNA sequences in that region.
Finding the repeated pair that has minimum distance (<1000 bp) from either integrase/recombinase or tRNA/tmRNA genes or both. If there are multiple repeated pairs, the pair that covers the largest region was considered as the potential att sites. If no integrase/recombinase or tRNA/tmRNA genes were found, then the initially predicted region was considered.
After identifying the att sites, the next step is verifying the att sites. If the att sites lie inside the initial prediction, the number of phage-like proteins was counted for the two gaps (between attL and the start of the initial prediction and between attR and the end of the initial prediction). If the function of one-quarter of the genes in those two gaps belongs to phage subsystems, the initial prediction was considered as the final prediction otherwise the region covered by att sites was considered as the final prediction. If the att sites were outside the initial prediction, the same procedure was followed.
After verifying the att sites, the predicted prophages were evaluated by checking the function of all proteins in that region. If there are more than five proteins whose functions belong to the phage subsystems or are unknown and the number of phage-like/unknown proteins is at least half of the total number of proteins in the predicted region, then the region was considered as a potential prophage. However, if a group of proteins, whose functions belong to the phage subsystems, was not considered in the classification step as a probable prophage, then this region was also considered as a potential prophage.
Calculation of false positives and false negatives
The manually curated phage subsystems were used to evaluate the accuracy of the approach. A two-step program was designed to automatically calculate the error rate of the prophage prediction (for those genomes which have no information about prophages in their original genome analysis paper). In the first step of the program, true positives (TP) and false positives (FP) were predicted. If the predicted region consists of at least six phage proteins or 50% of the proteins within the predicted region belong to phage subsystems or are unknown, the predicted region was considered a TP prophage otherwise the region was considered a FP and not a prophage (those limits were determined by empirically). Prophages considered as TPs were divided into two groups: (i) known prophages—if the region contains phage-like proteins; we considered that it would be identified by similarity based approaches and therefore denoted it as a ‘known’ and (ii) undefined prophages—if the region has no phage-like protein; thus this would unlikely to be called a prophage. In the second step of the program, a region was considered as a false negative (FN) if there were at least six consecutive genes, whose functions belonged to the phage subsystems and the region was unidentified as a potential prophage. However, hypothetical proteins were ignored in this case, because the presence of several hypothetical proteins was not sufficient to predict a region as a prophage region.
RESULTS
Transcriptional strand orientation
The orientation of transcriptional units along the genome highly correlates with the direction of replication (24). Near the replication origin, genes are oriented in such a way that the direction of transcription coincides with the direction of replication (25). In a bacterial genome, which typically has a single origin of replication within its circular DNA, two replication forks can proceed independently (26). Phage genes cluster along the genome as they are organized into transcriptional units that are co-regulated (27). This causes a large cluster of phage genes to be oriented in the same direction, even if it collides with DNA replication (27). To test this hypothesis, 600 complete phage genomes (both lytic and lysogenic) and 110 complete bacterial genomes were analyzed. For both phage and bacterial genomes, the longest stretch of consecutive genes in the same direction was calculated as a percentage of the number of genes in the genome (Supplemental Figure S2). Likewise, the number of gene transcription strand changes was calculated as a percentage of the number of genes in the genome. For phage genomes, most of the consecutive genes are encoded on the same strand (Figure 1). In contrast, for bacterial genomes, the longest consecutive cluster of genes (presumably a co-transcribed region) is a small fraction of the genome, and genes frequently change their transcriptional direction. For bacterial genomes, these clusters of genes that have the same transcriptional orientation are most likely operons although this cannot be determined from the sequence alone. Other studies have shown that the average bacterial operon size is three genes (28) but the average phage operon size has not been reported.
Figure 1.

Orientation of proteins in 110 bacterial genomes (triangles) and 600 phages (x). Most of the phages have a large group of proteins facing in same direction and fewer proteins change their transcriptional directions. Bacteria, in contrast, cluster fewer proteins in the same direction and have high number of transcriptional direction changes.
Customized AT and GC skew
Amino acid composition and codon usage
Several articles have discussed how the adaptation of phages towards their hosts plays an important role in viral evolution (29,30). By comparing the 190 prophages in 41 bacterial genomes, it was apparent that the overall amino acid usage in prophages and their hosts is very similar (Figure 2), although for some amino acids (notably Asp, Glu, Phe, Gly, Lys, Pro, Arg), codon usage differs between prophages and their hosts’ genomes (Figure 3). For Lys and Phe, the frequency of AAA (in Lys) and TTT (in Phe) is higher in prophages than in bacteria, which is probably caused by the different usage of nucleotides A and T in prophages. Similarly, there are six codons that encode arginine—CGT, CGC, CGA, CGG, AGA and AGG from which CGC is more frequently used and AGA is less frequently used in bacteria. Presumably, this skew maintains the balance of G and C nucleotides in Arg codons.
Figure 2.

Amino acid distribution in the predicted proteins encoded in 41 bacterial genomes (filled square) and their 190 prophages (open square). The amino acid utilization is similar for both but the standard deviation (vertical bars) is higher for prophages than for bacteria.
Figure 3.

Frequency of codon usage in 41 bacterial genomes (filled square) with 190 prophages (open square). For some amino acids (notably, Asp, Glu, Phe, Gly, Lys, Pro, Arg), codon usage differs between prophages and their hosts’ genomes.
The GC skew of bacterial chromosomes directly correlates with the direction of replication (31). Local changes or distortion in the cumulative skew distribution may result from the insertion of foreign DNA into the chromosome (17). Therefore, customized AT and GC skew profiles were designed. Unlike the conventional calculations of cumulative DNA skew (18), the customized skew was designed not only for identifying local distortions but also for quantifying the variation of the codon usage in the window of genes.
The customized AT and GC skews (see ‘Materials and Methods’ section) were calculated separately for 41 bacterial genomes and their 190 prophages. For all genomes except Xylella fastidiosa, prophages have different AT and GC skews (either positive or negative) than their hosts (Figure 4). If there were no bias between the two DNA strands for mutation or selection, the base composition within each strand should be such that A=T and G=C (32,33). This implies that the customized AT or GC skew of the whole bacterial genome would be very small. In contrast, it was hypothesized that the customized skew of prophages should be different than that of the whole bacterial genome. To test this hypothesis, two independent samples were constructed. The first sample consisted of the absolute difference between the customized AT/GC skew of prophage genes and the customized AT/GC skew of regions immediately flanking the prophage insertion. The manually curated prophages were used to construct the first sample, and so the sample size was 190. To construct the second sample, 800 different bacterial regions were randomly selected from the 41 bacterial genomes that have manually curated prophages. The absolute differences of the customized AT/GC skew of these regions and the customized AT/GC skew of the flanking genes of these regions was calculated for the second sample. A permutation test was used to analyze whether these two samples are statistically different (Supplemental Materials) (34). The customized AT skew was significantly different at the 1% level (using both the mean and the median of the sample) between prophage genes and their flanking genes when compared to random chromosomal segments and their flanking genes. The customized GC skew was also significantly different between these samples at the 1% level using the mean value of the sample (but only at 5% level using the median of the sample). Therefore, the calculation of the customized AT/GC skew is useful to distinguish prophage genes from the bacterial genome, but with very slightly different confidence levels (Supplemental Figure S3).
Figure 4.

(A) Customized AT skew for 41 complete bacterial genomes (filled square) and their prophages (open square). The x-axis is sorted (ascending order) based on the genomes’ GC content as shown below the figure. (B) Customized GC skew for 41 complete bacterial genomes (filled square) and their prophages (open square). The x-axis is sorted (ascending order) based on the genomes’ GC content.
Protein length
Anecdotal evidence from the study of mycobacterial phages (35) suggests that phages typically have shorter genes than bacterial genes. The reasons are not clear, but phage genomes may enrich for smaller genes because of selective constraints on genome size, i.e. for faster replication or more efficient packaging. The mean protein length was calculated for 41 bacterial genomes and their prophages (Figure 5) and the result supports the previous study. However, our testing demonstrates that the median length works better than the mean length for discriminating prophage and bacterial genes. As each characteristic was calculated for sliding window (of several genes), using median length calculation, a sharp change occurs at the beginning of a prophage region, but using mean length, the change occurs gradually. The difference between the median of all protein lengths in a genome and the median length of phage proteins in that genome is much higher than the same calculation for bacterial proteins (Supplemental Figure S4).
Figure 5.

Average length of bacterial proteins (filled square) and phage proteins (open square) for 41 bacterial genomes and their prophages. Phage proteins are smaller than bacterial proteins. The x-axis is not sorted.
Abundance of phage words
To find a signature pattern of prophages, the oligonucleotide composition between prophages and their hosts was analyzed for 190 prophages from 41 bacterial genomes. Each gene was split into 12 bp non-overlapping words, i.e. four non-overlapping codons (from empirical testing, we found that nine bp sequences had too few combinations to be discriminatory and 15 bp sequences were too rare for accurate statistical modeling). For a DNA sequence of length 12, there are 412 different possible combinations. However, only 27% of the words from these combinations are present in our data set of 41 bacterial genomes with prophages. In total, 25% of the words (4 223 854) are present in bacterial genes, 0.65% of the words (109 533) are present in phage genes and 1.34% of the words (226 228) are common to both phage and bacterial genes. To verify whether these 0.65% words (phage word library) represent the phageness and are uncommon in bacterial genomes, Shannon’s index and the frequency of the presence of these words (see ‘Materials and Methods’ section) were calculated for 600 complete phage genomes and 400 complete bacterial genomes. Shannon’s index was used to measure the presence of the different combination of phage words, while the frequency measurement was used for the presence of phage words. For all bacterial genomes, Shannon’s index (H) is <1 and the frequency of phage words (F) is <6%. In contrast, for phages, H varies from 0 to 5.5 and F varies from 0% to 45% (Figure 6). The relation between H and F was given by an equation F = 8.57 H + 0.047 for phages (regression coefficient R2 = 0.995) and F = 5.85 H + 0.014 for bacterial genomes (regression coefficient R2 = 0.993). The constant term for both equations is negligibly small, and the difference between the two slopes is statistically significant (P < 0.001; details in Supplemental Materials). The abundance of phage words was calculated as the slope (F/H) and the value distinguishes phages and bacterial genomes (Figure 6). This indicates that the words from the phage word library are more frequent in phage genomes than bacterial genomes.
Figure 6.

Comparison of the abundance of phage words in bacteria (triangles) and phage genomes (x). (A) The Shannon’s index (H) versus the frequency (F) of the presence of phage words for 600 complete phage genomes and 400 randomly chosen complete bacterial genomes. Both H and F are very small for bacterial genomes compared to phage genomes. The relationship between H and F for phages is F = 8.57 H + 0.047 with a regression coefficient R2 = 0.995 and for bacterial genome the relation is F = 5.85 H + 0.014 with a regression coefficient R2 = 0.993. (B) The ratio of the frequency and Shannon’s index, i.e. F/H for 600 complete phage genomes and 400 randomly chosen complete bacterial genomes. There is a statistically significant difference in F/H (abundance of phage words) between phages and bacteria.
Importance of different characteristics
All the characteristics described above were used to predict prophages in bacterial genomes. The importance of each characteristic varies between different organisms and depends on the training genomes (Table 2). If a training genome and a test genome are closely related, then for most cases, the abundance of phage words is the most important characteristic. The directionality of the transcriptional strand is a strong indicator in all cases, although short phages are missed when this criterion is used alone. Protein length gives better performance in closely related genomes and can work for distantly related genomes when the genome has similar protein length with the training genome. The customized AT and GC skew calculation works better for closely related genomes and gives better performance in bacteria with extreme AT or GC composition, rather those with approximately even distribution of bases. In general, identification of prophages, even without similarity, was strongly assisted by training sets of closely related genomes with well-characterized prophages.
Performance analysis
PhiSpy was used to predict prophages in 95 complete bacterial genomes using the training set of closely related genomes and 412 complete bacterial genomes using a universal generic training set (these predicted prophages are accessible at http://www.phantome.org/Downloads/Prophages/PhiSpy/ and Supplemental Table S3). A detailed flow chart of the performance analysis is described in the Supplemental Figure S5. In the 95 genomes, 320 potential prophages were identified. Among those, three prophages (in Streptococcus agalactiae NEM316) had no phage-like proteins and were considered as previously undefined prophages (Supplemental Table S2). For performance analysis, the predicted prophages were manually checked (based on the phage subsystems) for 50 genomes (out of the 95 genomes). Most of the genomes with manually annotated prophages (1) were not used in the performance analysis because these prophages were also used to parameterize one of the five criteria (abundance of phage words) developed to identify prophages, and so the result will be biased. We did not test whether phiSpy could detect those prophages used in training the classifier, as that would be a biased assessment. However, to compare with the published data, four genomes were considered which have manually annotated prophages (gray colored genomes in Table 1). For Streptococcus pyogenes M1 GAS and Streptococcus pyogenes MGAS315, the identified prophages matched with the published data. In Escherichia coli O157:H7 EDL933, 13 prophage regions were found to contain 17 prophages by phiSpy although 18 prophages were found in the original analysis of this genome (36). Of those 18 published prophages, there were four instances where two adjacent prophages were merged by phiSpy. The prophage that was not identified is a short prophage (∼8.26 kb) and only has four phage-like proteins. The same reason goes for the unidentified prophage in Neisseria meningitidis Z2491 (Table 1). The three unidentified prophages in Pseudomonas fluorescens Pf-5 are defective prophages (37).
To compare the performance of phiSpy with other phage finding tools, phiSpy, phage_finder, prophinder and the DRAD method were used to predict prophages in 50 genomes (using default settings). For DRAD, no prophages were identified. As shown in Table 1, phiSpy identified 94% of the prophages with a 6% FN rate and a 0.66% FP rate, whereas phage_finder predicted 82% of the prophages with a 18% FN rate and a 1.33% FP rate and prophinder identified 89% prophages with a 11% FN rate and a 12% FP rate (the predicted prophages from phiSpy, phage_finder and prophinder are available at http://www.phantome.org/Downloads/Prophages/PhiSpy/Manually_Verified/). Therefore, phiSpy can identify more prophages than other phage finding tools with the lowest FP rate and FN rate. For prophinder, most of the FP prophages have a low score. Most of the prophages that were not reported by phiSpy were mainly misclassified in the evaluation step, because there were few proteins annotated as phage proteins in those prophage regions. Also, some comparatively short prophages which were unidentified by phage_finder were also missed in our classification step but found in our evaluation step.
From 412 complete bacterial genomes where no closely related genomes with manually annotated prophages were available to construct training sets, 826 prophages were predicted by phiSpy and 284 of those were considered previously undefined, as they have no known phage genes. Therefore, phiSpy can detect potentially new prophages without relying on a training set. To check whether other phage finding applications can identify the prophages having hypothetical or unknown proteins, phage_finder and prophinder were used to predict prophages in those 412 bacterial genomes (Figure 7 and Supplementary Table S3). Phage_finder identified 378 prophages where 22 of them are undefined prophages and prophinder predicted 339 prophages where 43 of them are undefined prophages (Figure 7). There are 198 known prophages and zero undefined prophages identified in common between phiSpy, phage_phinder and prophinder. There are 52 prophages predicted by both prophinder and phiSpy but not predicted by phage_finder, 120 prophages predicted by both phiSpy and phage_finder but not predicted by prophinder, and only two prophages identified by both phage_finder and prophinder but not predicted by phiSpy. Hence, phiSpy can predict more known and undefined prophages compared to those tools. The prediction of novel prophages in genomes is only the first step: biological experiments are required to investigate whether these phage-like regions are viable or can be induced out of the chromosome. Recently, an approach was described to test prophage viability in Salmonella enterica that could be used to test some of these prophages (55).
Figure 7.

(A) Comparative analysis of all prophages identified in 412 complete bacterial genomes by phiSpy, phage_finder and prophinder. (B) Comparative analysis of undefined prophages (no phage-like proteins) identified from 412 complete bacterial genomes.
DISCUSSION
In this report, we describe the identification of prophage regions within bacterial genome sequences. We have advanced the current analysis of prophage identification by introducing five distinctive characteristics of prophages that do not depend on sequence similarities. These characteristics were applied for the initial prediction of potential prophages, and each of these predictions was evaluated by identifying the phage insertion point and the similarity of phage proteins. PhiSpy was used to predict prophages in 507 complete bacterial genomes and a total of 1146 potential prophages were identified, including 287 putative prophages that have no homology to existing phages and may be novel mobile genetic elements. However, the total number of identified prophages might be different from the actually identified prophages for two reasons: (i) if there are several short prophage regions in close proximity then one large phage region might be reported instead of several shorter ones and (ii) if more than one integrase was found in a single predicted prophage, more than one prophage might be reported.
Despite the use of multiple distinctive parameters to classify genes within a genomic region, the current random forest protocol does not allow the accurate determination of the phage start and end. To address this issue, we resorted to the analysis of phage attachment sites by detecting direct or inverted repeats, which are common at the insertion sites of most phages. As insertions are often flanked by several repeated sequences, two criteria were used to consider for all the candidate att sites: (i) proximity to tRNA or integrase genes, as phages can integrate into tRNA/tmRNA genes and the integrase gene is often at the end of the prophage (1) and (ii) inclusion of the greatest number of proteins thought to be included in phage subsystems, to provide further confidence in the prediction of the att sites.
The classification step of phiSpy predicts prophage regions more accurately if it is trained with genomes that are closely related to the test genome because: (i) differences in the GC% between the training genome and the test genome result in an incorrect weight for the customized AT/GC skew; (ii) differences in the protein length between the training genome and the test genome result in the wrong protein length prediction parameters; (iii) different operon sizes between the training genome and the test genome result in incorrect transcriptional strand orientation parameters; and (iv) finally, if the test genome (or a genome closely related to the test genome) was not included in the phage word library, two circumstances might occur: (a) some words in the phage library may match with the test genome’s word and (b) the genome might have some different prophages whose distinct words are absent in the library, which leads to a bad prediction for the calculation of the abundance of phage words.
The prophages not identified by PhiSpy in the classification step, might fall into one of three categories: (i) if there are few phage genes in a window whose characteristics were dominated by the bacterial genes of that window and those few phage genes were missed; (ii) if several short prophages are located very close together (this often happens in E coli or Salmonella), they were identified either as one long prophage or some of them were ignored in the processing of the final rank; (iii) while processing the final rank, sometimes the prediction of prophages was skewed leftwards by its windowing process (which proceeds left to right). The reason for the prophages identified in classification step but misclassified in evolution step is either there are few proteins annotated as phage proteins in the prophage regions or the function of the phage proteins are not yet annotated into phage subsystems.
Some of the characteristics of prophages used here have been described in previous studies, but none of the prophage identification tools applies them for identifying novel prophages. In this article, we have combined two approaches (similarity-based and composition-based analysis) and come-up with an automated application that can identify prophages with or without the homology to known phage genes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–3, Supplementary Figures 1–5 and Supplementary Methods.
FUNDING
Advances in Bioinformatics from the National Science Foundation (http://www.nsf.gov/) [DBI 0850356]. The funders had no role in study design, data collection and analysis or decision to publish the manuscript. Funding for open access charge: Advances in Bioinformatics from the National Science Foundation [DBI 0850356].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Barbara A. Bailey, for insightful discussion and helpful suggestions on statistical significance analysis.
REFERENCES
- 1.Casjens S. Prophages and bacterial genomics: what have we learned so far? Mol. Microbiol. 2003;49:277–300. doi: 10.1046/j.1365-2958.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- 2.Casjens S, Palmer N, van Vugt R, Huang WM, Stevenson B, Rosa P, Lathigra R, Sutton G, Peterson J, Dodson RJ, et al. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochaete Borrelia burgdorferi. Mol. Microbiol. 2000;35:490–516. doi: 10.1046/j.1365-2958.2000.01698.x. [DOI] [PubMed] [Google Scholar]
- 3.Canchaya C, Proux C, Fournous G, Bruttin A, Brüssow H. Prophage genomics. Microbiol. Mol. Biol. Rev. 2003;67:238–276. doi: 10.1128/MMBR.67.2.238-276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mc Grath S, Van SD. Bacteriophage: Genetics and Molecular Biology. Norfolk, UK: Caister Academic Press; 2007. [Google Scholar]
- 5.Aziz RK, Ismail S, Park HW, Kotb M. Post-proteomic identification of a novel phage-encoded streptodornase, Sda1, in invasive M1T1 Streptococcus pyogenes. Mol. Microbiol. 2004;54:184–197. doi: 10.1111/j.1365-2958.2004.04255.x. [DOI] [PubMed] [Google Scholar]
- 6.Aziz RK, Edwards R, Taylor WW, Low DE, McGeer A, Kotb M. Mosaic prophages with horizontally acquired genes account for the emergence and diversification of the globally disseminated M1T1 clone of Streptococcus pyogenes. J. Bacteriol. 2005;187:3311–3318. doi: 10.1128/JB.187.10.3311-3318.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell A. Episomes. Adv. Genet. 1962;11:101–145. [Google Scholar]
- 8.Landy A, Ross W. Viral integration and excision: structure of the lambda att sites. Science. 1977;197:1147–1160. doi: 10.1126/science.331474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimada K, Weisberg R, Gottesman M. Prophage lambda at unusual chromosomal locations. I. Location of the secondary attachment sites and the properties of the lysogens. J. Mol. Biol. 1972;63:483–503. doi: 10.1016/0022-2836(72)90443-3. [DOI] [PubMed] [Google Scholar]
- 10.Rausch H, Lehmann M. Structural analysis of the actinophage phi C31 attachment site. Nucleic Acids Res. 1991;19:5187–5189. doi: 10.1093/nar/19.19.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell A, Schneider S, Song B. Lambdoid phages as elements of bacterial genomes (integrase/phage21/Escherichia coli K-12/icd gene) Genetica. 1992;86:259–267. doi: 10.1007/BF00133724. [DOI] [PubMed] [Google Scholar]
- 12.Fouts D. Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006;34:5839–5851. doi: 10.1093/nar/gkl732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 14.Leplae R, Lima-Mendez G, Toussaint A. ACLAME: a classification of mobile genetic elements. Nucleic Acids Res. 2004;32:D45–D49. doi: 10.1093/nar/gkh084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lima-Mendez G, Van HJ, Toussaint A, Leplae R. Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics. 2008;24:863–865. doi: 10.1093/bioinformatics/btn043. [DOI] [PubMed] [Google Scholar]
- 16.Srividhya K, Alaguraj V, Poornima G, Kumar D, Singh GP, Raghavenderan L, Katta M, Mehta P, Krishnaswamy S. Identification of prophages in bacterial genomes by dinucleotide relative abundance difference. PLoS One. 2007;2:e1193. doi: 10.1371/journal.pone.0001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grigoriev A. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 1998;26:2286–2290. doi: 10.1093/nar/26.10.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lobry J. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996;13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- 19.Shannon CE. A mathematical theory of communication. Bell Syst. Tech. J. 1948;27:379–423. [Google Scholar]
- 20.Pierce JR. An Introduction to Information Theory: Symbols, Signals and Noise. 2nd edn. New York: Dover Publications, Inc.; 1980. [Google Scholar]
- 21.Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 22.Disz T, Akhter S, Cuevas D, Olson R, Overbeek R, Vonstein V, Stevens R, Edwards RA. Accessing the SEED genome databases via Web services API: tools for programmers. BMC Bioinformatics. 2010;11:319. doi: 10.1186/1471-2105-11-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 24.Salzber S, Salzberg A, Kerlavage A, Tomb J. Skewed oligomers and origins of replication. Gene. 1998;217:57–67. doi: 10.1016/s0378-1119(98)00374-6. [DOI] [PubMed] [Google Scholar]
- 25.Blattner FR, Plunkett GR, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 26.Lodish H, Berk A, Zipursky LS, Matsudaira P, Baltimore D, Darnell J. Molecular Cell Biology. New York: W. H. Freeman and Company; 2000. [Google Scholar]
- 27.Campbell A. Preferential orientation of natural lambdoid prophages and bacterial chromosome organization. Theor. Popul. Biol. 2002;61:503–507. doi: 10.1006/tpbi.2002.1604. [DOI] [PubMed] [Google Scholar]
- 28.Zheng Y, Szustakowski J, Fortnow L, Roberts R, Kasif S. Computational identification of operons in microbial genomes. Genome Res. 2002;12:1221–1230. doi: 10.1101/gr.200602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lucks JB, Nelson DR, Kudla GR, Plotkin JB. Genome landscapes and bacteriophage codon usage. PLoS Comput. Biol. 2008;4:e1000001. doi: 10.1371/journal.pcbi.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bahir I, Fromer M, Prat Y, Linial M. Viral adaptation to host: a proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 2009;5:311. doi: 10.1038/msb.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karlin S, Campbell A, Mråzek J. Comparative DNA a analysis across diverse genomes. Ann. Rev. Genet. 1998;32:185–226. doi: 10.1146/annurev.genet.32.1.185. [DOI] [PubMed] [Google Scholar]
- 32.Sueoka N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995;40:318–325. doi: 10.1007/BF00163236. [DOI] [PubMed] [Google Scholar]
- 33.Lobry JR. Properties of a general model of DNA evolution under no-strand bias conditions. J. Mol. Evol. 1995;40:326–330. doi: 10.1007/BF00163237. [DOI] [PubMed] [Google Scholar]
- 34.Good PI. Permutation, Parametric and Bootstrap Tests of Hypotheses. 3rd edn. New York: Springer; 2005. [Google Scholar]
- 35.Hatfull GF, Jacobs-Sera D, Lawrence JG, Pope WH, Russell DA, Ko CC, Weber RJ, Patel MC, Germane KL, Edgar RH, et al. Comparative genomic analysis of 60 mycobacteriophage genomes: genome clustering, gene acquisition, and gene size. J. Mol. Biol. 2010;397:119–143. doi: 10.1016/j.jmb.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perna NT, Plunkett G, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409:529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 37.Mavrodi DV, Loper JE, Paulsen IT, Thomashow LS. Mobile genetic elements in the genome of the beneficial rhizobacterium Pseudomonas fluorescens Pf-5. BMC Microbiol. 2009;9:8. doi: 10.1186/1471-2180-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson T, Kariyawasam S, Wannemuehler Y, Mangiamele P, Johnson S, Doetkott C, Skyberg J, Lynne A, Johnson J, Nolan L. The genome sequence of avian pathogenic Escherichia coli strain O1:K1:H7 shares strong similarities with human extraintestinal pathogenic E. coli genomes. J. Bacteriol. 2007;189:3228–3236. doi: 10.1128/JB.01726-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasko DA, Rosovitz MJ, Myers G, Mongodin EF, Fricke WF, Gajer P, Crabtree J, Sebaihia M, Thomson NR, Chaudhuri R, et al. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 2008;190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelson KE, Fouts DE, Mongodin EF, Ravel J, DeBoy RT, Kolonay JF, Rasko DA, Angiuoli SV, Gill SR, Paulsen IT, et al. Whole genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen Listeria monocytogenes reveal new insights into the core genome components of this species. Nucleic Acids Res. 2004;32:2386–2395. doi: 10.1093/nar/gkh562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hain T, Steinweg C, Kuenne CT, Billion A, Ghai R, Chatterjee SS, Domann E, Kärst U, Goesmann A, Bekel T, et al. Whole-genome sequence of listeria welshimeri reveals common steps in genome reduction with listeria innocua as compared to listeria monocytogenes. J. Bacteriol. 2006;188:7405–7415. doi: 10.1128/JB.00758-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Bannantine JP, Zhang Q, Amonsin A, May BJ, Alt D, Banerji N, Kanjilal S, Kapur V. The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc. Natl Acad. Sci. USA. 2005;102:12344–12349. doi: 10.1073/pnas.0505662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garnier T, Eiglmeier K, Camus J, Medina N, Mansoor H, Pryor M, Duthoy S, Grondin S, Lacroix C, Monsempe C, et al. The complete genome sequence of Mycobacterium bovis. Proc. Natl Acad. Sci. USA. 2003;100:7877–7882. doi: 10.1073/pnas.1130426100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brosch R, Gordon SV, Garnier T, Eiglmeier K, Frigui W, Valenti P, Santos SD, Duthoy S, Lacroix C, Garcia-Pelayo C, et al. Genome plasticity of BCG and impact on vaccine efficacy. Proc. Natl Acad. Sci. USA. 2007;104:5596–5601. doi: 10.1073/pnas.0700869104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eiglmeier K, Honoré N, Woods SA, Caudron B, Cole ST. Use of an ordered cosmid library to deduce the genomic organization of Mycobacterium leprae. Mol. Microbiol. 1993;7:197–206. doi: 10.1111/j.1365-2958.1993.tb01111.x. [DOI] [PubMed] [Google Scholar]
- 46.Morgan GJ, Hatfull GF, Casjens S, Hendrix WR. Bacteriophage Mu Genome Sequence: Analysis and Comparison with Mu-like Prophages in Haemophilus, Neisseria and Deinococcus. J. Mol. Biol. 2002;317:337–359. doi: 10.1006/jmbi.2002.5437. [DOI] [PubMed] [Google Scholar]
- 47.Silby MW, Cerdeño-Tárraga AM, Vernikos GS, Giddens SR, Jackson RW, Preston GM, Zhang X, Moon CD, Gehrig SM, Godfrey S, et al. Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol. 2009;10:R51. doi: 10.1186/gb-2009-10-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng W, Liou S, Plunkett G, Mayhew GF, Rose DJ, Burland V, Kodoyianni V, Schwartz DC, Blattner FR. Comparative genomics of Salmonella enterica Serovar typhi strains Ty2 and CT18. J. Bacteriol. 2003;185:2330–2337. doi: 10.1128/JB.185.7.2330-2337.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holden M, Feil EJ, Lindsay JA, Peacock SJ, Day N, Enright MC, Foster TJ, Moore CE, Hurst L, Atkin R, et al. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc. Natl Acad. Sci. USA. 2004;101:9786–9791. doi: 10.1073/pnas.0402521101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gill SR, Fouts DE, Archer GL, Mongodin EF, DeBoy RT, Ravel J, Paulsen IT, Kolonay JF, Brinkac L, Beanan M, et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 2005;187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 2008;190:300–310. doi: 10.1128/JB.01000-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferretti JJ, McShan WM, Ajdic D, Savic DJ, Savic G, Lyon K, Primeaux C, Sezate S, Suvorov AN, Kenton S, et al. Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc. Natl Acad. Sci. USA. 2001;98:4658–4663. doi: 10.1073/pnas.071559398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beres SB, Musser JM. Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS One. 2007;29:e800. doi: 10.1371/journal.pone.0000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee B, Park Y, Park D, Kang H, Kim J, Song E, Park I, Yoon U, Hahn J, Koo B, et al. The genome sequence of Xanthomonas oryzae pathovar oryzae KACC10331, the bacterial blight pathogen of rice. Nucleic Acids Res. 2005;33:577–586. doi: 10.1093/nar/gki206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanna LF, Matthews TD, Dinsdale EA, Hasty D, Edwards RA. Characterization of the ELPhiS Prophage from Salmonella enterica Serovar Enteritidis Strain LK5. Appl. Environ. Microbiol. 2012;78:1785–1793. doi: 10.1128/AEM.07241-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.