

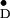
Abstract
Electron paramagnetic resonance (EPR) spectroscopy at 94 GHz is
used to study the dark-stable tyrosine radical
Y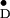 in single crystals of photosystem II
core complexes (cc) isolated from the thermophilic cyanobacterium
Synechococcus elongatus. These complexes contain at least
17 subunits, including the water-oxidizing complex (WOC), and 32
chlorophyll a molecules/PS II; they are active in
light-induced electron transfer and water oxidation. The crystals
belong to the orthorhombic space group
P212121, with four PS II dimers per
unit cell. High-frequency EPR is used for enhancing the sensitivity of
experiments performed on small single crystals as well as for
increasing the spectral resolution of the g tensor components and of
the different crystal sites. Magnitude and orientation of the g tensor
of Y
in single crystals of photosystem II
core complexes (cc) isolated from the thermophilic cyanobacterium
Synechococcus elongatus. These complexes contain at least
17 subunits, including the water-oxidizing complex (WOC), and 32
chlorophyll a molecules/PS II; they are active in
light-induced electron transfer and water oxidation. The crystals
belong to the orthorhombic space group
P212121, with four PS II dimers per
unit cell. High-frequency EPR is used for enhancing the sensitivity of
experiments performed on small single crystals as well as for
increasing the spectral resolution of the g tensor components and of
the different crystal sites. Magnitude and orientation of the g tensor
of Y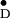 and related information on several
proton hyperfine tensors are deduced from analysis of angular-dependent
EPR spectra. The precise orientation of tyrosine
Y
and related information on several
proton hyperfine tensors are deduced from analysis of angular-dependent
EPR spectra. The precise orientation of tyrosine
Y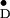 in PS II is obtained as a first step in
the EPR characterization of paramagnetic species in these single
crystals.
in PS II is obtained as a first step in
the EPR characterization of paramagnetic species in these single
crystals.
In oxygenic photosynthesis,
two photosystems (PS I and PS II) function in sequence to convert light
into energy-rich chemical compounds (1, 2). PS I uses energy from the
absorption of a photon to reduce NADP+ to NADPH, which is
required for CO2 reduction. The electrons for this process
are donated by PS II. On light excitation of PS II, an electron is
transferred from the primary donor P680, a chlorophyll
species, via an intermediate pheophytin Pheo a to the
plastoquinone acceptors QA and QB. Two
sequential univalent redox steps and concomitant protonation events
lead to plastohydroquinol QBH2, which leaves PS
II and provides electrons to PS I via the cytochrome
b6f complex (for review, see ref. 2). The
photooxidized cation radical P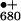 has the highest
oxidation potential of all cofactors known in nature (≥+1.1 V), which
is sufficient for water oxidation. P
has the highest
oxidation potential of all cofactors known in nature (≥+1.1 V), which
is sufficient for water oxidation. P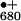 extracts an
electron from a redox active tyrosine YZ. The intermediate
tyrosine radical Y
extracts an
electron from a redox active tyrosine YZ. The intermediate
tyrosine radical Y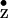 , in turn, oxidizes the
water-oxidizing complex (WOC), a tetranuclear manganese cluster. The
WOC passes through a cycle of four one-electron oxidation steps in
which water is oxidized and protons and O2 are released
(for reviews, see refs. 3–7). The exact water-splitting mechanism is
still unknown.
, in turn, oxidizes the
water-oxidizing complex (WOC), a tetranuclear manganese cluster. The
WOC passes through a cycle of four one-electron oxidation steps in
which water is oxidized and protons and O2 are released
(for reviews, see refs. 3–7). The exact water-splitting mechanism is
still unknown.
The cofactors involved in the electron transfer chain of PS II are
bound to two protein subunits, D1 and D2. From
amino acid sequence homology (8–10), two-dimensional electron
crystallography (11), and computer modeling (12), D1 and
D2 are assumed to be arranged analogously to the L and M
subunits in the reaction center of purple bacteria. This analogy has
been supported recently by x-ray crystallographic studies of the PS II
single crystals (13). Whereas YZ in D1 connects
P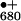 to the WOC in the electron transfer chain, the
homologous YD in D2 does not seem to take part
in the charge separation process. However, YD is also
coupled to the WOC and, under illumination, forms a dark-stable radical
Y
to the WOC in the electron transfer chain, the
homologous YD in D2 does not seem to take part
in the charge separation process. However, YD is also
coupled to the WOC and, under illumination, forms a dark-stable radical
Y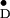 (Fig. 1).
The functional role of YD is not understood in detail; it
may be necessary for assembly of the PS II complex (14, 15). Recent
results also suggest that it may play a role in preventing
photoinhibition during activation of the PS II complex (16).
(Fig. 1).
The functional role of YD is not understood in detail; it
may be necessary for assembly of the PS II complex (14, 15). Recent
results also suggest that it may play a role in preventing
photoinhibition during activation of the PS II complex (16).
Figure 1.

Tyrosyl radical with numbering scheme and orientation of g tensor principal axes (collinear with the molecular axes). A hydrogen bond to the oxygen is indicated (14).
In the light-induced single electron transfer process and in the
water-splitting cycle, various paramagnetic species are formed (17)
that have been studied by conventional X-band (9 GHz) electron
paramagnetic resonance (EPR) techniques during the last decade. Most of
these species can be observed only in the freeze-trapped state in
frozen PS II solutions. X-band EPR experiments suffer from the limited
resolution of such spectra. With the advent of high-field EPR
spectroscopy (18), the Zeeman resolution was significantly increased,
allowing a more precise determination of the g tensor
components and thereby often permitting an unequivocal identification
of species and a spectral separation of overlapping radicals.
Additional electron-nuclear double resonance (ENDOR) experiments (19)
have led to the elucidation of the electron-nuclear hyperfine coupling
constants and the spin density distribution of the paramagnetic species
under study (for ENDOR on Y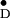 , see refs.
20–23). Another important structural element is the orientation of the
g and hyperfine tensor axes. These are related to the
molecular structure and can thus be used to determine the orientation
of the species in the protein. Partial orientation information can
already be gained from experiments on oriented membrane fragments (24,
25). Complete and accurate orientation information, however, is
obtained only from EPR studies of protein single crystals.
, see refs.
20–23). Another important structural element is the orientation of the
g and hyperfine tensor axes. These are related to the
molecular structure and can thus be used to determine the orientation
of the species in the protein. Partial orientation information can
already be gained from experiments on oriented membrane fragments (24,
25). Complete and accurate orientation information, however, is
obtained only from EPR studies of protein single crystals.
PS II core complexes (cc) have only recently been crystallized from the thermophilic cyanobacterium Synechococcus elongatus. The crystals are of the orthorhombic space group P212121 and contain four PS II dimers per unit cell. Each monomer contains at least 17 protein subunits, including the WOC and 32 chlorophyll a molecules. It has been shown that the crystals are active in light-induced electron transfer and water oxidation (26). The large number of sites in these crystals and the small g anisotropy of the organic radicals require a spectral resolution that cannot be achieved in conventional X-band (9 GHz) EPR experiments. W-band (94 GHz) EPR increases the Zeeman splitting as compared to conventional X-band EPR experiments by approximately one order of magnitude and should thereby permit overcoming of these difficulties. The increased Zeeman splitting also helps to separate g and hyperfine contributions to the spectrum. Another advantage of high-field EPR is increased sensitivity (18, 27), particularly important for samples of limited size. For the PS II single crystals, the volume is only about 35 nl, which presents a serious limitation for standard X-band EPR measurements.
Materials and Methods
Sample Preparation.
PS II cc from S. elongatus were purified according to Dekker et al. (28), with weak anion exchange chromatography used in the final stage of purification of the PS II cc (29). All chemicals were of analytical grade, and triply distilled deionized water (Millipore-Q) was used as solvent.
According to SDS/PAGE and matrix-assisted laser desorption ionization–time-of-flight mass spectrometry, the PS II cc of S. elongatus are composed of at least 17 subunits (30), of which 14 are located within the photosynthetic membrane: the RC proteins D1 and D2 (PsbA and PsbD), the heterodimeric cytochrome b559 (PsbE and PsbF), the two chlorophyll a-binding inner light-harvesting antenna proteins CP43 (PsbC) and CP47 (PsbB) and the smaller subunits PsbH to PsbN, and PsbX. The membrane-extrinsic cytochrome c550 (PsbV), the 12-kDa (PsbU), and the manganese stabilizing 33-kDa protein (PsbO) are located on the lumenal side of the PS II cc (31).
From this preparation, three-dimensional PS II single crystals could be grown (29) that are active in water oxidation (26) and diffract to a resolution of at least 3.8 Å in x-ray structure analysis. In the crystals, PS II occurs as a homodimer with noncrystallographic C2 symmetry.
The Y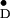 radical was generated in all samples
by exposure to ambient light before freezing.
radical was generated in all samples
by exposure to ambient light before freezing.
W-Band EPR.
Continuous-wave (cw) EPR experiments were performed by using a Bruker (Rheinstetten, Germany) Elexsys 680 spectrometer, operating at 94 GHz. PS II solutions were filled into synthetic quartz capillaries [Vitrocom (Mountain Lakes, NJ) CV7087S, 0.9 mm o.d., 0.7 mm i.d., length 33 mm, sealed on one side] to 3 mm height and frozen in liquid nitrogen.
Plate-shaped PS II single crystals (dimensions ≈0.5 × 0.5 × 0.15 mm3) were inserted into the capillaries with the plate normal perpendicular to the capillary axis. Alternatively, crystals were placed flat on the bottom end of capillaries (i.d. 0.5 mm), with the plate normal parallel to the capillary axis. Some crystals were mounted in a loop protruding from a W-band capillary to allow for x-ray diffraction experiments before the EPR measurements to determine the orientation of the crystallographic axes. The crystals were covered by a small amount of mother liquor and frozen in liquid nitrogen by using glycerol as a cryoprotectant. For orientation-dependent spectra, the samples were rotated about the capillary axis perpendicular to the magnetic field. The microwave frequency was measured by a built-in frequency counter and the magnetic field calibrated by measuring a Li:LiF sample (g = 2.002293) (32) at multiple frequencies.
X-Band-Pulsed ENDOR.
Pulsed ENDOR experiments were performed by using a Bruker ESP 380E X-band (9 GHz) spectrometer equipped with an ESP 360 D-P pulsed ENDOR accessory, an Oxford (Oxford, U.K.) CF935 helium cryostat, and an ENI (Rochester, NY) A-500 RF amplifier. A synthetic quartz tube (Wilmad PQ 727, o.d. 3 mm, i.d. 2 mm) was filled about 15 mm high with PS II cc solution and the sample frozen in liquid nitrogen.
Analysis.
cw EPR spectra were simulated by using a home-written program based on first-order perturbation theory. Crystal spectra are calculated as a superposition of spectra of the inequivalent sites, related by crystallographic (P212121) and noncrystallographic (C2 dimer axis) symmetries. The simulation of complete spectra extends the analysis of orientation dependence compared to other work, i.e. ref. 33, where g values have to be extracted from individual spectra first. Spectra of frozen solutions are similarly calculated from a large number of uniformly distributed orientations. Simulation parameters include g and hyperfine principal values and their orientations for one site in the unit cell, the orientation of the crystal in the magnetic field, and a linewidth parameter.
Slightly different microwave frequencies νmw for individual spectra lead to small shifts of the spectra on the magnetic field axis relative to each other. For analysis, the spectra were scaled according to B′0 = B0⋅94.000 GHz/νmw. The relative errors for the hyperfine couplings introduced by this step are negligible (<0.2%). The obtained g tensor principal values are not affected.
Results
cw EPR of PS II in Frozen Solution.
The frozen solution W-band EPR spectra of
Y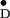 shown here (Fig.
2, Top) exhibit excellent
resolution, reflecting the very good homogeneity of the magnetic field
and the absence of g and hyperfine strain in the samples.
The g principal values obtained from simulations (error
Δg ≤ 2⋅10−5) are compared with
those reported in the literature (20, 25, 34–37) in Table
1.
shown here (Fig.
2, Top) exhibit excellent
resolution, reflecting the very good homogeneity of the magnetic field
and the absence of g and hyperfine strain in the samples.
The g principal values obtained from simulations (error
Δg ≤ 2⋅10−5) are compared with
those reported in the literature (20, 25, 34–37) in Table
1.
Figure 2.

W-band cw EPR spectra (black, experiment; light blue,
simulation) of Y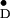 in frozen solution and
single crystals of PS II cc from S. elongatus.
(Left) Crystal rotated approximately about the
crystallographic a axis. (Right) Arbitrary
rotation axis. Colored lines indicate the calculated angular dependence
of the effective g value for each
Y
in frozen solution and
single crystals of PS II cc from S. elongatus.
(Left) Crystal rotated approximately about the
crystallographic a axis. (Right) Arbitrary
rotation axis. Colored lines indicate the calculated angular dependence
of the effective g value for each
Y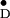 residue in the unit cell. Residues
belonging to the same PS II dimer share the same color. Simulation
parameters: gx/y/z =
2.00767/2.00438/2.00219, A
residue in the unit cell. Residues
belonging to the same PS II dimer share the same color. Simulation
parameters: gx/y/z =
2.00767/2.00438/2.00219, A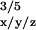 =
−26.1/−8/−19.5 MHz (rotated by ±20° around z
with respect to the g tensor),
A
=
−26.1/−8/−19.5 MHz (rotated by ±20° around z
with respect to the g tensor),
A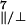 = 32.8/27.2 MHz
(A
= 32.8/27.2 MHz
(A along gx
direction) (see Tables 1 and 2). Experimental conditions: T
= 80 K, Pmw = 500 nW,
νmw = 94 GHz, acquisition time less than 5 minutes
per trace.
along gx
direction) (see Tables 1 and 2). Experimental conditions: T
= 80 K, Pmw = 500 nW,
νmw = 94 GHz, acquisition time less than 5 minutes
per trace.
Table 1.
Comparison of principal values of the g tensor for
Y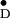 in
PS II
in
PS II
| gx | gy | gz | Δg* | Organism | Ref. |
|---|---|---|---|---|---|
| 2.00767 | 2.00438 | 2.00219 | 2⋅10−5 | S. elongatus | This work |
| 2.00740 | 2.00425 | 2.00205 | Not given | Synechocystis 6803 | 34 |
| 2.00737 | 2.00420 | 2.00208 | Not given | Spinach | 34 |
| 2.00745 | 2.00422 | 2.00212 | 2⋅10−4 | Spinach | 35 |
| 2.0074 | 2.0044 | 2.0023 | Not given | Spinach | 20 |
| 2.00756 | 2.00432 | 2.00215 | 1⋅10−4 | Spinach | 25 |
| 2.00782 | 2.00450 | 2.00232 | Not given | Spinach | 36 |
| 2.00752 | 2.00426 | 2.00212 | 7⋅10−5 | Spinach | 37 |
Δg denotes the accuracy of the given data.
The gx and gz components show a hyperfine splitting into four lines with a 1∶3∶3∶1 intensity ratio, whereas only a doublet splitting is resolved around the gy position. On the basis of earlier theoretical work on tyrosine radicals (38, 39), we assign this splitting to two highly anisotropic α proton hf couplings (positions 3 and 5) and a nearly isotropic β proton hf coupling (position 7a; see Fig. 1). Along the gx and gz direction, all three hyperfine couplings (hfc) of these protons are of comparable magnitude, whereas the hfcs of the two α protons are small and not resolved along the gy direction, so that only the coupling of the less anisotropic β proton remains. The hfcs of the second β proton and of the protons in positions 2 and 6 are too small to be resolved in EPR.
Pulsed ENDOR.
In order to obtain independent and precise information about the hfcs
of Y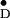 from S. elongatus, pulsed
ENDOR experiments at X-band (9 GHz) were performed on
Y
from S. elongatus, pulsed
ENDOR experiments at X-band (9 GHz) were performed on
Y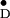 in frozen solution of the PS II cc.
in frozen solution of the PS II cc.
The X-band pulsed ENDOR spectrum of Y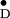 shown in Fig. 3 exhibits several well
resolved spectral features and is remarkably similar to
Y
shown in Fig. 3 exhibits several well
resolved spectral features and is remarkably similar to
Y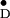 spectra from spinach (21). Only the
larger hfcs that are important for the analysis of the EPR spectra are
analyzed here. Our assignment and interpretation follow density
functional theory calculations for phenoxyl radicals (38, 39) and
earlier experimental work (20–23, 25, 36) summarized in Table
2. We interpret the structure in the
νRF = 28 … 32 MHz spectral range (labels 1 and
2) as an axial β proton hyperfine tensor with the principal values
A
spectra from spinach (21). Only the
larger hfcs that are important for the analysis of the EPR spectra are
analyzed here. Our assignment and interpretation follow density
functional theory calculations for phenoxyl radicals (38, 39) and
earlier experimental work (20–23, 25, 36) summarized in Table
2. We interpret the structure in the
νRF = 28 … 32 MHz spectral range (labels 1 and
2) as an axial β proton hyperfine tensor with the principal values
A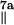 = 32.8 MHz and
A
= 32.8 MHz and
A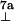 = 27.2 MHz. The respective
low-frequency lines are expected at νRF < 2 MHz and
are difficult to detect. On the basis of the strength of the coupling
and the moderate anisotropy, we assign this tensor to one of the β
protons of the CH2 group next to the phenoxyl ring
(position 7a). The other β proton in position 7b apparently lies
close to the nodal plane of the π system and therefore gives rise to
only small hyperfine couplings (40).
= 27.2 MHz. The respective
low-frequency lines are expected at νRF < 2 MHz and
are difficult to detect. On the basis of the strength of the coupling
and the moderate anisotropy, we assign this tensor to one of the β
protons of the CH2 group next to the phenoxyl ring
(position 7a). The other β proton in position 7b apparently lies
close to the nodal plane of the π system and therefore gives rise to
only small hyperfine couplings (40).
Figure 3.

X-band Davies-pulsed ENDOR spectrum of Y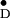 in frozen solution of PS II cc from S. elongatus. Principal
values of the obtained hyperfine coupling tensors are labeled by
numbers (see text). Experimental parameters: T = 5 K,
RF pulse length 8 μs, 64/128 ns microwave pulses,
νmw = 9.7 GHz, proton Larmor frequency,
νH = 14.8 MHz.
in frozen solution of PS II cc from S. elongatus. Principal
values of the obtained hyperfine coupling tensors are labeled by
numbers (see text). Experimental parameters: T = 5 K,
RF pulse length 8 μs, 64/128 ns microwave pulses,
νmw = 9.7 GHz, proton Larmor frequency,
νH = 14.8 MHz.
Table 2.
Comparison of hyperfine coupling principal values for
Y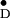 in PS II
in PS II
A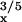
|
A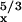
|
A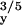 |
A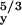
|
A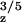
|
A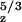
|
A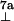
|
A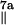
|
Organism | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| −25.5* | −26.8* | ≈ −8† | −19.0* | −20.1* | 27.2* | 32.8* | S. elongatus | This work | |
| −25.4 | −7.2 | −19.5 | 20.2 | 29.3 | Synechocystis 6803 | 23 | |||
| 29.4 | −9.0 | −19.6 | 21.6 | 23.2 | Spinach | 20 | |||
| 24 | 3 | 19 | 27/28 | 31 | Spinach | 25 | |||
| 14.8 | 18.8 | 20.3 | 23.0 | 27.0 | 30.5 | Spinach | 36 | ||
| 12.3 | 18.8 | 20.3 | 23.0 | 27.0 | 30.5 | Spinach | 21 | ||
| −25.6 | −27.5 | −8.0 | −19.1 | −20.5 | 27.2 | 31.5 | Spinach | 22 | |
| −25.6 | −27.5 | −8.0 | −19.1 | −20.5 | 28.5 | 33.0 | Chlamydomonas reinhardtii | 22 | |
| −25.6 | −27.5 | −8.0 | −19.1 | −20.5 | 24.5 | 29.0 | Phormidium laminosum | 22 | |
Values given in MHz.
Signs are based on theory, i.e. ref. 38. Experimental error <0.2 MHz.
Estimate from simulations of single crystal EPR spectra.
The split peaks around νRF ≈25 and 5 MHz (labels 5/5′ and 6/6′) are interpreted as the center components of two slightly inequivalent ring α proton hfcs (positions 3 and 5; Fig. 1). The largest components of these hyperfine tensors overlap with the signal from the proton in position 7a; a close examination of the slope of the line at νRF ≈ 27.5 MHz reveals small shoulders (labels 3 and 4) that we interpret as the edges of the 3,5 proton tensors. The smallest components of the 3,5 hfcs lie in the νRF = 10 … 20 MHz range and are hard to elucidate. The observed inequivalence of these hfcs reflects a slightly asymmetric spin density distribution, which is probably caused by an asymmetric hydrogen bond to the tyrosine oxygen (14, 38) (see Fig. 1).
In the νRF = 10 … 20 MHz range, several additional hfcs are observed. These are ascribed to the remaining protons in positions 2, 6, and 7b, a hydrogen bond proton from a histidine ligand to the oxygen (14, 41), and possibly to further protons of the environment (matrix). An unambiguous assignment is rather difficult. These smaller couplings, however, are not resolved in the EPR spectra. They contribute only to the EPR linewidth and, therefore, are not considered further here.
cw EPR on Single Crystals.
Orientation-dependent EPR spectra of two single crystals for different
rotation axes are shown in Fig. 2. The spectra are complicated because,
(i) there are eight magnetically inequivalent
Y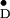 radicals in the crystal unit cell, and
(ii) some large anisotropic hyperfine couplings are resolved
and result in significant overlap of spectral features from the
different sites. The splitting varies because of the hfc anisotropy of
the respective nuclei. Furthermore, from the molecular symmetry (Fig.
1), it is expected that hyperfine and g tensor axes do not
coincide.
radicals in the crystal unit cell, and
(ii) some large anisotropic hyperfine couplings are resolved
and result in significant overlap of spectral features from the
different sites. The splitting varies because of the hfc anisotropy of
the respective nuclei. Furthermore, from the molecular symmetry (Fig.
1), it is expected that hyperfine and g tensor axes do not
coincide.
The orientation-dependent spectra exhibit a 180° periodicity under rotation because of inversion symmetry of the observed magnetic interactions. For special orientations of the crystal relative to the magnetic field, a degeneracy can occur, reducing the number of magnetically inequivalent radicals. For the spectra shown on the Left in Fig. 2, the crystal was rotated almost exactly about a crystallographic axis. The external magnetic field, therefore, was always perpendicular to that axis. This special orientation leads to a pairwise degeneracy of spectral contributions from different sites. When the field is parallel to one of the other crystallographic axes (at orientations 0°, 90°, and 180°), all four crystallographic sites are magnetically degenerate, and only the two dimer halves give rise to a line splitting.
On the Right of Fig. 2, similar spectra for another arbitrary orientation of the single crystal with respect to the rotation axis are shown. Here, the degeneracies are lifted, and the spectra exhibit clearly more than four magnetically inequivalent radicals, thereby confirming the presence of PS II dimers in the unit cell. Three more sets of spectra for differently mounted single crystals (not shown here) were obtained and used in the analysis to determine a consistent parameter set.
In all cases, the spectra are far too complex to directly trace the g values of the eight sites. Therefore, the spectra were simulated and the simulation parameters refined by a least-squares fitting procedure. A complete parameter set for this problem consists of 6n + 17 parameters (where n is the number of considered hfc tensors), describing the orientation of the crystal in the laboratory frame (3 parameters), the orientation of the noncrystallographic C2 symmetry axis (2 parameters), and principal values and orientations of all symmetrical tensors (g, Gaussian linewidth, and hfc tensors). To obtain numerically robust results from the fitting procedure, the number of free parameters had to be reduced as follows: (i) The principal values of g were taken from simulations of our frozen solution W-band EPR spectra and kept fixed. (ii) The orientation of the noncrystallographic symmetry axis was taken from X-ray diffraction results. (iii) Only the hfcs of the protons in positions 3, 5, and 7a were included in the simulation. The principal values of these tensors were taken from our pulsed ENDOR experiments. (iv) The hyperfine tensors for protons in positions 3 and 5 were assumed to be related by molecular symmetry, neglecting the small differences in the couplings observed by ENDOR. (v) Only an isotropic linewidth parameter was used.
First, simulations were performed to obtain an estimate for the smallest component of the A3/5 tensor that was not available from ENDOR, to determine all hfc tensor orientations, and to find the best linewidth parameter. In the final refinement of the simulation, only six free parameters (orientation of g in the crystal axis system and orientation of the crystal with respect to the laboratory system) were used (Table 3).
Table 3.
Orientation (direction cosines) of g tensor axes of
Y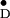 in the two dimer halves for one
site* in single crystals of PS II cc from
S. elongatus
in the two dimer halves for one
site* in single crystals of PS II cc from
S. elongatus
| x1 | y1 | z1 | x2 | y2 | z2 | C2† | |
|---|---|---|---|---|---|---|---|
| a | −0.681 | −0.201 | −0.704 | +0.177 | +0.257 | +0.950 | +0.282 |
| b | −0.440 | +0.881 | +0.174 | −0.557 | −0.769 | +0.312 | +0.558 |
| c | +0.585 | +0.428 | −0.688 | +0.811 | −0.585 | +0.007 | −0.781 |
Orientations for other sites can be obtained by using the symmetry operations of the P212121 space group; a, b, and c are the crystal axes.
C2 denotes the dimer axis for the considered crystal site.
For judging the quality of the simulation results, two effects have to be taken into account: (i) For orientations where the shift of spectral contributions by two sites is on the order of half the effective hyperfine coupling, the hyperfine structures are smeared out. This effect is very sensitive to the linewidth. The neglect of unresolved but anisotropic hyperfine interactions that contribute to the linewidth is therefore reflected by different relative amplitudes of features in the experimental and simulated spectra. The spectral positions of those features, however, are virtually unaffected and are well reproduced in the simulations. (ii) Although the overall quality of the simulations depends on the accurate values of all spectroscopic parameters, the orientation of the g tensor obtained from a fit is rather insensitive to changes in the hyperfine parameters. On the basis of the variation of the fit results for several sets of given hfcs and g principal values, we estimate the error for the orientation of the g tensor to be less than 3°.
For the P212121 space group, it is in general impossible to unambiguously identify the symmetry axes obtained from EPR spectra with the crystallographic axes by magnetic resonance methods. The existence of an additional noncrystallographic symmetry element, the 2-fold rotation axis C2 of each site, however, allows resolution of this ambiguity. The direction cosines of the C2 axis differ with respect to each crystallographic axis. Because the simulations depend on these values, the crystallographic axes can be identified by exclusion of those tentative assignments that fail to yield a good simulation of the experimental spectra. To verify this indirect assignment of the crystal axes. EPR experiments were complemented by x-ray diffraction experiments performed on the same crystal mounted in the EPR tube. These experiments confirmed the assignment derived from the EPR spectra.
The noncrystallographic C2 symmetry has an additional consequence. In general, even when the orientation of the crystallographic axis system is known, it is unclear to which site in the unit cell the obtained g tensor orientation belongs. This is because all other sites can be generated from an arbitrarily chosen site by the same set of symmetry operations. Because the C2 axis is noncrystallographic, it is different for each crystal site and can therefore be used as a site label. This resolves ambiguities in correlating the g tensor orientation with structural models obtained from x-ray crystallography.
Discussion
In this work, the g tensor principal values of
Y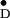 in PS II cc could be determined with
high accuracy. These values are compared to data taken from the
literature in Table 1. Our results, taking error margins into account,
differ slightly from those reported by most other authors. This could
be because of differences in PS II preparations from different
organisms. However, given the variation of reported g values
for the same system, i.e. spinach, it is more likely that small
differences are the result of different field calibration methods.
in PS II cc could be determined with
high accuracy. These values are compared to data taken from the
literature in Table 1. Our results, taking error margins into account,
differ slightly from those reported by most other authors. This could
be because of differences in PS II preparations from different
organisms. However, given the variation of reported g values
for the same system, i.e. spinach, it is more likely that small
differences are the result of different field calibration methods.
The EPR lines observed in this work are rather narrow (minimum
linewidth about 0.25 mT), reflecting negligible g strain and
indicating a well-defined environment of the
Y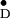 radical. This is assumed to result from
a tight hydrogen bond (42–44) between Y
radical. This is assumed to result from
a tight hydrogen bond (42–44) between Y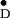 and a histidine residue in the protein (12, 14, 41, 45) that affects
the g values. The g tensor, particularly
gx, is a sensitive probe for hydrogen bond
strength and orientation (34, 46, 47). This is of particular interest
because the hydrogen bond is believed to have a significant effect on
redox potential and radical reactivity (48). The symmetry-related
Y
and a histidine residue in the protein (12, 14, 41, 45) that affects
the g values. The g tensor, particularly
gx, is a sensitive probe for hydrogen bond
strength and orientation (34, 46, 47). This is of particular interest
because the hydrogen bond is believed to have a significant effect on
redox potential and radical reactivity (48). The symmetry-related
Y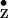 radical in PS II is assumed to be in a
more heterogeneous environment with a less well defined hydrogen bond
(49). A comparative analysis of the binding situation of the tyrosine
radicals, therefore, will help to understand the properties of
Y
radical in PS II is assumed to be in a
more heterogeneous environment with a less well defined hydrogen bond
(49). A comparative analysis of the binding situation of the tyrosine
radicals, therefore, will help to understand the properties of
Y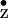 that are essential for the function of
the PS II complex.
that are essential for the function of
the PS II complex.
The analysis of orientation-dependent EPR spectra in the single crystals of PS II yielded precise data on the orientation of the g tensor in the unit cell. Because the g tensor is closely related (50) to the geometrical structure of phenoxyl type radicals (Fig. 1), the orientation of the phenoxyl ring itself can be derived. This information can be used to complement x-ray diffraction data in developing a detailed structural model of the PS II cc.
The dimeric form of PS II found in our experiments has also been
observed in electron microscopy studies of two-dimensional crystals
from spinach PS II (11, 51, 52). This suggests that PS II appears as a
dimer in native membranes as well. It can furthermore be assumed that
the dimer structure in the crystals used in this work is identical to
that in the native membrane. Because the C2 axis
of the dimer is perpendicular to the membrane plane, the orientation of
Y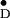 relative to the membrane normal can be
derived by calculating the angles between the g tensor axes
and the C2 axis (see Table 3). This permits
comparison of the single crystal data with recent EPR experiments on
oriented membrane fragments (25). Our single crystal data mostly
confirm the orientation derived from those samples (Fig.
4). The small difference in orientation
may be caused by some disorder of the oriented membranes.
relative to the membrane normal can be
derived by calculating the angles between the g tensor axes
and the C2 axis (see Table 3). This permits
comparison of the single crystal data with recent EPR experiments on
oriented membrane fragments (25). Our single crystal data mostly
confirm the orientation derived from those samples (Fig.
4). The small difference in orientation
may be caused by some disorder of the oriented membranes.
Figure 4.

Orientation of the phenoxyl group of Y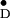 in
PS II from S. elongatus with respect to the membrane normal
(parallel to the C2 symmetry axis), as derived
from the single crystal EPR spectra. The angle α = 84° is
between the gy direction and the membrane normal
n; β = 26° is the tilt of the phenoxyl ring plane
with respect to n. Work on oriented membrane fragments from
spinach yielded α = 74° and β = 26° (25).
in
PS II from S. elongatus with respect to the membrane normal
(parallel to the C2 symmetry axis), as derived
from the single crystal EPR spectra. The angle α = 84° is
between the gy direction and the membrane normal
n; β = 26° is the tilt of the phenoxyl ring plane
with respect to n. Work on oriented membrane fragments from
spinach yielded α = 74° and β = 26° (25).
There is considerable variation in the hyperfine data of
Y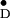 and their interpretation in the
literature (Table 2). Our results are in very good agreement with Rigby
et al. (22). For an understanding of the electronic
structure and the reactivity of the tyrosine radical, it is crucial to
arrive first at a consistent interpretation of the hyperfine structure.
As an example, the slight inequivalence of the protons in positions 3
and 5 can be used, together with calculations, to derive information
about the orientation of the hydrogen bond (38). The pronounced
difference between protons in positions 7a and 7b can be related to the
orientation of the phenoxyl head groups with respect to the protein
backbone (40). These structural details are likely to have an effect on
the reactivity and function of phenoxyl type radicals (48).
and their interpretation in the
literature (Table 2). Our results are in very good agreement with Rigby
et al. (22). For an understanding of the electronic
structure and the reactivity of the tyrosine radical, it is crucial to
arrive first at a consistent interpretation of the hyperfine structure.
As an example, the slight inequivalence of the protons in positions 3
and 5 can be used, together with calculations, to derive information
about the orientation of the hydrogen bond (38). The pronounced
difference between protons in positions 7a and 7b can be related to the
orientation of the phenoxyl head groups with respect to the protein
backbone (40). These structural details are likely to have an effect on
the reactivity and function of phenoxyl type radicals (48).
The experiments performed in this work show that high-field EPR provides the sensitivity needed for investigating very small samples. Using the upper limit of 100% yield for the YD radical in the samples, we estimate a maximum of 1013 spins in a typical single crystal. An estimate shows that, with optimum modulation and filtering, well resolved spectra with signal-to-noise ratios of >100 are achievable in about 3 minutes. It should therefore be possible to obtain spectra with a signal-to-noise ratio of 10 and similar hyperfine resolution even from a crystal of 1-nl volume (3⋅1011 spins) within less than 15 minutes. This opens the possibility of working on samples of which only very small quantities or tiny crystals are available.
Apart from sensitivity considerations, high-field/high-frequency EPR also provides excellent spectral resolution. This permits investigation of single crystals containing paramagnetic species with small g anisotropy, even when the number of magnetically inequivalent sites is rather large and/or the spectra exhibit a complex hyperfine structure, as in the case of the tyrosine radical.
In further work, we want to access more detailed hyperfine information
by performing high-field ENDOR experiments on PS II single crystals and
frozen solutions. The achievable orientation selection of ENDOR spectra
for tyrosine radicals in frozen solution samples has been demonstrated
before (53). High-field ENDOR on crystalline samples will provide
complete orientational information of all hfc tensors, in particular
for the H-bond proton. Similar experiments are envisioned for the
Y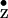 radical with the aim of understanding
the structural and functional differences between
Y
radical with the aim of understanding
the structural and functional differences between
Y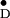 and Y
and Y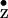 in
detail. Furthermore, experiments on the other paramagnetic
intermediates in PS II and on the S states of the WOC are planned. Work
on Y
in
detail. Furthermore, experiments on the other paramagnetic
intermediates in PS II and on the S states of the WOC are planned. Work
on Y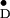 in the PS II single crystals has
paved the way for these future studies.
in the PS II single crystals has
paved the way for these future studies.
Acknowledgments
We gratefully acknowledge N. Krauss (Freie Universität Berlin) for performing the x-ray diffraction experiments and M. Kammel (Technische Universität Berlin) for assistance in the early stages of the experiments. This work was supported by Deutsche Forschungsgemeinschaft (Lu-315/15-1 and Sfb 498 TP C3 and C5) and Fonds der Chemischen Industrie.
Abbreviations
- ENDOR
electron nuclear double resonance
- hfc
hyperfine coupling constant
- PS II
Photosystem II
- cc
core complex(es)
- cw, continuous wave.
References
- 1.Ort D R, Yocum C F. In: Advances in Photosynthesis. Ort D R, Yocum C F, editors. Vol. 4. Dordrecht, The Netherlands: Kluwer; 1996. pp. 1–9. [Google Scholar]
- 2.Witt H-T. Ber Bunsenges Phys Chem. 1996;100:1923–1942. [Google Scholar]
- 3.Messinger J. Biochim Biophys Acta. 2000;1459:481–488. doi: 10.1016/s0005-2728(00)00187-0. [DOI] [PubMed] [Google Scholar]
- 4.Renger G. Biochim Biophys Acta. 2001;1503:210–228. doi: 10.1016/s0005-2728(00)00227-9. [DOI] [PubMed] [Google Scholar]
- 5.Yachandra V K, Sauer K, Klein M P. Chem Rev. 1996;96:2927–2950. doi: 10.1021/cr950052k. [DOI] [PubMed] [Google Scholar]
- 6.Britt R D. In: Advances in Photosynthesis. Ort D R, Yocum C F, editors. Vol. 4. Dordrecht, The Netherlands: Kluwer; 1996. pp. 137–164. [Google Scholar]
- 7.Debus R J. Biochim Biophys Acta. 1992;1102:269–352. doi: 10.1016/0005-2728(92)90133-m. [DOI] [PubMed] [Google Scholar]
- 8.Trebst A. Z Naturforsch. 1986;41c:240–245. [Google Scholar]
- 9.Michel H, Deisenhofer J. Biochemistry. 1988;27:1–7. [Google Scholar]
- 10.Tang X-S, Fushimi K, Satoh K. FEBS Lett. 1990;273:257–260. doi: 10.1016/0014-5793(90)81098-9. [DOI] [PubMed] [Google Scholar]
- 11.Rhee K-H, Morris E P, Barber J, Kühlbrandt W. Nature (London) 1998;396:283–286. doi: 10.1038/24421. [DOI] [PubMed] [Google Scholar]
- 12.Ruffle S V, Donnelly D, Blundell T L, Nugent J H A. Photosynth Res. 1992;34:287–300. doi: 10.1007/BF00033446. [DOI] [PubMed] [Google Scholar]
- 13.Zouni A, Witt H T, Kern J, Fromme P, Krauss N, Saenger W, Orth P. Nature (London) 2001;409:739–743. doi: 10.1038/35055589. [DOI] [PubMed] [Google Scholar]
- 14.Tang X-S, Chisholm D A, Dismukes G C, Brudvig G W, Diner B. Biochemistry. 1993;32:13742–13748. doi: 10.1021/bi00212a045. [DOI] [PubMed] [Google Scholar]
- 15.Kirilovsky D L, Boussac A G P, van Mieghem F J E, Ducruet J-M R C, Setif P R, Yu J, Vermass W F J, Rutherford A W. Biochemistry. 1992;31:2099–2107. doi: 10.1021/bi00122a030. [DOI] [PubMed] [Google Scholar]
- 16.Magnuson A, Rova M, Mamedov F, Fredriksson P-O, Styring S. Biochim Biophys Acta. 1999;1411:180–191. doi: 10.1016/s0005-2728(99)00044-4. [DOI] [PubMed] [Google Scholar]
- 17.Miller A-F, Brudvig G W. Biochim Biophys Acta. 1991;1056:1–18. doi: 10.1016/s0005-2728(05)80067-2. [DOI] [PubMed] [Google Scholar]
- 18.Lebedev Y S. Appl Magn Reson. 1994;7:339–362. [Google Scholar]
- 19.Feher G. Phys Rev. 1956;103:834–835. [Google Scholar]
- 20.Hoganson C W, Babcock G T. Biochemistry. 1992;31:11874–11880. doi: 10.1021/bi00162a028. [DOI] [PubMed] [Google Scholar]
- 21.Gilchrist M L, Ball J A, Randall D W, Britt R D. Proc Natl Acad Sci USA. 1995;92:9545–9549. doi: 10.1073/pnas.92.21.9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rigby S E J, Nugent J H A, O'Malley P J. Biochemistry. 1994;33:1734–1742. doi: 10.1021/bi00173a016. [DOI] [PubMed] [Google Scholar]
- 23.Warncke K, Babcock G T, McCracken J. J Am Chem Soc. 1994;116:7332–7340. [Google Scholar]
- 24.Rutherford A W. Biochim Biophys Acta. 1985;807:189–201. [Google Scholar]
- 25.Dorlet P, Rutherford A-W, Un S. Biochemistry. 2000;39:7826–7834. doi: 10.1021/bi000175l. [DOI] [PubMed] [Google Scholar]
- 26.Zouni A, Jordan R, Schlodder E, Fromme P, Witt H-T. Biochim Biophys Acta. 2000;1457:103–105. doi: 10.1016/s0005-2728(00)00100-6. [DOI] [PubMed] [Google Scholar]
- 27.Prisner T F, Rohrer M, Möbius K. Appl Magn Reson. 1994;7:167–183. [Google Scholar]
- 28.Dekker J P, Boekema E J, Witt H T, Rögner M. Biochim Biophys Acta. 1988;936:307–318. [Google Scholar]
- 29.Zouni A, Lüneberg C, Fromme P, Schubert W D, Saenger W, Witt H-T. In: Photosynthesis: Mechanisms and Effects. Garab G, editor. II. Dordrecht, The Netherlands: Kluwer; 1998. pp. 925–928. [Google Scholar]
- 30.Barry B A, Boerus R J, de Paula L C. In: The Molecular Biology of Cyanobacteria. Bryant D A, editor. Dordrecht, The Netherlands: Kluwer; 1994. pp. 217–257. [Google Scholar]
- 31.Han K C, Shen J R, Ikeuchi M, Inoue Y. FEBS Lett. 1994;355:121–124. doi: 10.1016/0014-5793(94)01182-6. [DOI] [PubMed] [Google Scholar]
- 32.Stesmans A, van Gorp G. Rev Sci Instrum. 1989;60:2949–2952. [Google Scholar]
- 33.Geßner C, Trofanchuk O, Kawagoe K, Higuchi Y, Yasuoka N, Lubitz W. Chem Phys Lett. 1996;256:518–524. [Google Scholar]
- 34.Un S, Tang X-S, Diner B A. Biochemistry. 1996;35:679–684. doi: 10.1021/bi9523769. [DOI] [PubMed] [Google Scholar]
- 35.Un S, Brunel L-C, Brill T M, Zimmermann J-L, Rutherford A W. Proc Natl Acad Sci USA. 1994;91:5262–5266. doi: 10.1073/pnas.91.12.5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farrar C T, Gerfen G J, Griffin R G, Force D A, Britt R D. J Phys Chem B. 1997;101:6634–6641. [Google Scholar]
- 37.Gulin V I, Dikanov S A, Tsvetkov Y D, Evelo R G, Hoff A J. Pure Appl Chem. 1992;64:903–906. [Google Scholar]
- 38.O'Malley P J, Ellson D. Biochim Biophys Acta. 1997;1320:65–72. [Google Scholar]
- 39.Himo F, Gräslund A, Eriksson L A. Biophys J. 1997;72:1556–1567. doi: 10.1016/S0006-3495(97)78803-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carrington A, McLachlan A D. Introduction to Magnetic Resonance. New York: Harper & Row; 1969. [Google Scholar]
- 41.Campbell K A, Peloquin J M, Diner B A, Tang X-S, Chisholm D A, Britt R D. J Am Chem Soc. 1997;119:4787–4788. [Google Scholar]
- 42.Barry B A, Babcock G T. Chem Scr. 1988;A28:117–121. [Google Scholar]
- 43.Evelo R G, Hoff A J, Dikanov S A, Tyrishkin A M. Chem Phys Lett. 1989;161:479–484. [Google Scholar]
- 44.Babcock G T, Barry B A, Debus R J, Hoganson C W, Atamian M, McIntosh L, Sithole I, Yocum C F. Biochemistry. 1989;28:9557–9565. doi: 10.1021/bi00451a001. [DOI] [PubMed] [Google Scholar]
- 45.Debus R J, Barry B A, Babcock G T, Macintosh L. Proc Natl Acad Sci USA. 1988;85:427–430. doi: 10.1073/pnas.85.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engström M, Himo F, Gräslund A, Minaev B, Vahtras O, Agren H. J Phys Chem A. 2000;104:5149–5153. [Google Scholar]
- 47.Bleifuss G, Pötsch S, Hofbauer W, Gräslund A, Lubitz W, Lassmann G, Lendzian F. In: Magnetic Resonance and Related Phenomena. Ziessow D, Lubitz W, Lendzian F, editors. II. Berlin: Technische Universität Berlin; 1998. pp. 879–880. [Google Scholar]
- 48.Babcock G T, Espe M, Hoganson C, Lydakis-Simantiris N, McCracken J, Shi W, Styring S, Tommos C, Warncke K. Acta Chem Scand. 1997;51:533–540. doi: 10.3891/acta.chem.scand.51-0533. [DOI] [PubMed] [Google Scholar]
- 49.Force D A, Randall D W, Britt R D, Tang X S, Diner B. J Am Chem Soc. 1995;117:12643–12644. [Google Scholar]
- 50.Fasanella E L, Gordy W. Proc Natl Acad Sci USA. 1969;62:299–301. doi: 10.1073/pnas.62.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rhee K-H, Morris E P, Zheleva D, Hankamer B, Kühlbrandt W, Barber J. Nature (London) 1997;389:522–526. [Google Scholar]
- 52.Boekema E J, van Breemen J F L, van Roon H, Dekker J P. J Mol Biol. 2000;301:1123–1133. doi: 10.1006/jmbi.2000.4037. [DOI] [PubMed] [Google Scholar]
- 53.Bennati M, Farrar C T, Bryant J A, Inati S J, Weis V, Gerfen G J, Riggs-Gelasco P, Stubbe J, Griffin R G. J Magn Reson. 1999;138:232–243. doi: 10.1006/jmre.1999.1727. [DOI] [PubMed] [Google Scholar]