

Background: Little is known about potential regulation of non-sirtuin HDACs by cellular metabolites.
Results: HDAC activity is stimulated by conjugated CoA derivatives and NADPH and is inhibited by free CoA.
Conclusion: Cellular metabolites required for anabolism directly stimulate HDAC activity.
Significance: Cellular HDAC activity may be modulated in response to the metabolic state of a cell.
Keywords: Chromatin, Coenzyme A, Epigenetics, Histone Deacetylase, Histone Deacetylase Inhibitors, Histone Modification, Histone Deacetylase Activity
Abstract
Histone deacetylases (HDACs) function in a wide range of molecular processes, including gene expression, and are of significant interest as therapeutic targets. Although their native complexes, subcellular localization, and recruitment mechanisms to chromatin have been extensively studied, much less is known about whether the enzymatic activity of non-sirtuin HDACs can be regulated by natural metabolites. Here, we show that several coenzyme A (CoA) derivatives, such as acetyl-CoA, butyryl-CoA, HMG-CoA, and malonyl-CoA, as well as NADPH but not NADP+, NADH, or NAD+, act as allosteric activators of recombinant HDAC1 and HDAC2 in vitro following a mixed activation kinetic. In contrast, free CoA, like unconjugated butyrate, inhibits HDAC activity in vitro. Analysis of a large number of engineered HDAC1 mutants suggests that the HDAC activity can potentially be decoupled from “activatability” by the CoA derivatives. In vivo, pharmacological inhibition of glucose-6-phosphate dehydrogenase (G6PD) to decrease NADPH levels led to significant increases in global levels of histone H3 and H4 acetylation. The similarity in structures of the identified metabolites and the exquisite selectivity of NADPH over NADP+, NADH, and NAD+ as an HDAC activator reveal a previously unrecognized biochemical feature of the HDAC proteins with important consequences for regulation of histone acetylation as well as the development of more specific and potent HDAC inhibitors.
Introduction
HDACs4 catalyze the removal of acetyl groups from lysine residues of a wide array of substrate proteins in addition to histones, including transcription factors and other nuclear and cytoplasmic proteins. Based on phylogenetic sequence analysis, HDACs are classified into class I (HDAC1–3 and -8), class II (HDAC4–7 and -9), class III, also known as sirtuins (SIRT1–7), and class IV (HDAC11) (1, 2). HDACs are ubiquitously expressed and play roles in regulation of cell growth, differentiation, and death (1, 3). They show deregulated expression in pathological states such as cancer and have therefore garnered significant interest as therapeutic targets (4). There are currently more than 80 clinical trials underway to determine the therapeutic efficacies of several HDAC inhibitors that have shown anti-neoplastic functions in cell culture and animal studies. Two HDAC inhibitors, Vorinostat and Romidepsin, have been approved for use in treatment of cutaneous T-cell lymphoma (5, 6). Other HDAC inhibitors have been used successfully as mood stabilizers and anti-epileptics and have shown promise in treatment of diverse neurological diseases such as Huntington and Alzheimer disease, amyotrophic lateral sclerosis, and spinal muscular atrophy (7, 8). Therefore, it is critical to fully understand the molecular mechanisms that regulate the activities of HDACs for the development of more effective HDAC inhibitors and more informed use of the current drugs in the clinic.
HDACs are generally found in multiprotein complexes, the functions of which can be regulated through several mechanisms. These complexes can be recruited to specific genomic loci by DNA-binding proteins, thereby bringing HDACs to close proximity of their histone substrates (9–11). HDAC-containing complexes are localized in both the nucleus and cytoplasm but can shuttle between the two subcellular compartments, affecting their accessibility to relevant substrates (12, 13). Modulating the stability of interactions between HDACs and other members of the complexes in which they reside can also affect the overall activity of HDAC complexes (14, 15).
Other regulatory mechanisms may affect the HDAC enzymatic activity more directly. Acetylation of HDAC1 protein reduces its enzymatic activity both in vivo and in vitro (16). Sumoylation of HDAC1 increases its enzymatic activity, and phosphorylation of HDAC1 stimulates both its activity and complex formation (17, 18). The catalytic activity of class III HDACs (Sirtuins) depends on the presence of the oxidized form of nicotinamide adenosine dinucleotide (NAD+) (19–21). Because the availability of NAD+ is linked to cellular metabolism, the metabolic state of the cell could be a direct regulator of sirtuins (22). Such direct regulation by a metabolic cofactor may transmit information on the cellular energy state to the chromosome, influencing nuclear functions such as gene expression and DNA replication.
However, little is known about direct regulation of non-sirtuin HDAC activity by metabolic intermediates. A first example of this kind of regulation is the binding and inhibition of HDAC1 and -2 by the endogenous lipid mediator sphingosine 1-phosphate (23). Because HDACs release free acetate anions from chromatin, we asked whether intermediates of nitrogen and carbon metabolism that generate or consume two-carbon units as acetate directly regulate HDAC activity.
Here, we show that coenzyme A (CoA) derivatives, such as acetyl-CoA, butyryl-CoA, and malonyl-CoA, as well as NADPH stimulate the activity of class I HDACs on histones, whereas free CoA inhibits HDAC activity in vitro. Detailed kinetic analysis (24) showed that CoA derivatives and NADPH cause mixed activation of recombinant HDAC2, affecting both the affinity of the enzyme for the substrate as well as its catalytic activity. Inhibition by free CoA is substrate-independent and is characterized by decreased reaction velocity while at the same time increasing the affinity of the enzyme for histones. We further show that HDAC activation by NADPH may be relevant in vivo as inhibition of NADPH production increases global histone acetylation. Many of the identified metabolites are products of catabolic pathways of glucose and amino acids that together with NADPH, a source of reducing power, are required for anabolic reactions that fuel cell growth and replication. Our data therefore suggest that cellular HDAC activity may be tightly linked to cellular biosynthetic capacity. Furthermore, identification of natural activators and inhibitors of HDACs that contain a nucleotide-like moiety may enhance our understanding of structure-function relationship and better inform the design and development of HDAC inhibitors.
EXPERIMENTAL PROCEDURES
Experiments in Figs. 1–3 were performed with recombinant HDACs obtained from US Biological. All metabolic compounds were purchased from Sigma as sodium or lithium salts.
FIGURE 1.

Coenzyme A derivatives and NADPH increase the in vitro activity of HDAC1 and HDAC2. A, recombinant HDAC1 and -2 were incubated with saturating amounts (50 μm) of 3H-labeled histones in the absence or presence of the indicated metabolites (1 mm). Released 3H-labeled acetate was extracted and measured by scintillation counting. The graph represents the fold change in HDAC activity compared with basal activity. B, same as in A but in the absence or presence of 1 mm of the indicated nicotinamide dinucleotides. C, in vitro HDAC activity of 1 μg of nuclear extract from each of human breast cancer cell lines MCF7 and MDA-MB-231 and the prostate cancer cell lines LNCaP and PC3 was determined and reported as in A. D, HDAC1- and HDAC2-complexes were immunoprecipitated from MDA-MB-231 whole cell extract, and HDAC activity was assessed in the presence or absence of 0.5 and 1.5 mm of either crotonyl-CoA or free CoA. Immunoprecipitation with nonspecific rabbit IgG and no enzyme controls was used to determine assay background. E, immunoprecipitated HDAC1 and -2 complexes from MCF7, MDA-MB-231, and the embryonic kidney cell line HEK293 were used to assess the in vitro activity in the presence and absence of NADPH. Immunoprecipitation with nonspecific rabbit IgGs, no- enzyme and a protein A-Dynabeads only controls were used to determine background levels. F, recombinant HDAC1 and -2 were incubated with saturating amounts of acetylated substrate Fluor de Lys in the absence or presence of the indicated metabolites. HDAC activity was determined by fluorimetry. The graph represents the fold change in HDAC activity compared with basal activity in absence of metabolites. Error bars indicate standard deviation of three independent experiments. A two-tailed Student's t test was used to calculate p values (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
FIGURE 2.

Kinetics of HDAC2 activation by NADPH and acetyl-CoA. A, determination of the effective concentration of NADPH. 25 nm recombinant HDAC2 and 50 μm 3H-labeled histones were incubated with increasing amounts of NADPH, and in vitro HDAC activity was assessed. The graph represents the fold change in HDAC activity compared with basal activity in absence of NADPH. EC50 indicates the effective concentration for NADPH. B, as in A but for acetyl-CoA. C, analysis of activation kinetics for HDAC2 and NADPH. 25 nm recombinant HDAC2 was incubated with increasing amounts of 3H-labeled histones (0–2000 μm) in the absence or presence of increasing amounts of NADPH. The graph represents the rate of histone deacetylation (μm histones deacetylated per min). D, as in C but for acetyl-CoA. E, log Km and log Vmax values with increasing amounts of NADPH, as determined from B. F, as in E but for acetyl-CoA. G, curve fitting of data in C using the Hill equation. H, as in G but for acetyl-CoA. I, HDAC enzyme activity at very low substrate concentrations with increasing concentration of NADPH is shown as a bar chart. J, as in I but for acetyl-CoA. Error bars represent the standard deviation of three independent experiments.
FIGURE 3.

Kinetics of HDAC2 inhibition by coenzyme A. A, determination of the effective concentration of CoA. 25 nm recombinant HDAC2 and 50 μm 3H-labeled histones were incubated with increasing amounts of CoA, and in vitro HDAC activity was determined. The graph depicts fold change in HDAC activity over basal activity. B, analysis of kinetics of inhibition by CoA. 25 nm recombinant HDAC2 was incubated with increasing amounts of 3H-labeled histones (0–2000 μm) without or in the presence of 25 or 75 μm CoA. The graph shows the rate of histone deacetylation (μm histones deacetylated per min). C, same as B, but in the presence of 500, 1000, and 2000 μm CoA. D, log Km and log Vmax values with increasing amounts of CoA, as determined from B and C. E, curve fitting of data in B using the Hill equation. F, curve fitting of data in C using the Hill equation. Error bars represent the standard deviation of three independent experiments.
Generation of Recombinant WT HDAC1 and HDAC1 Mutants
An EcoRI-HDAC1-His6-SalI fragment was generated by PCR using a commercially available cDNA for HDAC1 (OriGene). Recombinant HDAC baculovirus was generated using the Bac-to-Bac expression system (Invitrogen), following the manufacturer's instructions. HDAC1 mutants were created by site-directed mutagenesis on the pFastbac-HDAC1 plasmid using QuikChange Site-directed mutagenesis kit (Agilent).
Purification of Recombinant HDAC Protein
Sf9 cell pellets were washed with ice-cold PBS and resuspended in hypotonic buffer (20 mm HEPES-KOH, pH 8.0, 10 mm NaCl, 10% glycerol, 2.5 mm β-mercaptoethanol, 1× protease inhibitor mixture (Roche Applied Science)). The cells were incubated on ice for 30 min and then disrupted using a Dounce homogenizer. Nuclei were spun down at 7000 × g for 10 min, and the cytoplasmic supernatant (containing the majority of the recombinant HDAC protein) was transferred to a fresh tube and re-centrifuged at 22,000 × g for 30 min to remove debris. The recombinant protein was purified using the TALON Metal Affinity Resin (Clontech) following the manufacturer's instructions and dialyzed in HDAC storage buffer (20 mm HEPES, 150 mm KCl, 30% glycerol, and 2.5 mm β-mercaptoethanol).
Isolation of 3H-Labeled Histones from HeLa Cells
HeLa cells were grown exponentially in suspension to a density of 6 × 105 cells/ml. Histones were labeled and extracted as described previously (25). Concentration of histones was determined by BCA protein assay (Pierce), and their molarity was calculated using the average molecular weight of histones H2A, H2B, H3, and H4.
HDAC Assay
Recombinant HDACs or nuclear extracts were incubated with 3H-labeled histones and metabolic intermediates (quantities as indicated in figures) for 30 min at 37 °C in Buffer D (13.3 mm HEPES-KOH, pH 7.9, 7 mm MgCl2, 13.3% glycerol, 66.7 mm KCl, and 66.7 μm EDTA). The reaction was stopped with 0.2 volume of 5× Stop Solution (0.6 m acetic acid, 3.6 m HCl), and the reaction product (3H-labeled acetate) was extracted with 2 volumes of ethyl acetate. Extracted 3H-labeled acetate was quantified using a liquid scintillation counter (TriCarb2800TR, PerkinElmer Life Sciences). HDAC assays with fluorescent substrate were performed using the Fluor de LysTM-Green HDAC assay kit (Enzo Life Sciences).
Nuclear Extract
Cell cultures were grown in DMEM to 80% confluency. ∼5 × 106 cells were washed twice with ice-cold PBS and resuspended in 500 μl of Buffer A-NE (10 mm HEPES-KOH, pH 7.9, 10 mm KCl, 0.1 mm EDTA, and freshly added 0.4% IGEPAL, 1 mm DTT, 0.5 mm PMSF, and 1× protease inhibitor mixture (Roche Applied Science)). Nuclei were centrifuged at 16,000 rcf for 3 min at 4 °C and resuspended in 150 μl of Buffer B-NE (20 mm HEPES-KOH, pH 7.9, 400 mm NaCl, 1 mm EDTA, 10% glycerol, 1% IGEPAL, and freshly added 1 mm DTT, 0.5 mm PMSF, and 1× protease inhibitor Mixture (Roche Applied Science)) and incubated at 4 °C for 2 h with vigorous shaking. Nuclear extract was separated from debris by centrifugation at 16,000 rcf for 10 min at 4 °C and subsequently dialyzed twice for 2 h into Buffer D-NE (20 mm HEPES-KOH, pH 7.9, 100 mm KCl, 0.1 mm EDTA, 20% glycerol) at 4 °C. Protein concentration was detected by BCA protein assay (Pierce).
Histone Extraction for Western
Cell cultures were grown in DMEM plus serum to 80% confluency, and 5 × 106 cells were washed twice with ice-cold PBS and resuspended in 750 μl of Buffer A-H (10 mm HEPES-KOH, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.34 m sucrose, 10% glycerol, and freshly added 1 mm DTT and 1× protease inhibitors (Roche Applied Science)). After addition of 0.1% Triton, cells were lysed for 8 min on ice. Nuclei were pelleted at 1500 rcf for 5 min at 4 °C and washed with 1 ml of Buffer A-H for 1 min at 4 °C. Nuclei were pelleted at 1500 rcf for 2.5 min at 4 °C and resuspended in 5 pellet volumes of extraction buffer (432 mn H2SO4, 28 mm β-mercaptoethanol, 10% glycerol). Histones were extracted for 15 min on ice. After centrifugation at 16,000 rcf for 10 min, histones were precipitated from the supernatant by addition of 20% TCA followed by centrifugation at 16,000 rcf for 10 min at 4 °C. The pellet was washed once with 1 ml of 70% EtOH, dried, and resuspended in water.
Immunoprecipitation
Cells were grown in DMEM to ∼90% confluency, washed twice with ice-cold PBS, and lysed in 100 μl of lysis buffer (20 mm HEPES-KOH, pH 7.9, 100 mm KCl, 1 mm EDTA, 0.5% Nonidet P-40, 1× protease inhibitor mixture (Roche Applied Science) and 1 mm DTT) per 106 cells for 30 min on ice and centrifuged at 16,000 rcf for 10 min at 4 °C. 1 ml of clarified whole cell extract was incubated with 2 μg of αHDAC1, αHDAC2 (Bethyl Laboratories A300-713A and A300-705A, respectively), or rabbit IgG (Millipore) for 1 h at room temperature. 70 μl of Dynabeads protein A (Invitrogen) were washed with lysis buffer and used for precipitation for 1 h at 4 °C. Immunoprecipitated protein was washed three times for 10 min at room temperature with 1 ml of lysis buffer and one time with HDAC assay buffer, and HDAC activity was assayed.
Cell Fractionation
Cell fractionation was performed as described previously (26).
Data Analysis
EC50 values for Fig. 2, A and B, were determined with SigmaPlot, using the nonlinear regression analysis for a four-parameter logistic curve. Curve fitting and determination of Km, Vmax, and Hill values for Fig. 2, C–J, were performed with the enzyme kinetics module using the “Enzyme Activator-Nonessential” option and Padgraph Prism (SigmaPlot). Parameters for Fig. 3A were determined by fitting experimental data to a function describing a double-sigmoid curve (Equation 1), as described previously (27),
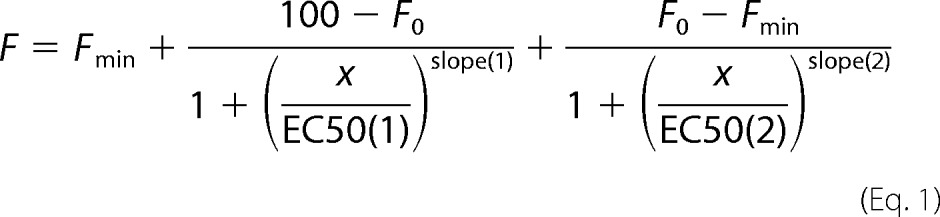 |
Using the curve-fitting tool of MATLAB and assuming that Fmin = 0, the data best fitted Equation 2,
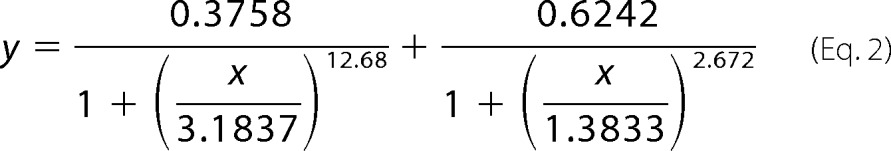 |
where x indicates log10-transformed CoA concentrations; y indicates fold activation, and the denominators of the fractions in parentheses are the log10-transformed EC50 values of F1 and F2. Curve fitting and determination of Km, Vmax, and Hill values for Fig. 3, B–F, were performed with the enzyme kinetics module using the “Single Substrate-Single Inhibitor” function and Padgraph Prism (SigmaPlot), and the curves with the highest R2 were chosen as best fit.
RESULTS
In Vitro HDAC Activity Is Regulated Directly by Specific Cellular Metabolites
Because histone acetylation requires acetyl-CoA and deacetylation releases acetate anions, we asked if metabolites of pathways that generate or consume two-carbon units directly regulate the activities of class I HDACs. To assay HDAC activity, 25–50 nm recombinant HDAC1 or HDAC2 were used in absence or presence of 1 mm of each metabolite shown in Fig. 1A using 50 μm 3H-labeled HeLa histones as substrate. As expected, addition of 1 mm sodium butyrate, a known inhibitor of HDACs, inhibited the activities of HDAC1 and HDAC2 (Fig. 1A). We found that the in vitro activities of HDAC1 and HDAC2 increased by ∼1.5-fold with addition of acetyl-, acetoacetyl-, and succinyl-CoA, by ∼2-fold with butyryl-, isobutyryl-, glutaryl-, HMG-, and malonyl-CoA, and by 2–3-fold with methylmalonyl-, crotonyl-, and β-methylcrotonyl-CoA relative to no compound control (Fig. 1A). However, the long chain fatty acid derivative palmitoyl-CoA inhibited HDAC1 and HDAC2 activities (Fig. 1A). All activating metabolites shared the presence of the CoA module. However, free CoA reduced the activities of HDAC1 and HDAC2 by 63 and 52%, respectively. Succinate had no effect on the in vitro HDAC activity. Interestingly, although sodium butyrate and free CoA inhibited HDAC1 and more so HDAC2 activities, butyryl-CoA increased the activities of the same HDACs by 2-fold (Fig. 1A).
The chemical structure of coenzyme A includes an adenosine nucleotide phosphorylated on the ribose moiety. Considering that a phosphorylated ribose module is present also in the adenosine nucleotide of NADP(H), we tested if NADP(H) could similarly affect HDAC activity. We found that 1 mm NADPH, but not NADP+, stimulated class I HDAC activity ∼2-fold (Fig. 1B). Neither NAD+ nor NADH showed any significant effect when assayed at the same concentrations (Fig. 1B), indicating that the phosphorylated ribose moiety may be required for the NADPH ability to activate HDACs. Taken together with the above, we conclude that a variety of natural CoA derivatives that function as biosynthetic metabolites as well as NADPH, the major reducing power for biosynthesis, are able to stimulate the in vitro activities of recombinant HDACs.
CoA Derivatives and NADPH Enhance HDAC Activity in Nuclear Extracts
To test if the CoA derivatives, NADPH and free CoA, affect HDAC activity in their native complexes, we determined the effects of the metabolites on HDAC activity in nuclear extracts from four different cancer cell lines. As shown in Fig. 1C, HDAC activities of nuclear extracts from two breast cancer (MCF7 and MDA-MB-231) and two prostate cancer (LNCaP and PC3) cell lines were increased by ∼1.5–2.2-fold upon addition of 500 μm of the indicated CoA derivatives or NADPH. Addition of the same amount of free CoA slightly reduced HDAC activity of the nuclear extracts. We therefore conclude that the CoA derivatives or NADPH enhance activities of HDACs in their natural molecular environment.
CoA Derivatives and NADPH Enhance the Activity of HDAC1- and HDAC2-containing Complexes
Next, we immunoprecipitated HDAC1 or HDAC2 from whole cell lysates of different cancer cell lines and assayed their response to the CoA compounds. Addition of crotonyl-CoA increased activities of HDAC1 and -2 complexes immunoprecipitated from the MDA-MB-231 cells by up to 2.5-fold, whereas free CoA caused significant inhibition (Fig. 1D). Addition of NADPH also increased HDAC activity of HDAC1 and -2 complexes from three different cell lines from 1.6- to 2.6-fold (Fig. 1E). Altogether, our data indicate that CoA derivatives and NADPH can stimulate class I HDAC activity in vitro and in vivo.
Stimulation of HDAC Activity by Metabolites Depends on Histone Substrates
Because allosteric activation could depend on the type of substrate used (28), we wished to determine whether activation of HDAC1 and HDAC2 by CoA derivatives or NADPH depends on histones as substrate. Thus, we used Fluor de Lys instead of histones as substrate in HDAC activity assays. Fluor de Lys is a small synthetic substrate for histone deacetylases, which is converted to a fluorophore upon deacetylation. Addition of 500 or 1000 μm malonyl-CoA, HMG-CoA, or NADPH had essentially no effect on HDAC1 and HDAC2 activities when Fluor de Lys was used as substrate (Fig. 1F). However, free CoA and trichostatin A inhibited deacetylation of Fluor de Lys by these deacetylases (Fig. 1F). These data indicate that activation of HDAC1 or HDAC2 by CoA derivatives or NADPH is substrate-dependent, although their inhibition by free CoA is not.
Half-maximal Effective Concentration (EC50) of Activators Is the Micromolar Range
To determine at which range of concentration NADPH and acetyl-CoA function as metabolic activators of HDACs, we assayed the extent of deacetylation of 50 μm histones by 25 nm HDAC2 in the presence of increasing concentrations of each activator, ranging from 50 to 2500 μm. The response curve for NADPH showed increased activation up to 2500 μm, yielding an ∼2.3-fold activation with an EC50 = 541.9 μm (Fig. 2A). Similar to NADPH, acetyl-CoA increased activation up to ∼2-fold at 2500 μm, yielding an EC50 = 602.9 μm (Fig. 2B). These data indicate that NADPH is a somewhat stronger activator of HDAC2 compared with acetyl-CoA in vitro.
Activation by NADPH and Acetyl-CoA Is Due to V-type Activation Counteracted by K-type Inhibition
To understand the nature of HDAC2 activation by NADPH and acetyl-CoA, we used our in vitro assay to analyze the enzymatic kinetics in the presence or absence of these compounds. 25 nm of recombinant HDAC2 was incubated with increasing concentrations of labeled histones, ranging from 0.5 to 50 μm, and in the presence of 0, 250, 500, 1000, and 2000 μm of each activator. The resulting data were fitted using the SigmaPlot enzymatic module (nonessential activation) and expressed as micromolars of histones deacetylated per min (see under “Experimental Procedures”). Fig. 2, C and D, shows the Michaelis-Menten graphs for HDAC2 activation by NADPH and acetyl-CoA, respectively. Vmax and Km values for HDAC2 in the absence of NADPH are 1.41e−2 μm/min and 12.73 μm, respectively, and the activation constant Ka(NADPH) is 224.71 μm. Similarly, Vmax and Km values for HDAC2 in the absence of acetyl-CoA are 1.08e−2 μm/min and 8.88 μm (Fig. 2D). The slight difference of Vmax and Km values in these two experiments is likely due to different batches of commercially acquired HDAC2 protein. Activation by acetyl-CoA is slightly lower than that by NADPH, having a Ka(AcCoA) of 251.75 μm.
To determine how Vmax and Km change at each concentration of activators, we determined the parameters W and Q. W is the ratio of Vmax in the presence of saturating effector to Vmax in the absence of effector (Vmax(inf)/Vmax(0)). Q is the opposite ratio of the corresponding Km values (Km(0)/Km(inf)) (Table 1) (24). The product WQ is >1 for enzyme activation, which is the case for both NADPH and acetyl-CoA (Table 1). In the case of NADPH, W >1 but Q <1, indicating that the enzyme velocity is increased, but affinity for substrate is decreased (Fig. 2E). In the case of acetyl-CoA, the curves indicate a more complex pattern. Both Vmax and Km values increase with increasing effector concentrations up to 500 μm, when a plateau is reached for Vmax but Km decreases. This indicates that up to 500 μm of the effector increases the reaction velocity more than it causes a decrease in affinity for the substrate, after which further activation is mainly achieved by regaining affinity for the substrate (Fig. 2F). Interestingly, at very low substrate concentrations of 0.5 μm histones, the presence of NADPH slightly inhibits HDAC activity, indicating that under these conditions K-type inhibition may prevail (Fig. 2G). We did not observe inhibition by acetyl-CoA at the lowest substrate concentrations tested (Fig. 2H).
TABLE 1.
Summary of enzyme kinetics parameters
| Parameter | NADPH |
Acetyl-CoA |
Coenzyme A (<100 μm) |
Coenzyme A (>500 μm) |
||||
|---|---|---|---|---|---|---|---|---|
| Value | S.E. | Value | S.E. | Value | S.E. | Value | S.E. | |
| Vmax(0) (μm/min) | 1.41e−2 | 7.56e−4 | 1.08e−2 | 7.12e−4 | 2.19e-2 | 8.09e-4 | 1.48e−2 | 4.39e−4 |
| Km(0) (μm) | 12.73 | 1.75 | 8.88 | 1.69 | 20 | 1.5920 | 13.06 | 0.98 |
| Vmax(inf) (μm/min) | 0.03731 | 0.002393 | 0.01587 | 0.000512 | 0.002117 | 0.000202 | ||
| Km(inf) (μm) | 24.85 | 3.28 | 8.866 | 0.8436 | 2.323 | 1.145 | ||
| Ka/Ki | 224.71 | 39.25 | 251.75 | 92.44 | 135.87 | 70.58 | 440.47 | 20.58 |
| α | 2.19 | 0.43 | 1.26 | 0.35 | 0.24 | 0.10 | ||
| β | 3.00 | 0.25 | 1.69 | 0.17 | 0.22 | 5.7e-2 | ||
| W | 2.605447 | 1.522989 | 0.093178 | |||||
| Q | 0.530785 | 0.855274 | 9.074473 | |||||
| QW | 1.382931 | 1.302572 | 0.84554 | |||||
Activation by NADPH or Acetyl-CoA Does Not Affect Cooperativity
To determine whether NADPH or acetyl-CoA enhances enzyme activity by causing cooperative binding of the HDACs to histones, we used the kinetic data from Fig. 2, C and D, to calculate the Hill values (29) using Graphpad Prism. As shown in Fig. 2, I and J, and Table 2, Hill values at all effector concentrations are close to 1, indicating that enzyme-substrate binding is not cooperative at any effector concentration.
TABLE 2.
Summary of Hill values
| Effector concentration | NADPH |
Acetyl-CoA |
Effector concentration | Coenzyme A |
|||
|---|---|---|---|---|---|---|---|
| Value | S.E. | Value | S.E. | Value | S.E. | ||
| μm | μm | ||||||
| 0 | 0.957 | 0.02331 | 0.9463 | 0.04577 | 0 | 0.9464 | 0.02707 |
| 250 | 1.008 | 0.02885 | 1.03 | 0.06653 | 25 | 0.9812 | 0.01854 |
| 500 | 1.05 | 0.01242 | 0.9971 | 0.02882 | 75 | 1.022 | 0.05057 |
| 1000 | 1.062 | 0.01937 | 1.038 | 0.05608 | 500 | 1.032 | 0.05405 |
| 2000 | 1.083 | 0.02547 | 1.031 | 0.01536 | 1000 | 1.04 | 0.02853 |
| 2000 | 1.005 | 0.1117 | |||||
Kinetics of Inhibition of HDAC Activity by Free CoA
We next investigated the nature of HDAC2 inhibition by free CoA in a similar manner as for the activators above. Using the same curve fitting function of SigmaPlot as for Fig. 2, A and B, did not yield any statistically significant result. We noticed that the experimental data were best fitted with a double sigmoidal curve. The resulting curve is the sum of two sigmoid functions, where the Vmax of function 1 (F1) equals Vmin of function 2 (F2) (Fig. 3A and under “Experimental Procedures”) (R2 = 0.9992). At low concentrations of CoA (<200 μm), the data are best described with a curve determined by F2 with an IC50(2) of 24.17 μm. Concentrations between 100 and 200 μm CoA correspond to a plateau of the double sigmoidal curve. Concentrations >200 μm yielded a second inhibitory response, best fitted with F1 with an IC50(1) of 1526.5 μm. Although the latter concentration is unlikely to be reached within the cellular environment, the IC50(2) is well in the range of well known HDAC inhibitors such as sodium butyrate, valproate, or phenyl butyrate with IC50 values ranging from 27 to 106 μm for HDAC1 or HDAC2 (30).
This dual nature of the dose response may reflect different modes of inhibition at different concentrations of the inhibitor. We therefore explored a wider range of CoA concentrations (25–2000 μm as compared with 250–2000 μm in Fig. 2, C and D) to generate the Michaelis-Menten plots. An attempt to fit all the data into a single graph did not result in statistically significant modeling. However, when data up to 75 μm CoA were separated from those above 500 μm, curves were fitted using the Michaelis-Menten equation (R2 >95%) and kinetic parameters could be determined (Fig. 3, B and C, and Table 1).
Plotting logVmax and logKm values against increasing amounts of free CoA shows that both Vmax and Km values diminish, giving a WQ value of 0.846 (Fig. 3D and Table 1). Inhibition is therefore a result of V-type inhibition (W = 0.093; Table 1) countered by Q-type activation (Q = 9.07, Table 1), indicating strong reduction of reaction velocity, yet increasing the affinity of the enzyme for the substrate. Interestingly, at lower CoA concentrations, Km values diminish more rapidly than Vmax; this relation inverts at higher concentrations of free CoA (>500 μm), which possibly reflects the reason for the double sigmoidal shape of the curve in Fig. 3A. We did not observe changes for binding cooperativity as Hill values were found to be close to 1 (Fig. 3, E and F, and Table 2).
Structural Basis of HDAC Activation by Metabolites
The structures of NADPH and CoA contain a phosphorylated adenine nucleotide. Thus, we searched for possible structural motifs in either HDAC1 or HDAC2 that could accommodate a phosphorylated adenine nucleotide. HDAC1 and HDAC2 are almost entirely made up of an HDAC domain flanked by relatively short N- and C-terminal unstructured regions. HDAC domains consist of an array of β-sheets sandwiched between two rows of α-helices that resemble a Rossmann-fold (31). This structural arrangement is typical of proteins that interact with nucleotides. We therefore searched for chemical similarities as well as sequence and structural alignments between HDAC1 and -2 and proteins that are known to bind either CoA derivatives or nicotinamide adenosine dinucleotides such as cytochrome P450 reductase or NADPH oxidases (see Table 3). We surmised that regions of similarity may include candidate residues that contribute to adenine nucleotide binding. We then mutated such residues in HDAC1 to either change their charge or bulkiness or to the corresponding sequence of class II HDACs to potentially maintain HDAC activity but affect “activatability.” In total, we generated 17 mutant HDAC1 proteins with single or multiple point mutations (see Table 3 for a description of and rational for construction of the mutants).
TABLE 3.
HDAC1 mutant description and design rational
| Mutant (HDAC1) | Method of design | Substitution |
|---|---|---|
| N134A | Chemical alignmenta | Decreased bulkiness |
| G138I | Chemical alignmenta | Increased bulkiness |
| K283A | Chemical alignmenta | Neutralized charge |
| L298D | Chemical alignmenta | Hydrophyllic, charge |
| G300I | Chemical alignmenta | Increased bulkiness |
| S263A/S267G | Sequence alignment to Sir2 homologue Af1 | Corresponding residues in HDAC3 |
| S263A/S265T/S267G | Sequence alignment to Sir2 homologue Af1 | Corresponding residues in HDAC3 and -8 |
| S265T/S267G | Sequence alignment to Sir2 homologue Af1 | Corresponding residues in HDAC3 |
| D181N | Structural alignmentb | Charge |
| F198L/K200R | Structural alignmentb | Corresponding residues in HDAC4, -5, and -9 |
| K200A | Structural alignmentb | Reduced bulkiness, eliminating charge |
| K200Q | Structural alignmentb | Eliminating charge |
| E203A | Structural alignmentb | Reduced bulkiness, eliminating charge |
| E203Q | Structural alignmentb | Eliminating charge |
| E203D | Structural alignmentb | Reduced bulkiness |
| T208S | Structural alignmentb | Corresponding residue in HDAC4, -5, and 9 |
| R229T | Structural alignmentb | Corresponding residue in HDAC4, -5, and 9 |
| R229Q | Structural alignmentb | Corresponding residue in HDAC8 |
a Chemical sequence alignment was performed using the IBM Sequence pattern discovery tool. NAD(H)/NADP(H)-binding proteins for comparison were HSCARG, SIRT1, aldose reductase, catalase, flavin reductase, cytochrome P450 reductase, NADPH oxidases 1–4.
b Structural alignment was performed using VAST (ncbi.nml.nih.gov). NAD(H)/NADP(H)-binding proteins for comparison were the same as in Footnote a.
Recombinant wild type (WT) HDAC1 and the various mutants were generated using the Sf9 insect cells (Fig. 4A) and tested for activity and activatability by either β-methylcrotonyl-CoA or NADPH, as these compounds had the strongest effect on the WT enzyme. All mutations reduced HDAC activity when compared with the corresponding WT with six mutants retaining 30–80% of WT HDAC activity and 13 mutants less than 25%. Six mutants had essentially no catalytic activity and were considered catalytically dead (Fig. 4B). Interestingly, although the remaining mutants affected the overall activity, most maintained their activatability by either NADPH or β-methylcrotonyl-CoA (Fig. 4C). Only one mutant, G300I, showed slightly decreased activatability, albeit not reaching statistical significance relative to WT (1.8 versus 2.2-fold, p value = 0.09; Fig. 4C). Although the G300I mutant has low basal HDAC activity, other mutants such as G138I showed even lower basal activity, yet retained their activatability (Fig. 4, B and C). Fig. 4D shows the crystal structure of HDAC2 (32) with the Gly-300 residue highlighted in blue. This glycine is located deep within the catalytic pocket, close to the catalytic residues highlighted in Fig. 4D in red. The G300I mutation replaces the small glycine with a bulky isoleucine, which may reduce the size of the catalytic pocket. This position is unlikely to be the site of interaction with the activator, yet G300I may hinder the effects of the activator on the enzyme-substrate interaction.
FIGURE 4.

D181N and G300I mutations in HDAC1 alter its response to activating metabolites. A, Coomassie staining of recombinant HDAC1 WT and mutant proteins. B, HDAC activity of recombinant mutant proteins was determined using saturating amounts of 3H-labeled histones. The graph represents the relative HDAC activity of mutant proteins compared with WT levels. C, recombinant WT and mutant HDAC1 proteins were incubated with saturating amounts of 3H-labeled histones in the absence or presence of 2 mm NADPH, and HDAC activity was assessed. The graph represents fold change in HDAC activity compared with basal histone deacetylation. Error bars indicate the standard deviation of three independent experiments. A two-tailed Student's t test was used to calculate p values (**, p < 0.01; ***, p < 0.001). D, image of a fragment of the crystal structure of HDAC2 (32) in which the location of the corresponding residues of Asp-181 and Gly-300 are indicated in green and dark blue, respectively. Catalytic residues (His-145, His-146, Asp-181, and His-183) are indicated in red.
A second mutation, D181N, changed the charge on the surface of the protein and, similar to G300I, caused a drastic reduction of the basal HDAC activity. Yet, this mutation made the mutant consistently more responsive to the activator compared with WT HDAC1 (3.1-fold versus 2.2-fold, respectively, p value = 0.01; Fig. 4, B and C). The corresponding residue in HDAC8, Asp-183, has been shown to take part in the catalytic charge-relay system (33). This raises the possibility that the activators may render catalysis less dependent on the negative charge of this aspartate. Although we were unable to identify a mutation that would retain the basal activity of the enzyme but abolish its activatability, our analyses of the HDAC mutants show that the enzyme can potentially be modified in a manner that alters its response to the activators. Further analyses are required to define the region(s) of the HDAC proteins that interact with the identified activators.
Inhibition of Glucose-6-phosphate Dehydrogenase (G6PD) Increases Histone Acetylation in Vivo
Our next aim was to determine whether regulation of HDACs by metabolites is relevant in an in vivo environment. NADPH is known to be present in the nucleus, where HDAC1 and -2 are active. G6PD, the rate-limiting enzyme of the pentose phosphate pathway, is a main producer of NADPH. We surmised that because inhibition of this protein decreases cellular NADPH levels (34), this may lead to decreased HDAC activity and increased levels of histone acetylation. We chose two breast cancer cell lines, MDA-MB-231 and MDA-MB-453, that express G6PD to different extents and should therefore differ in their response to G6PD inhibition. Total cell extracts from each line were separated into cytoplasmic), nucleoplasmic, and chromatin fractions and analyzed by Western blotting (Fig. 5A). As expected, tubulin was detected exclusively in the cytoplasm, whereas an antibody specific for histone H3 lysine 18 acetylation (H3K18ac) stained chromatin only (Fig. 5A). G6PD was expressed at significantly higher levels in MDA-MB-453 cells. Interestingly, a minor fraction of G6PD was detected in both nucleoplasm and chromatin fractions of the MDA-MB-453 cells (Fig. 5A), indicating that some G6PD may be associated with chromatin in the nucleus of MDA-MB-453 cells.
FIGURE 5.

Inhibition of G6PD increases histone acetylation in MDA-MB-453 breast cancer cells. A, G6PD expression in MDA-MB-231 and MDA-MB-453 cancer cells. Equal amounts of total cell lysate (T), cytoplasmic (Cy), nucleoplasmic (Nc), and chromatin-bound (Ch) fractions were analyzed by Western blotting using anti-G6PD, -tubulin, and -H3K18ac antibodies. The band intensities were quantified using the LI-COR Odyssey and indicated below each lane. B, MDA-MB-231 and MDA-MB-453 cells were incubated with or without 150 μm DHEA for 24 h. Histones were extracted, and acetylation was detected by Western blotting with site-specific antibodies, as indicated. Total histones were visualized by Coomassie staining. Detection and quantification were performed as in A. The numbers indicate the ratio of the integrated intensity relative to the no DHEA control.
We incubated both MDA-MB-231 and MDA-MB-453 cells with 5-dehydroepiandrosterone (DHEA; 100 μm), a known inhibitor of G6PD (35), or solvent only (DMSO) for 12 h, extracted histones, and analyzed their acetylation levels by Western blotting. We examined three acetylation sites in each of histones H4 and H3. As shown in Fig. 5B, DHEA-treated MDA-MB-231 cells did not show increased histone acetylation compared with DMSO-treated cells. In contrast, MDA-MB-453 cells, which had high levels of G6PD, showed increased histone acetylation at all sites analyzed with the strongest effects on H4K8ac and H4K12ac. We conclude that inhibition of G6PD by DHEA causes increased global histone acetylation.
DISCUSSION
We have identified several natural metabolites that function as allosteric activators of HDAC activity in vitro. These metabolites all share an adenosine phosphate moiety and include several CoA derivatives and NADPH. Some of the CoA derivatives are intermediates of pathways that break down carbohydrates or amino acids. For instance, glutaryl-CoA, crotonyl-CoA, and HMG-CoA are generated in the pathway of lysine degradation, whereas β-methylcrotonyl-CoA and succinyl-CoA are degradation products of leucine and methionine, respectively. Acetyl-CoA is a central molecule in the intermediary metabolism and can be generated from degradation of glucose, pyruvate, amino acids, as well as β-oxidation of fatty acids. Other identified CoA derivatives are involved in anabolic reactions such as malonyl-CoA and HMG-CoA, which are the main precursor for fatty acid and cholesterol syntheses, respectively. NADPH, the main reducing agent for biosynthetic reactions, also increased HDAC activity in vitro. Interestingly, ATP was also shown to activate class I HDACs, albeit possibly indirectly through ATP-dependent molecular chaperons that co-precipitated with the HDAC complexes (15) The structure of the HDAC domain contains a Rossmann-like fold, a motif that is found commonly in proteins such as dehydrogenases that bind nucleotides. Thus, the HDAC structure may have evolved to respond to one or more metabolites with nucleotide containing moieties.
The allosteric stimulation of HDAC activity by the CoA derivatives and NADPH followed a mixed activation kinetics in which both Km and Vmax values were increased. Following a method of analysis described by Reinhart (24) established that activation was a product of V-type activation counteracted by Q-type inhibition. So the compounds increased the overall HDAC activity by increasing the reaction velocity despite some reduction in the affinity of the enzyme for the substrate. The reasons for the decrease in the Km value at the highest concentration of acetyl-CoA are unclear but could be due to contamination by free CoA, which lowers the Km value of HDACs for histones or nonspecific effects on HDAC-histone interactions.
The metabolites failed to stimulate HDAC activity when Fluor de Lys, a synthetic small peptidic substrate, was used instead of histones in the HDAC reaction, suggesting that the activating metabolites must affect the substrate-enzyme interaction(s) in the regions of substrate other than the acetylated residue itself. Although the level of activation was at most 2–3-fold, histone acetylation is rapidly turned over in vivo, and therefore, small changes in the balance between acetylation and deacetylation activities can potentially lead to large differences in steady state histone acetylation levels.
Unlike the thioester-conjugated CoA derivatives, the unconjugated free CoA molecule inhibited HDAC activity on both histones and Fluor de Lys. The inhibition by free CoA is the V-type inhibition with reduced enzyme velocity but increased affinity of the enzyme for the substrate. Overall, the similar extent of decreased Vmax and Km values closely resembles an uncompetitive inhibitory kinetics. This occurs when Vmax and Km values decrease at the same rate. Uncompetitive inhibition is rare in enzymology and is based on the principle that the inhibitor binds preferentially to the enzyme-substrate complex and not to the free enzyme. Inhibition of G6PD by DHEA is, in fact, an example of uncompetitive inhibition (36). The rarity of uncompetitive inhibition may be due to potential accumulation of a pathway's intermediates to toxic levels, as the inhibition cannot be overcome by increased substrate concentrations. Thus, in cases in which competitive inhibition is negligible, uncompetitive inhibition can have large effects on enzymatic activity (37).
Interestingly, although free CoA and butyrate, a well known HDAC inhibitor, inhibited HDAC activity, butyryl-CoA stimulated HDAC activities of HDAC1 and -2. This finding suggests that the ability of butyrate to inhibit HDACs may stem from structural mimicry of natural HDAC activators.
Our kinetic analyses indicate that IC50 values for either inhibitors or activators is in the micromolar range. This is comparable with the intracellular levels reported for metabolites such as free CoA or acetyl-CoA, which can reach cytoplasmic concentrations of 100 and 60 μm, respectively. It is therefore possible that intracellular concentrations of these metabolites can reach local concentrations in the effective range. Such a scenario has been reported for the in vitro activity of the Sir2 histone deacetylase, which requires NAD+ at up to 1 mm (19).
Our limited mutational analysis of HDAC1 did not identify a mutant that retained WT levels of activity, even when the mutations were based on the corresponding residues in HDAC1 orthologues. This may not be surprising, as most of the enzyme consists of the HDAC domain itself. However, two mutations, one within the catalytic pocket and one on the surface of HDAC1, did alter its response to the activator. Additional studies, including perhaps the structure of HDAC1 with an activator, could reveal the residues that are critical for binding of activators by HDACs.
Inhibition of G6PD has been shown to decrease levels of NADPH (34). As predicted by our in vitro findings, pharmacological inhibition of G6PD increased histone acetylation in MDA-MB-453 cells, presumably by decreasing NADPH levels and thus, reducing HDAC activity. Like NADPH, many of the metabolites identified in this study are needed for syntheses of macromolecules needed for growth. Whether all or some of these compounds, or other structurally related molecules, are true activators of HDACs in vivo needs further investigation, but their presence may signal availability of biosynthetic capacity to the HDACs. The alteration of HDAC activity may in turn result in changes in gene expression patterns that promote cellular growth. Whatever the case may be, modulation of in vitro HDAC activity by anabolic metabolites with a nucleotide moiety could aid the design and development of more effective HDAC inhibitors.
Acknowledgments
We thank Feng Guo for helpful discussion, Melania Ebrahimi for help with HDAC assays, and Carol Eng for help with HeLa cell cultures. We are grateful to all members of the Kurdistani laboratory for intellectual participation throughout this study.
This work was supported in part by a California Institute for Regenerative Medicine award and Beckman Young Investigator and Howard Hughes Medical Institute Early Career awards (to S. K. K.).
- HDAC
- histone deacetylase
- G6PD
- glucose-6-phosphate dehydrogenase
- rcf
- relative centrifugal force
- DHEA
- 5-dehydroepiandrosterone.
REFERENCES
- 1. de Ruijter A. J., van Gennip A. H., Caron H. N., Kemp S., van Kuilenburg A. B. (2003) Histone deacetylases (HDACs). Characterization of the classical HDAC family. Biochem. J. 370, 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gregoretti I. V., Lee Y. M., Goodson H. V. (2004) Molecular evolution of the histone deacetylase family. Functional implications of phylogenetic analysis. J. Mol. Biol. 338, 17–31 [DOI] [PubMed] [Google Scholar]
- 3. Blander G., Guarente L. (2004) The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 73, 417–435 [DOI] [PubMed] [Google Scholar]
- 4. Weichert W. (2009) HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 280, 168–176 [DOI] [PubMed] [Google Scholar]
- 5. Tan J., Cang S., Ma Y., Petrillo R. L., Liu D. (2010) Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hagelkruys A., Sawicka A., Rennmayr M., Seiser C. (2011) The biology of HDAC in cancer. The nuclear and epigenetic components. Handb Exp. Pharmacol. 206, 13–37 [DOI] [PubMed] [Google Scholar]
- 7. Leunissen C. L., de la Parra N. M., Tan I. Y., Rentmeester T. W., Vader C. I., Veendrick-Meekes M. J., Aldenkamp A. P. (2011) Antiepileptic drugs with mood-stabilizing properties and their relation with psychotropic drug use in institutionalized epilepsy patients with intellectual disability. Res. Dev. Disabil. 32, 2660–2668 [DOI] [PubMed] [Google Scholar]
- 8. Chuang D. M., Leng Y., Marinova Z., Kim H. J., Chiu C. T. (2009) Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 32, 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim D. W., Lassar A. B. (2003) Smad-dependent recruitment of a histone deacetylase/Sin3A complex modulates the bone morphogenetic protein-dependent transcriptional repressor activity of Nkx3.2. Mol. Cell. Biol. 23, 8704–8717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y., Ng H. H., Erdjument-Bromage H., Tempst P., Bird A., Reinberg D. (1999) Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13, 1924–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ballas N., Grunseich C., Lu D. D., Speh J. C., Mandel G. (2005) REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121, 645–657 [DOI] [PubMed] [Google Scholar]
- 12. Wang A. H., Kruhlak M. J., Wu J., Bertos N. R., Vezmar M., Posner B. I., Bazett-Jones D. P., Yang X. J. (2000) Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol. Cell. Biol. 20, 6904–6912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grozinger C. M., Schreiber S. L. (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. U.S.A. 97, 7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arévalo-Rodríguez M., Cardenas M. E., Wu X., Hanes S. D., Heitman J. (2000) Cyclophilin A and Ess1 interact with and regulate silencing by the Sin3-Rpd3 histone deacetylase. EMBO J. 19, 3739–3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson C. A., White D. A., Lavender J. S., O'Neill L. P., Turner B. M. (2002) Human class I histone deacetylase complexes show enhanced catalytic activity in the presence of ATP and co-immunoprecipitate with the ATP-dependent chaperone protein Hsp70. J. Biol. Chem. 277, 9590–9597 [DOI] [PubMed] [Google Scholar]
- 16. Qiu Y., Zhao Y., Becker M., John S., Parekh B. S., Huang S., Hendarwanto A., Martinez E. D., Chen Y., Lu H., Adkins N. L., Stavreva D. A., Wiench M., Georgel P. T., Schiltz R. L., Hager G. L. (2006) HDAC1 acetylation is linked to progressive modulation of steroid receptor-induced gene transcription. Mol. Cell 22, 669–679 [DOI] [PubMed] [Google Scholar]
- 17. David G., Neptune M. A., DePinho R. A. (2002) SUMO-1 modification of histone deacetylase 1 (HDAC1) modulates its biological activities. J. Biol. Chem. 277, 23658–23663 [DOI] [PubMed] [Google Scholar]
- 18. Pflum M. K., Tong J. K., Lane W. S., Schreiber S. L. (2001) Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J. Biol. Chem. 276, 47733–47741 [DOI] [PubMed] [Google Scholar]
- 19. Imai S., Armstrong C. M., Kaeberlein M., Guarente L. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 [DOI] [PubMed] [Google Scholar]
- 20. Landry J., Sutton A., Tafrov S. T., Heller R. C., Stebbins J., Pillus L., Sternglanz R. (2000) The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U.S.A. 97, 5807–5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smith J. S., Brachmann C. B., Celic I., Kenna M. A., Muhammad S., Starai V. J., Avalos J. L., Escalante-Semerena J. C., Grubmeyer C., Wolberger C., Boeke J. D. (2000) A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. U.S.A. 97, 6658–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chalkiadaki A., Guarente L. (2012) Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 8, 287–296 [DOI] [PubMed] [Google Scholar]
- 23. Hait N. C., Allegood J., Maceyka M., Strub G. M., Harikumar K. B., Singh S. K., Luo C., Marmorstein R., Kordula T., Milstien S., Spiegel S. (2009) Regulation of histone acetylation in the nucleus by sphingosine 1-phosphate. Science 325, 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reinhart G. D. (2004) Quantitative analysis and interpretation of allosteric behavior. Methods Enzymol. 380, 187–203 [DOI] [PubMed] [Google Scholar]
- 25. Carmen A. A., Rundlett S. E., Grunstein M. (1996) HDA1 and HDA3 are components of a yeast histone deacetylase (HDA) complex. J. Biol. Chem. 271, 15837–15844 [DOI] [PubMed] [Google Scholar]
- 26. Wysocka J., Reilly P. T., Herr W. (2001) Loss of HCF-1-chromatin association precedes temperature-induced growth arrest of tsBN67 cells. Mol. Cell. Biol. 21, 3820–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cairns S. P., Robinson D. M., Loiselle D. S. (2008) Double-sigmoid model for fitting fatigue profiles in mouse fast- and slow-twitch muscle. Exp. Physiol. 93, 851–862 [DOI] [PubMed] [Google Scholar]
- 28. Kaeberlein M., McDonagh T., Heltweg B., Hixon J., Westman E. A., Caldwell S. D., Napper A., Curtis R., DiStefano P. S., Fields S. (2005) Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 280, 17038–17045 [DOI] [PubMed] [Google Scholar]
- 29. Hill A. V. (1909) The mode of action of nicotine and curari, determined by the form of the contraction curve and the method of temperature coefficients. J. Physiol. 39, 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bradner J. E., Mak R., Tanguturi S. K., Mazitschek R., Haggarty S. J., Ross K., Chang C. Y., Bosco J., West N., Morse E., Lin K., Shen J. P., Kwiatkowski N. P., Gheldof N., Dekker J., DeAngelo D. J., Carr S. A., Schreiber S. L., Golub T. R., Ebert B. L. (2010) Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc. Natl. Acad. Sci. U.S.A. 107, 12617–12622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dowling D. P., Di Costanzo L., Gennadios H. A., Christianson D. W. (2008) Evolution of the arginase fold and functional diversity. Cell. Mol. Life Sci. 65, 2039–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bressi J. C., Jennings A. J., Skene R., Wu Y., Melkus R., De Jong R., O'Connell S., Grimshaw C. E., Navre M., Gangloff A. R. (2010) Exploration of the HDAC2 foot pocket. Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 20, 3142–3145 [DOI] [PubMed] [Google Scholar]
- 33. Vannini A., Volpari C., Gallinari P., Jones P., Mattu M., Carfí A., De Francesco R., Steinkühler C., Di Marco S. (2007) Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 8, 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tian W. N., Braunstein L. D., Pang J., Stuhlmeier K. M., Xi Q. C., Tian X., Stanton R. C. (1998) Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J. Biol. Chem. 273, 10609–10617 [DOI] [PubMed] [Google Scholar]
- 35. Ziboh V. A., Dreize M. A., Hsia S. L. (1970) Inhibition of lipid synthesis and glucose-6-phosphate dehydrogenase in rat skin by dehydroepiandrosterone. J. Lipid Res. 11, 346–354 [PubMed] [Google Scholar]
- 36. Levy H. R. (1979) Glucose-6-phosphate dehydrogenases. Adv. Enzymol. Relat. Areas Mol. Biol. 48, 97–192 [DOI] [PubMed] [Google Scholar]
- 37. Cornish-Bowden A. (1986) Why is uncompetitive inhibition so rare? A possible explanation, with implications for the design of drugs and pesticides. FEBS Lett. 203, 3–6 [DOI] [PubMed] [Google Scholar]