


Abstract
More than 98% of a typical vertebrate genome does not code for proteins. Although non-coding regions are sprinkled with short (<200 bp) islands of evolutionarily conserved sequences, the function of most of these unannotated conserved islands remains unknown. One possibility is that unannotated conserved islands could encode non-coding RNAs (ncRNAs); alternatively, unannotated conserved islands could serve as promoter-distal regulatory factor binding sites (RFBSs) like enhancers. Here we assess these possibilities by comparing unannotated conserved islands in the human and mouse genomes to transcribed regions and to RFBSs, relying on a detailed case study of one human and one mouse cell type. We define transcribed regions by applying a novel transcript-calling algorithm to RNA-Seq data obtained from total cellular RNA, and we define RFBSs using ChIP-Seq and DNAse-hypersensitivity assays. We find that unannotated conserved islands are four times more likely to coincide with RFBSs than with unannotated ncRNAs. Thousands of conserved RFBSs can be categorized as insulators based on the presence of CTCF or as enhancers based on the presence of p300/CBP and H3K4me1. While many unannotated conserved RFBSs are transcriptionally active to some extent, the transcripts produced tend to be unspliced, non-polyadenylated and expressed at levels 10 to 100-fold lower than annotated coding or ncRNAs. Extending these findings across multiple cell types and tissues, we propose that most conserved non-coding genomic DNA in vertebrate genomes corresponds to promoter-distal regulatory elements.
INTRODUCTION
In completely sequenced vertebrate genomes, only ∼1.5% of genomic DNA codes for proteins (1–3). An additional 3.5% of the genome lacks coding sequences but is nonetheless conserved across vertebrate phylogeny, strongly suggesting its functional importance (1–3). This conserved, non-coding 3.5% of the genome clusters into >700 000 unannotated conserved islands, 90% of which are <200 bp (‘Materials and Methods’ section). The vast majority of these conserved islands have no known function.
Two possible functions for unannotated conserved islands are (i) to encode enhancers and other distal regulatory sequences and (ii) to encode non-coding RNAs (ncRNAs). Indeed, tens of thousands of vertebrate conserved islands have already been found to overlap enhancers (4–7), which function at a distance to regulate the expression of associated genes. Most of these enhancers were identified in a genome-wide manner based on the presence of the co-activator p300/CBP and of H3K4me1-modified histones (8). Similarly, ∼10 000 conserved islands have been found to overlap promoters or exonic sequences of ncRNAs (9–11), whose function may be to act in cis or in trans to regulate gene expression (12–15). However, it remains unclear how many conserved islands will ultimately prove to have enhancer-related, ncRNA-related or other functions, since both enhancers and ncRNAs remain to be completely identified. Unfortunately, comprehensive identification of enhancers and ncRNAs will ultimately require genome-scale experiments to be conducted on all cell types in a vertebrate body, since both enhancers and ncRNAs function in subsets of tissues and cell types (5,11,16).
A conceptual and experimental challenge in distinguishing whether a conserved island is an enhancer or a promoter of an ncRNA is that many enhancers produce short (<2 kb) ncRNAs called enhancer RNAs (eRNAs) (7,17–20). Genomic sequence conservation at an enhancer has traditionally been thought to reflect the importance of regulatory factor binding sites (RFBSs), which recruit transcription factors initially to the enhancer and ultimately, through DNA looping, to associated promoters (21–23). However, the synthesis of eRNAs raises the possibility that conservation of sequences at enhancers may also reflect their importance for promoting eRNA transcription or for encoding functions of eRNA transcripts. Since eRNAs, much like other ncRNAs, could in theory act in cis or in trans to regulate gene expression, any given enhancer could have important functions both as a traditional enhancer and as a promoter of a non-coding eRNA. Thus, neither the presence of RNA Polymerase II (RNAPII) nor evidence of transcriptional initiation at an unannotated conserved island is on its own sufficient to determine whether its sequence conservation reflects conservation of enhancer function, conservation of ncRNA promoter function or both.
Here we estimate, using genome-wide approaches, how many conserved islands function as enhancers (and other distal regulatory elements) and how many encode ncRNAs. We comprehensively define distal regulatory elements using ChIP-Seq and DNAseI-hypersensitivity (24) assays based on data from published sources (1,18). We also comprehensively define transcribed regions of the genome by applying a novel transcript calling algorithm to RNA-Seq data obtained from total cellular RNA. Applying these approaches to mouse cortical neurons and a human (HeLa) cell line, we find that whereas hundreds of unannotated conserved islands are transcribed in these cell types into ncRNAs, tens of thousands of unannotated conserved islands can be identified as distal regulatory elements. These conserved, promoter-distal regulatory elements are distinguishable from conventional ncRNA promoters based on the low expression level, and non-polyadenylated status of the transcripts they synthesize, as well as by their lack of the promoter-specific H3K4me3 mark (25–27). We find similar ratios of conserved ncRNAs to conserved distal regulatory elements when expanding our analysis to 10 different human cell lines. Our results suggest that the underlying reason for the conservation of most unannotated conserved bases in vertebrate genomes is their importance within promoter-distal regulatory elements.
MATERIALS AND METHODS
Our goal was to compare three kinds of genomic loci: conserved sequences, RFBSs and transcribed regions. To identify unannotated transcribed regions we developed a novel algorithm, Haar-wavelet Transcript Calling (HaTriC), which is described in the Supplementary Methods. Our strategy was to first identify each kind of locus and second to relate them to one another.
Conserved islands
The conserved islands were obtained from the PhastCons scores (as compared with 30 other vertebrates) (2) using a coarse-graining procedure that identified bins of at least 10 bps where the average score was >0.9 (Supplementary Figure S3). Summary statistics for the conserved islands can be found in Supplementary Table S4 and Supplementary Figure S3. The assignment of conserved islands to various non-overlapping categories was carried out as follows:
If the conserved island overlapped an exon, then we categorized it as either an ‘Exon of annotated protein-coding gene’ or ‘Exon of annotated ncRNA’ depending on the coding potential of the gene.
If there was an annotated Transcription start site (TSS) within 1kb of either end of the conserved island (and the conserved island did not overlap an annotated exon), it was classified as a ‘Promoter of annotated protein-coding gene’ or ‘Promoter of annotated non-coding gene’.
If there was an enhancer or a RFBS within 100 bp of the conserved island, then we categorized it as an ‘Enhancer’ or ‘Other (unannotated) RFBS’ conserved island.
If there was at least 33% overlap (Supplementary Figure S10) with a matrix attachment region (MAR), then the conserved island was classified as a ‘MAR’.
All the remaining conserved islands were assigned to the ‘Intronic conserved island’ conserved or the ‘Extragenic conserved island’ category based on the overlap with annotated introns.
Calculating how many conserved islands overlap exons of unannotated transcripts is complicated by the fact that HaTriC does not provide the exon–intron structure of transcribed regions. Hence, we used a statistical approach where we assumed that the unannotated transcribed regions have the same distribution of exon numbers, exon lengths and exon conservation as the long annotated ncRNAs (Supplementary Table S7). Using these assumptions, it is possible to estimate the number of conserved islands explained by unannotated transcripts in a given cell type (Figure 1 and 3A).
Figure 1.

More conserved islands overlap enhancers and RFBSs than unannotated ncRNAs. The bars show the number of unannotated conserved islands that overlapped an enhancer, other promoter-distal RFBS or novel (HaTriC-defined) transcript (in mouse neurons). For Enhancers and RFBSs, a conserved island had to overlap an enhancer or RFBS to be counted. For novel ncRNAs, we used a statistical approach to estimate the number of conserved islands within exons of transcribed regions (Supplementary Methods). Lists of enhancer loci were taken from (5,18). RFBSs were defined as TF binding sites (mouse neurons) or DHSs (HeLa) that were not at promoters or enhancers.
Figure 3.

Across many cell types, more conserved islands overlap RFBSs than ncRNAs. (A) Number of novel ncRNAs, ucRFBSs, and insulators discovered with each additional tissue or cell type investigated. The number of conserved islands assigned to each category initially increases as a power-law. (B) Extrapolation to additional cell types of the number of unannotated conserved islands explained by ncRNAs, unannotated RFBSs and insulators (based on the slopes in the left panel). Assuming that there are a total of 100, 200, 500, 1000 or 1500 distinct cell-types or conditions, we calculated the total number of conserved islands that would overlap ncRNAs, insulators or other RFBSs.
Regulatory factor binding sites
RFBSs were identified from publicly available DNAseI hypersensitivity and ChIP-Seq datasets. To understand how RFBSs are related to transcribed regions, we categorized them based on their proximity to promoters, enhancers, introns, exons and novel transcribed regions. We classified RFBSs as conserved or non-conserved based on overlap with conserved islands. Each RFBS was assigned to a category according to the scheme outlined below, and the results are reported in Supplementary Table S3. We start with the full set of peaks, and once a peak has been assigned to a category, it cannot be assigned to any further categories.
If the peak was within 1kb of an annotated TSS, it is considered ‘Promoter of annotated protein-coding gene’ or ‘Promoter of annotated ncRNA’ for annotated coding genes and ncRNAs, respectively.
If the peak was within 1kb of an enhancer it is classified as either ‘Intragenic enhancer’ or ‘Extragenic enhancer’.
If the peak was within 1kb of the start of a novel transcribed region (identified by HaTrIC, but not present in annotation) it is assigned to the ‘Promoter of novel ncRNA’ category.
If the peak overlaps an annotated protein-coding gene but is further than 1kb from the start, it is assigned to the ‘Overlaps exon of annotated protein-coding gene’ or ‘Overlaps intron of annotated protein-coding gene’ depending on its overlap with exons. Similarly, peaks overlapping annotated ncRNAs are considered either ‘Overlaps exon of annotated ncRNA’ or ‘Overlaps intron of annotated ncRNA’.
If the peak does not fit into any of the previous categories it is classified as ‘Unannotated extragenic’.
Transcribed regions
To understand transcription across the genome, we combined annotation, de novo transcript-calling and a targeted search near enhancers and RFBSs (Table 1) to produce a set of transcribed regions. To characterize the transcriptome, we assigned each read uniquely to a transcribed region. To define the transcribed regions in the most accurate way possible, as reported in Supplementary Table S7, Supplementary Figure S4 and Table 1 in the main text, we combined the annotation, the HaTriC transcript-caller, and a targeted search close to enhancers and RFBSs. Below, we describe how each category of transcribed regions was defined, as well as the criteria for assigning reads to each category. We considered the categories sequentially, and at each step we identified (and removed from further analysis) all reads that overlapped regions in the current category.
Annotated protein-coding genes were first separated into non-overlapping clusters. From each cluster the longest region, gi, was extracted as a representative of that cluster (this was done to avoid double counting, and the majority of clusters contain only one gene). If the average read density of gi fell below a threshold, the region was ignored and the reads were retained and made available for inclusion in another category. Next, we applied HaTriC and merged the identified regions that had been categorized as corresponding to a part of a gene or uniquely to a gene with their overlapping genes. When two regions gi and
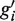 are merged, they are removed and a new region
are merged, they are removed and a new region 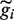 is created. The new region contains the union of the reads from gi and
is created. The new region contains the union of the reads from gi and 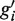 , and it extends from the 5′-most end of gi and
, and it extends from the 5′-most end of gi and 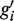 to the 3′-most end of gi and
to the 3′-most end of gi and 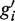 . The transcribed regions
. The transcribed regions 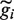 were frequently longer than the annotation would have predicted.
were frequently longer than the annotation would have predicted.Having removed all reads corresponding to annotated coding genes, we carried out the same procedure for annotated non-coding genes. When counting the number of reads in the two categories relating to annotated genes (as reported in Table 1, but not for the transcript read density reported in Supplementary Figures S4, S7 and Figure 2), we also assigned all sense reads found within 10 kb upstream or downstream of
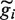 to the (protein-coding or ncRNA) genic category. As reported by van Bakel et al. (28), these regions often have a read density that is above the background levels found in more distal regions.
to the (protein-coding or ncRNA) genic category. As reported by van Bakel et al. (28), these regions often have a read density that is above the background levels found in more distal regions.Next, we searched for promoter AS transcribed regions, i.e., divergent transcribed regions (29,30). We started by searching all windows located 2 kb upstream of all annotated TSSs. If a window contained >r0 reads, it was considered significant. Most transcribed regions that were not detected by HaTriC are <2 kb (see Supplementary Figure S4), but to account for longer regions we extended the search to the next 2 kb window upstream of the TSS if the TSS proximal window contained >r0 reads. Additional windows were investigated until a window containing <r0 reads was found. For a set of adjacent 2 kb windows, the length of the transcribed region is defined as the maximum distance between all pairs of reads found in these windows. We refer to the procedure where subsequent 2 kb windows are scanned as a ‘window-based search’. The threshold was set to r0 = 9 reads in a 2 kb window for the mouse neurons and r0 = 5 reads for the HeLa cells, corresponding to an FDR of 0.001. The regions detected using the window-based search were merged with all unannotated regions proximal to known genes identified by the transcript caller.
This category corresponds to long unannotated transcripts and hence we assign all regions categorized by HaTriC as unannotated and distal to known genes to this class. Since there are occasionally low numbers of reads close to the starts and ends of the unannotated transcribed regions (similar to how promoter AS reads are found near annotated TSSs), we carried out a window-based search upstream and downstream of the transcribed regions. Any reads found from the window-based search was included in the total read count reported in Table 1 and Supplementary Table S1.
We first applied the window-based search to the AS strand downstream of all RFBSs overlapping annotated genes. The regions obtained using the window-based method are then merged with the ones found by HaTriC and categorized as anti-sense (AS) with respect to known genes.
For extragenic enhancers, we applied the window-based search in the downstream directions on both strands. For intragenic enhancers, only the AS downstream window was considered. The regions obtained using the window-based method are merged with all eRNA regions identified by HaTriC.
Since the H3K4me3 mark is strongly associated with active promoters, we wanted to make sure that we did not miss any significant transcription initiated from these loci. For all extragenic RFBSs that were within 2 kb of a H3K4me3 peak, we used the window-based method on both strands to extract a set of transcribed regions.
For the remaining extragenic RFBSs that did not have a H3K4me3 peak nearby, we again used the window-based method on both strands to extract a set of transcribed regions.
Finally, for HeLa cells where we also have access to CTCF data, we applied the window-based method on both strands at CTCF peaks.
Table 1.
Comprehensive accounting of RNA-Seq reads by genomic locus
| Transcript category | Percentage of RNA-Seq reads | No. of loci | Percentage of genome |
|---|---|---|---|
| Protein-coding gene | 71.451 | 12 108 | 21.27 |
| Annotated non-coding gene | 1.381 | 2564 | 0.92 |
| snRNAs, tRNAs, scRNAs, srpRNAs, rRNAs | 26.465 | 3625 | 0.01 |
| Promoter AS transcript | 0.354 | 4844 | 0.63 |
| Other (HaTric-defined) AS transcript | 0.038 | 660 | 0.11 |
| Novel (HaTric-defined) transcript | 0.076 | 255 | 0.08 |
| Extragenic eRNA | 0.013 | 622 | 0.04 |
| Intragenic eRNA | 0.008 | 331 | 0.01 |
| Other RFBSs-associated RNA | 0.062 | 793 | 0.04 |
| Associated with other H3K4me3 peaks | 0.017 | 367 | 0.01 |
| Total | 99.8643 | 26 169 | 23.1147 |
The vast majority of RNA-Seq reads in mouse neurons fall within 10 categories of genomic loci (rows), identified and classified using a combination of gene annotation, HaTriC transcript calling, and chromatin state (Supplementary Methods). Here categories of expressed loci are characterized based on their fraction of the total number of RNA-Seq reads, their number of genomic loci and their fraction of genomic base-pairs. Transcribed loci were required to have nine RNA-Seq reads and a read density of at least 1 per kb. Annotated gene categories include UTRs and introns. Annotated non-coding genes include those annotated in the UCSC, RefSeq, Ensembl, lincRNA and macroRNA collections, excluding snRNAs, tRNAs, scRNAs, srpRNAs and rRNAs. A ‘Promoter AS transcript’ is an AS transcript with its 5′-end within 2 kb of an annotated TSS. An ‘Other (HaTriC-defined) AS transcript’ is an AS transcript (overlapping an annotated gene) with its 5′-end further than 2 kb from any annotated TSS. An ‘Other RFBS-associated RNA’ starts within 2 kb of a RFBS not identified as an enhancer. snRNAs, tRNAs, scRNAs, srpRNAs and rRNAs are defined by repeatMasker. We note that rRNAs are under-represented here relative to within a cell due to their removal from total RNA samples by hybridization prior to sequencing. Similar results for HeLa cells are presented in Supplementary Table S8.
Figure 2.

The transcriptional profile at ucRFBSs is more similar to that of enhancers than promoters. Comparison of several properties of transcribed regions (as defined in ‘Materials and Methods’) that overlap at least one conserved island. TSSs were defined by annotation for annotated genes or by HaTriC for unannotated genes. Only expressed loci are included; thresholds for defining expressed loci were nine RNA-Seq reads and a read density of at least 1 per kb. (A) Transcribed regions at enhancers and ucRFBSs are short and expressed at lower levels than annotated genes, as shown by the average expression near TSSs in each category. For sites without obvious strand orientation (e.g. H3K4me3 sites), forward and reverse (rev) genomic strands are plotted separately; otherwise, only sense reads are plotted. (B) ucRFBSs and enhancers are associated with fewer 5′ ends of 5′-sequenced ESTs than promoters. (Note the different y-scales.) (C) The ratio of polyA+ to total RNA reads is much lower at enhancers and ucRFBSs relative to annotated RNAs. The x-axis is the ratio of normalized polyA+ reads divided by the number of normalized total RNA reads at a locus, and the y-axis is the cumulative density (CDF). (D) ucRFBSs and enhancers are expressed at lower levels than annotated RNAs. (E) Transcribed regions at ucRFBSs and enhancers are shorter than those at protein-coding genes. (F) ucRFBSs and enhancers are not bound by the initiation-specific H3K4me3 mark. (G) Genomic sequence conservation at promoters extends outward further than genomic sequence conservation at ucRFBSs and enhancers. (H) The CpG content at ucRFBSs and enhancers is lower than that at promoters.
The Supplementary Methods contain details on how the annotations of the mouse and human genomes were assembled. There is also a list of all the datasets used in this study (Supplementary Table S6). As far as possible, given the availability of datasets, we performed parallel analyses in mouse neurons and human HeLa cells.
RESULTS
Assigning reads to transcribed regions
Our strategy to understand the function of conserved elements required a comprehensive accounting of the transcriptome, including an ability to comprehensively identify non-coding transcripts and distinguish ncRNAs from protein-coding genes. To achieve this understanding, we developed an algorithm to define transcribed regions of the genome de novo (i.e., without relying on annotation) using short-read RNA-Seq data. We applied this algorithm to strand-specific RNA-Seq from mouse cortical neurons and HeLa cells. Although other RNA-Seq studies have applied de novo transcript detection in both yeast (31) and mouse (11,32), with a few exceptions [e.g., (28,33,34)], these and other studies of gene expression have only detected or defined mature polyadenylated transcripts. Here we consider polyadenylated transcripts as well as non-polyadenylated transcripts, since unannotated conserved sequences could give rise to either type of transcript.
To define transcribed regions of the genome de novo, we developed a computational approach, Haar-wavelet Transcript Calling (HaTriC). HaTriC is an iterative algorithm that combines evidence from multiple length scales to determine if a given region is actively transcribed (see Supplementary Methods for full description). Each iteration of the HaTriC algorithm involves the following three steps: (i) At each genomic locus, the change in RNA-Seq read density between upstream and downstream regions is computed [as Haar-wavelet coefficients, similar to (35)]. (ii) The genomic loci with the biggest changes in RNA-Seq density are selected. These loci represent a set of candidate boundaries of transcribed regions. (iii) Each candidate transcribed region, defined as the sequence between two candidate boundaries, is classified as either transcribed or not transcribed based on its average read density. This classification is straightforward, since the densities of the candidate regions have a bimodal distribution, with the higher density mode corresponding to transcribed regions (Supplementary Figure S1). By applying the above procedure iteratively, excluding the regions that were called as transcribed in previous rounds, we are able in each successive round to detect transcribed regions with lower read density. As regions with high read density are removed, the distribution of candidate regions' read densities goes from bimodal to unimodal, at which point the iteration terminates because no additional transcribed regions are detected. The parameters for the algorithm (‘Materials and Methods’ section) are optimized by maximizing the fraction of transcribed regions (on one strand of one chromosome) that have transcriptional initiation marker H3K4me3-modified histones (25–27) bound at their start.
To identify transcribed regions, we applied HaTriC to RNA-Seq reads obtained from sequencing ribosomal RNA-depleted total RNA from mouse neurons (∼140 million reads) (18) and HeLa cells (∼50 million reads). We obtained ∼10 000 transcribed regions in each cell type (Supplementary Table S1). These transcribed regions do not necessarily represent specific RNA transcripts [as do the transcripts defined in some other studies, e.g., (11)] but can instead correspond to multiple overlapping transcripts synthesized from the same strand. For example, the RNA-Seq reads falling within a transcribed region that corresponds to a gene typically reflect pre-mRNA transcripts (corresponding to reads aligning both to introns and exons) as well as mature mRNA transcripts (corresponding to exons only). The purpose of this approach is to identify regions that are transcribed rather than to define precisely the exon–intron structure of specific transcripts.
To evaluate the quality of transcribed regions identified by HaTriC, we compared transcribed regions with annotated protein-coding genes. Comparing the transcribed regions with the RefSeq (36), UCSC (37) and Ensembl (38) gene annotations, we found, in accordance with another recent study (28), that most transcribed regions either overlap coding genes on the correct strand (76%) or are found within 10 kb of the start of an annotated coding gene (18%), despite the fact that these two sets of regions together represent just 15% of the genome (Supplementary Table S1). The majority of the transcribed regions that overlap annotated genes (78%) match the gene annotation in an unambiguous manner (Supplementary Methods, Supplementary Table S1). These results demonstrated sufficient accuracy in calling transcribed regions to allow us to begin to categorize regions lacking any annotation.
Few conserved islands correspond to unannotated ncRNAs
We asked to what extent novel, HaTriC-defined ncRNAs coincide with evolutionarily conserved sequences. To identify the conserved sequences, we segregated the genome into conserved and non-conserved regions using a simple coarse-graining procedure performed on PhastCons conservation scores derived from alignments of 30 vertebrate species (2) (‘Materials and Methods’ section). In each genome (human and mouse), we identified 1 million conserved islands, most of them <200 bp (Supplementary Figure S3). We define ∼300 000 of these as annotated conserved islands based on their overlap with the promoters or exons of annotated coding or non-coding genes (RefSeq, UCSC, Ensembl, lncRNA and macroRNA annotations combined (9,11,36,37,38), Supplementary Table S4). The remaining 700 000 (presumed non-coding) conserved islands we define as unannotated conserved islands (55% extragenic, 45% intragenic).
We addressed how many unannotated conserved islands might encode promoters or exons of novel extragenic ncRNAs. Our strategy was to asssess the overlap of unannotated conserved islands with expressed sequences. To identify novel ncRNAs, we applied HaTriC and found ∼200 ncRNAs that were already annotated in RefSeq, Ensembl, UCSC, the macroRNA or lincRNA datasets and ∼250 unannotated extragenic ncRNAs (Supplementary Table S1) (Although 200 annotated ncRNAs may seem like a small number to find, this result is consistent with the reported lower levels of expression and greater tissue specificity of ncRNAs (39), especially those lacking strong experimental support.). Annotated and novel ncRNAs accounted for 2% of transcribed regions called by HaTriC and an estimated 3% of the total number of RNA-Seq reads but <0.01% of the length of the genome (Table 1 and Supplementary Table S1). Given the small number of novel ncRNA loci that are actively transcribed in either mouse neurons or HeLa cells, relatively few unannotated conserved islands are estimated to function as promoters (<400) or exons (<1800) of novel extragenic ncRNAs in these cells (Figure 1 and ‘Materials and Methods’ section). These estimates depend on assumptions about the gene structure of novel ncRNAs and the overlap of their exons with conservation islands, since HaTriC does not define intron-exon structure directly. Our assumptions are based on the gene structure of annotated ncRNAs in RefSeq, UCSC and Ensembl. A recently published survey of human lincRNAs (39) provided an independent set of gene structures that have less exonic overlap with conserved islands (Supplementary Table S7). Thus, our above estimates may overemphasize the extent of conservation explained by novel ncRNAs.
We also addressed how many unannotated conserved islands might encode novel non-coding AS transcripts, which have been proposed to regulate sense gene expression (40–43). Previously described AS transcripts include < 2 kb promoter AS transcripts (34,44,45), synthesized upstream of genic promoters; <2 kb eRNAs from intronic enhancers (18,19); and other, sometimes longer AS transcripts (46). It remains unclear how common these transcripts are, how highly expressed they are relative to annotated genes, and how frequently they overlap unannotated conserved islands. We found a substantial number of AS transcribed regions, accounting for 15% of all transcribed regions detected by HaTriC (Supplementary Table S1). Most AS transcribed regions correspond to promoter AS transcripts (Table 1). Because AS transcripts are generally short (Supplementary Figure S4c,d), the total fraction of the genome transcribed into AS in these cell types is small. Thus, in vertebrate genomes as in yeast (44,45), AS transcription is composed predominantly of short (<2 kb), lowly expressed transcripts synthesized from promoters. Accordingly, AS transcripts do not explain the function of many unannotated conserved islands, since AS transcripts originate predominantly from conserved islands that are already annotated as promoter regions.
Our analysis suggests that few unannotated conserved islands encode ncRNAs or serve as their promoters. One reason we could be underestimating the overlap of conservation with ncRNAs is that our ability to detect ncRNAs spanning the 45% of conserved islands that are intronic is limited to detection of anti-sense ncRNAs. This limitation arises because our method does not allow us to distinguish specific ncRNAs that overlap pre-mRNAs or mRNAs on the same strand. However, the similar numbers of H3K4me3 binding sites at extragenic and intronic loci (Supplementary Table S3) suggest that this limitation is unlikely to result in a dramatic revision to our findings. A second potential reason that we could be underestimating the extent of overlap between conserved islands and ncRNAs would be if there were additional, less highly expressed novel ncRNAs not detected by HaTriC. Like any algorithm for identifying transcribed regions or transcripts, HaTriC has the least statistical power to detect and define lowly expressed transcripts, especially if the transcripts are short. For example, few eRNAs are detected by HaTriC (Supplementary Table S1). To evaluate the extent of this challenge, we asked how many RNA-Seq reads could be explained by different sources of annotation, including annotated transcripts HaTriC-defined transcripts, and transcripts associated with enhancers or other promoter-distal RFBSs (Table 1 and Supplementary Methods). We found that 99.8% of reads can be explained by these combined sources of genome annotation. Of the remaining <0.2% of unaccounted reads, >99% could be explained by a negative binomial background model, suggesting a low level of technical noise (Supplementary Figure S2).
We cannot fully rule out the alternative possibility that these reads are derived from tens of thousands of very lowly expressed ncRNAs. We estimate, however, that the expression levels of such transcripts would be <1 transcript per 100 cells (Supplementary Methods). This estimate, detailed in the supplement, is based on a reference point of 240 000 mRNAs per cell (47). While we note that this reference point is imprecise, our overall copy number distributions accord well with those obtained using reference points based on digital in situ hybridization (48) (data not shown). Even if our copy number estimates were off by an order of magnitude, these results imply that if the unexplained reads do not represent technical noise, they may well represent biological (transcriptional) noise (28,49).
Many conserved islands overlap promoter-distal regulatory sites
Having observed that few unannotated conserved islands appear to coincide with ncRNAs, we investigated an alternative hypothesis, that most unannotated conserved islands function as promoter-distal RFBSs. Such sites include enhancers, insulators and silencers (1,50). Each of these types of RFBS is marked by DNAseI Hypersensitivity Sites (DHSs), since the relatively open chromatin associated with RFBSs is vulnerable to DNAseI digestion (1,51,52). Thus, to identify RFBSs, we examined DHSs [Supplementary Table S2a (1)]. We found that DHSs overlapped ∼9000 unannotated conserved islands, suggesting that a large number of unannotated conserved islands function as RFBSs. This overlap is highly significant (P-value <10−16, hypergeometric test where the intersection is 9000 between 700 000 conserved islands and 60 000 DHSs, assuming a total of 30 000 000 potential loci), consistent with the idea that sequence conservation at overlapping unannotated conserved island/DHS loci reflects the importance of regulatory factor binding to DNA.
In theory, DHSs should correspond to all RFBSs bound in a given cell type, but in practice any DHS experiment will miss some RFBSs. In an independent approach to finding RFBSs, we turned to ChIP-Seq experiments to identify binding sites for a number of different regulatory factors. The factors immunoprecipitated were NPAS4, CREB, SRF and CBP in mouse neurons (18) and AP2α, AP2γ, MAX, cFOS, cMYC, E2F4 and E2F6 in HeLa cells (1). Analysis of the HeLa data revealed that ∼90% of the binding sites for these factors overlapped a DHS, indicating that the DHSs represent a sensitive means of detecting RFBSs (Supplementary Table S2b) (53). Unsurprisingly in light of this high degree of overlap, ∼12 000 conserved RFBSs identified by factor binding were distributed across promoters, enhancers and unclassified RFBSs in much the same way as those identified by DNAse hypersensitivity (Figure 1; Supplementary Table S2a and S3a). Thus, regardless of the method used to identify RFBSs, many more unannotated conserved islands overlap with RFBSs than with ncRNAs. Moreover, the finding that a much larger fraction of the non-coding genomic elements serve as binding sites for regulatory factors rather than exons or promoters of ncRNAs is true for non-conserved parts of the genome as well. For the mouse neurons, we estimate that there are ∼40 000 non-conserved enhancers and RFBSs, compared with only 2700 promoters and exons for ncRNAs.
The large number of unannotated conserved islands found to overlap RFBSs led us to investigate what regulatory function unannotated conserved RFBSs (ucRFBSs) might serve. Insulators, which represent one major class of regulatory site, are involved in partitioning active and inactive regions of the genome (50,54). We asked how many ucRFBSs identified in HeLa cells were insulators, defined by the presence of the protein CCCTC-binding factor (CTCF). We found that ∼3 000 ucRFBSs could be classified as insulators on this basis (last three rows in Supplementary Table S3b).
Although the remaining ucRFBSs are distal both to annotated and HaTriC-defined promoters, we considered the possibility that they might be weak, unannotated ncRNA promoters not detected by HaTriC. We found that <500 ucRFBSs could be classified as promoters based on the presence of the promoter-specific H3K4me3 mark (Figure 2F, Supplementary Table S3c). Nonetheless, a larger number of ucRFBSs could represent inactive promoters that drive high levels of transcription in other cell types. We investigated this possibility by examining sequence, conservation and expression profiles of ucRFBSs (Figure 2). First, we reasoned that if ucRFBSs act as promoters in any tissue, they should share sequence characteristics with known promoters. Contrary to this prediction, ucRFBSs (like enhancers) lack high densities of CpG dinucleotides that are frequently found at coding promoters (Figure 2H). Second, we reasoned that promoters and promoter-distal regulatory sequences might be distinguished based on their extent of conservation. We found that the lengths of conserved islands at ucRFBSs more closely resemble those found at enhancers than those found at promoters, again suggesting that most ucRFBSs do not act as promoters in any tissue or cell-type (Figure 2G). Finally, if ucRFBSs act as promoters in any tissue, they should be enriched for 5′ ends of (5′-sequenced) ESTs (37), which have been sequenced at low depth from a wide variety of different tissues. However, the overlap between ucRFBSs and ESTs is similar to that observed between ESTs and previously defined enhancers and is dramatically less than the overlap between 5′ EST ends and promoters (Figure 2B). Moreover, only promoters of protein-coding genes have more spliced than unspliced ESTs (Supplementary Figure S9). These results suggest that ucRFBSs do not act as conventional coding or ncRNA promoters.
We hypothesized that many of the remaining ucRFBSs might represent enhancers that were missed by enhancer-identification algorithms (5,8,18) because their level of enrichment of p300/CBP or H3K4me1 was below the chosen threshold. To investigate this possibility, we examined CBP and H3K4me1 binding at ucRFBSs in mouse neurons. The majority of ucRFBSs had low but significant levels of CBP and H3K4me1 enrichment, suggesting that many of these unclassified sites could indeed be enhancers (data not shown and Supplementary Figure S5). However, a subset of these ucRFBSs may represent regulatory sites that are not enhancers. We conclude that ucRFBSs are a mixture of insulators, enhancers and perhaps other classes of promoter-distal regulatory sites (e.g., locus-control regions and silencers).
Regulatory sites express non-polyadenylated, unspliced transcripts at 10 to 100-fold lower levels than mRNAs
Our case studies of HeLa and mouse neuron cells suggest that most unannotated conserved islands overlap promoter-distal RFBSs (e.g. enhancers), rather than ncRNAs. However, recent genome-wide studies (18,19) have found that certain promoter-distal RFBSs (i.e. enhancers) are associated with low levels of transcription, producing ncRNAs. These results blur the distinction between promoter-distal regulatory sites and ncRNA promoters, raising questions about how well these two classes of loci can truly be distinguished. However, we find that that these enhancers and ucRFBSs can be clearly distinguished experimentally from ncRNA promoters for the following reasons. First, while both enhancers and ucRFBSs produce RNAs, these RNAs are expressed at much lower levels than those produced from traditional coding or non-coding promoters (Figure 2A,D, P-value <10−9, KS-test on distributions from Figure 2). The approximate expression levels of transcripts emanating from ucRFBSs averaged <1 transcript per 100 cells (Supplementary Figure S4a,b, Supplementary Methods), or ∼10 to 100-fold lower than the copy number of an average genic mRNA. Second, relative to ncRNAs, the transcripts emanating from promoter-distal RFBSs are less likely to be spliced or polyadenylated (18,19) (Figure 2B,C,S9, P-value <10−16, KS-test on distributions from Figure 2). Third, while the length distributions of ncRNAs and ucRFBSs overlap, ucRFBSs are shorter (<2 kb) than the typical lincRNA (P-value <10−2, KS-test on distributions from Figure 2). Fourth, sequence conservation at ucRFBSs in most cases does not extend beyond the specific location where regulatory factors bind, whereas coding and ncRNAs promoters exhibit conservation over a longer genomic region (Figure 2G). Thus, conserved distal regulatory sites differ significantly from ncRNA promoters based on their chromatin and transcriptional profiles.
As more cell types are examined, more ucRFBSs are found
To the extent that we can ascribe functions to unannotated conserved islands in our case study of one mouse and one human cell type, the ascribed functions are overwhelmingly related to binding of regulatory factors to DNA, with relatively few unannotated conserved islands corresponding to ncRNAs (Figure 1). However, our case study explains only ∼20 000 out of 700 000 unannotated conserved islands, presumably because the remainder function only in cell types (or cellular conditions) other than those examined here. Since we are only able so far to investigate a small number of cell-types, our predictions about the functional role of the majority of conserved islands requires an extrapolation of our findings to additional cell types. In this extrapolation, how many additional unannotated conserved islands may be attributed to RFBSs and ncRNAs as more cell types are examined will depend on the relative cell-type specificity of RFBSs and ncRNAs.
To extrapolate how many additional conserved islands could be ascribed to conserved unannotated RFBSs (ucRFBSs) and ncRNAs as additional cell types are examined, we identified ncRNAs (using H3K4me3-binding) and ucRFBSs (using DHS sites) in 10 additional human cell lines using data from the ENCODE project (1) (‘Materials and Methods’ section). As we examined additional cell types, we discovered novel ucRFBSs and ncRNAs at similar rates (Figure 3A), implying that ucRFBSs and ncRNAs have similar cell-type specificity. However, this conclusion relies on the accuracy of using H3K4me3 to identify ncRNA promoters. To confirm that unannotated H3K4me3 loci are a reasonable proxy for ncRNA promoters, we used HaTriC to identify 800 novel ncRNAs in 10 human tissues, using RNA-Seq data generated from total RNA (Supplementary Table S5). The approximate number of novel ncRNAs found from each additional ENCODE cell line (using H3K4me3) or human tissue (using HaTriC) was similar (∼100), suggesting that unannotated H3K4me3 sites are a reasonable proxy for ncRNA promoters [as was previously found by others (11)]. In fact relatively weak H3K4me3 sites are frequently found in locations where no transcription can be detected by RNA-Seq (Figure 2, cyan line), suggesting that our method of promoter identification by H3K4me3 may over-represent the number of ncRNA promoters. Because the rate of additional ncRNAs and RFBSs found with each new cell type examined is roughly equal, examining additional cell types does not radically alter our conclusion that more unannotated conserved islands have RFBS-related than ncRNA-related function.
We considered finally how many unannotated conserved islands might ultimately be assigned to RFBS-related or ncRNA-related functions once RFBSs and ncRNAs have been identified in all vertebrate cell types. The adult human body contains ∼400 cell types (55). However, this number is likely to be an underestimate in the sense that it does not account for rare (and unknown) adult cell types, developmental stage-specific cell types, or for the ability of cells to adopt different gene expression and chromatin states depending on environmental cues. The true number is very difficult to approximate, but it is nonetheless instructive to extrapolate how conserved ncRNA and RFBS discovery might scale with increasing numbers of cell types. We therefore arbitrarily took 2 000 as a potential upper limit on the number of cell types across human development and adulthood. Extrapolating our findings using the ENCODE cell lines to an estimated 1000–2000 cell type-conditions, we find it to be plausible that 20% of conserved islands encode ncRNAs or their promoters, whereas 80% of conserved islands function as promoter-distal RFBSs (Figure 3B). Even though there are many uncertainties associated with this estimate, it is to the best of our knowledge the first quantitative attempt to address the question of how many conserved non-coding sequences function as distal regulatory elements versus encode ncRNAs. We expect that access to RNA-Seq and ChIP-Seq data from additional cell-types, as well as more accurate multi-species alignments will ultimately allow similar approaches to produce more accurate estimates.
DISCUSSION
The function of the ∼60% of conserved bases in vertebrate genomes that are non-coding is unknown (1,3). We provide genome-wide evidence suggesting that the reason most of these DNA bases are conserved across vertebrate evolution is due to their importance as promoter-distal regulatory elements, including enhancers. In individual cell types (HeLa cells or mouse neurons), we find that 4-fold more conserved islands are associated with promoter-distal regulatory elements rather than with ncRNAs. As we examine additional cell types, we find in each additional cell type four times as many conserved islands corresponding to promoter-distal regulatory elements as to ncRNAs. Extrapolating these results to an estimated 1500 cell type-conditions in a vertebrate body, it is plausible that almost ∼300 000 unannotated conserved islands function as promoter-distal regulatory elements, while ∼60 000 encode functions related to ncRNAs. At the same time, we cannot rule out the alternative possibility that many or even most unannotated conserved islands may have novel, as-yet unknown functions that are unrelated to RFBSs, ncRNAs, or even gene expression. For example, we used the H-rule (56) to estimate (Supplementary Figure S10) that 4% of conserved islands could correspond to MARs, which anchor chromatin to the nuclear matrix (57).
When characterizing the transcriptome, a crucial experimental parameter is the total number of reads captured in the experiment, since the extent of sequencing determines one's ability to detect lowly expressed transcripts. To evaluate how the sequencing depth affects our results, we down-sampled the RNA-Seq data. For mouse neurons >90% of the novel ncRNAs are discovered using only 50% of the original reads (Supplementary Figure S8), and extrapolation suggests that additional novel transcripts would be discovered at a rate of ∼2 regions/10 million reads. Most of the regions that would be discovered from additional sequencing would express transcripts at even lower abundance than the regions that have been discovered at the current sequencing depth.
This low degree of extragenic transcription is in contrast with the results reported in a recent study by Mercer et al. (58). Using Capture-Seq, a combination of microarray capture and RNA-Seq, they identified 257 novel transcripts from 0.77 Mb of human fibroblast genome. Extrapolation of their result suggests that there could be ∼1 million lowly expressed extragenic transcripts in the entire genome, whereas extrapolation of our results suggests a number that is around three orders of magnitude lower. This difference is likely due in large part to their much higher sensitivity, which allows them to detect transcripts expressed at an average of 0.0006 mRNAs per cell.
Detection and expression level thresholds are crucial to understanding the ongoing debate about the extent of intergenic transcription. Initial estimates based on tiling arrays (1) suggested that the majority of the genome was transcribed in at least one cell type. Even though this finding has been challenged (28), other more recent studies (58,59) have argued that there is ubiquitous transcription, albeit at levels as low as an average of 0.0006 transcripts per cell. At the given sequencing depth, where we estimate our detection threshold at about one mRNA per cell, our findings are consistent with van Bakel et al. (28). Thus, while higher detection sensitivity allows for identification of further transcripts, these transcripts are expressed at very low levels. The weaker the expression level of a protein-coding or ncRNA, the less it tends to be conserved (60), suggesting that deeper the transcriptome is sequenced, the less overlap identified transcripts will have with evolutionary conservation.
Despite most of the genome not being transcribed, we do find thousands of short regions that produce low levels of transcripts associated with promoters, insulators, enhancers and other regulatory sites. These transcripts tend to be unspliced and non-polyadenylated (Figure 2B,C), suggesting that they do not leave the nucleus. Even if these transcripts themselves are not functionally important (49), the act of transcribing them may nonetheless be necessary. For example, much of the observed ncRNA transcription may be important for establishing and maintaining chromatin states such as histone acetylation or methylation [e.g., (61,62)]. In this case, the low levels of associated transcripts may reflect either the low stability of the non-functional transcripts that are produced or the low frequency of transcriptional initiation required for chromatin maintenance.
We propose that most sequence conservation in the non-coding genome reflects the importance of RFBSs. This proposition may appear to conflict with the observation that binding of certain regulatory factors (CEBPA, HNF4A) in liver tissue is poorly (<10% of sites) conserved between any two mammals (63). However, it has recently been argued that many of non-conserved binding sites are less likely to be functional and that the degree of conservation is significantly increased if expression of nearby genes is taken into consideration (64). More broadly, the extent of conservation of binding that is observed in any particular experiment likely depends on the particular tissues or regulatory factors examined, as is suggested by the higher conservation of binding of the transcription factor Twist across Drosophila species (65). Thus, in order to determine conclusively whether a particular conserved genomic sequence is associated with conserved binding, it will be necessarily to examine multiple bound factors at that locus across multiple tissues. Together our findings suggest that while many RFBSs come and go over an evolutionary time scale, a core set of conserved RFBSs account for most of the non-coding sequence conservation in vertebrate genomes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–8, Supplementary Figures 1–10, Supplementary Methods and Supplementary References [66–85].
FUNDING
National Institutes of Health (NIH) [1DP2OD006461-01, 1R21NS070250-01A1, NS028829]; National Science Foundation (NSF) [BCS-0954570]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Tae-Kyung Kim for preparing the ChIP-Seq libraries for the mouse neurons, Jian Gu and Kristithe Lea for preparing the HeLa RNA-Seq libraries and the ENCODE project for making their data publicly available.
REFERENCES
- 1.ENCODE Project Consortium. Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, et al. Identification and analysis of functional elements in % of the human genome by the encode pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Margulies EH, Cooper GM, Asimenos G, Thomas DJ, Dewey CN, Siepel A, Birney E, Keefe D, Schwartz AS, Hou M, et al. Analyses of deep mammalian sequence alignments and constraint predictions for % of the human genome. Genome Res. 2007;17:760–774. doi: 10.1101/gr.6034307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 5.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. Chip-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by erna. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 9.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 10.Ponjavic J, Ponting CP, Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007;17:556–565. doi: 10.1101/gr.6036807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guttman M, Amit I, Garber M, French C, Lin M, Feldser D, Huarte M, Zuk O, Carey B, Cassady J, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai M-C, Hung T, Argani P, Rinn JL, et al. Long non-coding RNA hotair reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011 doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersson Ørom U, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotech. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Routledge SJE, Proudfoot NJ. Definition of transcriptional promoters in the human beta globin locus control region. J. Mol. Biol. 2002;323:601–611. doi: 10.1016/s0022-2836(02)01011-2. [DOI] [PubMed] [Google Scholar]
- 18.Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei C-L, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–634. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science. 1998;281:60–63. doi: 10.1126/science.281.5373.60. [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Barkess G, Qian H. Chromatin looping and the probability of transcription. Trends Genet. 2006;22:197–202. doi: 10.1016/j.tig.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Dean A. On a chromosome far, far away: LCRs and gene expression. Trends Genet. 2006;22:38–45. doi: 10.1016/j.tig.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brinkman AB, Roelofsen T, Pennings SWC, Martens JHA, Jenuwein T, Stunnenberg HG. Histone modification patterns associated with the human X chromosome. EMBO Rep. 2006;7:628–634. doi: 10.1038/sj.embor.7400686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 27.Robertson AG, Bilenky M, Tam A, Zhao Y, Zeng T, Thiessen N, Cezard T, Fejes AP, Wederell ED, Cullum R, et al. Genome-wide relationship between histone H3 lysine 4 mono- and tri-methylation and transcription factor binding. Genome Res. 2008;18:1906–1917. doi: 10.1101/gr.078519.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Bakel H, Nislow C, Blencowe BJ, Hughes TR. Most “dark matter” transcripts are associated with known genes. PLoS Biol. 2010;8:e1000371. doi: 10.1371/journal.pbio.1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, Young RA, Sharp PA. Divergent transcription from active promoters. Science. 2008;322:1849–1851. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yassour M, Kaplan T, Fraser HB, Levin JZ, Pfiffner J, Adiconis X, Schroth G, Luo S, Khrebtukova I, Gnirke A, et al. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc. Natl Acad. Sci. USA. 2009;106:3264–3269. doi: 10.1073/pnas.0812841106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotech. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Q, Kim YC, Lu J, Xuan Z, Chen J, Zheng Y, Zhou T, Zhang MQ, Wu C-I, Wang SM. Poly A-Transcripts expressed in hela cells. PLoS One. 2008;3:e2803. doi: 10.1371/journal.pone.0002803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He Y, Vogelstein B, Velculescu VE, Papadopoulos N, Kinzler KW. The antisense transcriptomes of human cells. Science. 2008;322:1855–1857. doi: 10.1126/science.1163853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ben-Yaacov E, Eldar Y. A fast and flexible method for the segmentation of aCGH data. Bioinformatics. 2008; 24:139–145. doi: 10.1093/bioinformatics/btn272. [DOI] [PubMed] [Google Scholar]
- 36.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (refseq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, et al. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010;38:D613–D619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flicek P, Aken BL, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Coates G, Fairley S, et al. Ensembl's 10th year. Nucleic Acids Res. 2010;38:D557–D562. doi: 10.1093/nar/gkp972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Czech B, Malone CD, Zhou R, Stark A, Schlingeheyde C, Dus M, Perrimon N, Kellis M, Wohlschlegel JA, Sachidanandam R, et al. An endogenous small interfering RNA pathway in drosophila. Nature. 2008;453:798–802. doi: 10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539–543. doi: 10.1038/nature06908. [DOI] [PubMed] [Google Scholar]
- 42.Zhou X, Sunkar R, Jin H, Zhu J, Zhang W. Genome-wide identification and analysis of small RNAs originated from natural antisense transcripts in oryza sativa. Genome Res. 2008;19:70–78. doi: 10.1101/gr.084806.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahmoudi S, Henriksson S, Corcoran M, Méndez-Vidal C, Wiman KG, Farnebo M. Wrap53, a natural p53 antisense transcript required for p53 induction upon DNA damage. Mol. Cell. 2009;33:462–471. doi: 10.1016/j.molcel.2009.01.028. [DOI] [PubMed] [Google Scholar]
- 44.Neil H, Malabat C, d'Aubenton Carafa Y, Xu Z, Steinmetz L, Jacquier A. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009;457:1038–1042. doi: 10.1038/nature07747. [DOI] [PubMed] [Google Scholar]
- 45.Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz L. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 47.Islam S, Kjällquist U, Moliner A, Zajac P, Fan J-B, Lönnerberg P, Linnarsson S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011;21:1160–1167. doi: 10.1101/gr.110882.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hebenstreit D, Fang M, Gu M, Charoensawan V, van Oudenaarden A, Teichmann SA. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol. Syst. Biol. 2011;7:497. doi: 10.1038/msb.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat. Struct. Mol. Biol. 2007;14:103–105. doi: 10.1038/nsmb0207-103. [DOI] [PubMed] [Google Scholar]
- 50.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu. Rev. Genom. Hum. Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 51.Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Ann. Rev. Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 52.Wu C, Bingham PM, Livak KJ, Holmgren R, Elgin SC. The chromatin structure of specific genes: I. Evidence for higher order domains of defined DNA sequence. Cell. 1979;16:797–806. doi: 10.1016/0092-8674(79)90095-3. [DOI] [PubMed] [Google Scholar]
- 53.Bergman CM, Carlson JW, Celniker SE. Drosophila DNase I footprint database: a systematic genome annotation of transcription factor binding sites in the fruitfly, drosophila melanogaster. Bioinformatics. 2005;21:1747–1749. doi: 10.1093/bioinformatics/bti173. [DOI] [PubMed] [Google Scholar]
- 54.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vickaryous MK, Hall BK. Human cell type diversity, evolution, development, and classification with special reference to cells derived from the neural crest. Biol. Rev. Camb. Philos. Soc. 2006;81:425–455. doi: 10.1017/S1464793106007068. [DOI] [PubMed] [Google Scholar]
- 56.Evans K, Ott S, Hansen A, Koentges G, Wernisch L. A comparative study of S/MAR prediction tools. BMC Bioinformatics. 8:71. doi: 10.1186/1471-2105-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Glazko GV, Koonin EV, Rogozin IB, Shabalina SA. A significant fraction of conserved noncoding DNA in human and mouse consists of predicted matrix attachment regions. Trends Genet. 2003;19:119–124. doi: 10.1016/S0168-9525(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 58.Mercer T, Gerhardt DJ, Dinger ME, Crawford J, Trapnell C, Jeddeloh JA, Mattick JS, Rinn JL. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat. Biotechnol. 2011 doi: 10.1038/nbt.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, Ponting CP, Stadler PF, Morris KV, Morillon A, et al. The reality of pervasive transcription. PLoS Biol. 2011;9:e1000625. doi: 10.1371/journal.pbio.1000625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Managadze D, Rogozin IB, Chernikova D, Shabalina SA, Koonin EV. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs. GB. 2011;3:1390–1404. doi: 10.1093/gbe/evr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, et al. The Paf1 complex is required for histone H3 methylation by compass and Dot1p: linking transcriptional elongation to histone methylation. Mol. Cell. 2003;11:721–729. doi: 10.1016/s1097-2765(03)00091-1. [DOI] [PubMed] [Google Scholar]
- 62.Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of set1 histone methylase by elongating pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 63.Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. Five-vertebrate chip-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hemberg M, Kreiman G. Conservation of transcription factor binding events predicts gene expression across species. Nucleic Acids Res. 2011;39:7092–7102. doi: 10.1093/nar/gkr404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.He Q, Bardet AF, Patton B, Purvis J, Johnston J, Paulson A, Gogol M, Stark A, Zeitlinger J. High conservation of transcription factor binding and evidence for combinatorial regulation across six drosophila species. Nat. Genet. 2011;43:414–420. doi: 10.1038/ng.808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.