

Abstract
An examination into the derivatization of various natural products using newly developed α-fluorination methodology is disclosed. An activated ketene enolate, generated from an acid chloride, is allowed to react with an electrophilic fluorine source (NFSi). Quenching the reaction with a nucleophilic natural product produces biologically relevant α-fluorinated carbonyl derivatives of select chemotherapeutics, antibiotics, and other pharmaceuticals.
The fields of asymmetric catalysis and natural product derivatization have both made powerful contributions to organic synthesis and practical drug discovery alike.1 In this note, we seek to link the two important disciplines through a tandem process coupling catalytic, asymmetric fluorination with the functionalization of natural products and select analogues to produce interesting new compounds.
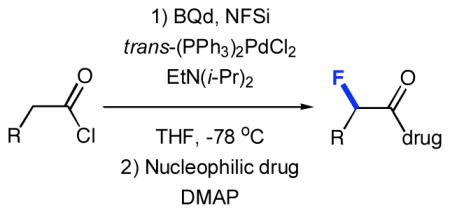
The fluorination of biologically active molecules and pharmaceuticals has received increased attention in recent years due to the extensive therapeutic benefits that often result from specific C-H to C-F transformations. Bioabsorption, binding affinity, chemical reactivity, and increased metabolic durability are some of the properties that can be enhanced by strategic fluorination.2 Recently, we reported a versatile system for the catalytic, asymmetric α-fluorination of acid chlorides. Activated, chiral ketene enolates (2, Figure 1), generated in situ from acid chlorides, Hünig’s base, benzoylquinidine (BQd), and trans-(PPh3)2PdCl2, were allowed to react with N-fluorobenzenesulfonimide (NFSi)3 at −78 °C in THF. Upon quenching with an appropriate nucleophile, α-fluorinated esters, amides, acids, and thioesters were produced with excellent enantioselectivity and in good yield.4,5 Herein, we present a new facet of our asymmetric fluorination methodology involving the site-specific derivatization of biologically relevant compounds.6
Figure 1.

General scheme for asymmetric fluorination
Reaction conditions are as follows: A) trans-(PPh3)2PdCl2, BQd, Hünig’s base, NFSi, THF, −78 °C, 8 h. B) NuH, −78 °C to room temperature.
(R)-(+)-Aminoglutethimide, a potent inhibitor of P450 scc and aromatase, was investigated first because of its commercial availability, single nucleophilic site, and known biological activity.7 Under standard reaction conditions, p-methoxyphenylacetyl chloride was fluorinated with NFSi and quenched with (R)-(+)-aminoglutethimide to give 3a (Table 1) in nearly quantitative yield and 99% diastereomeric excess (de). Kinetic resolution due to the interaction of the chiral nucleophile with putative intermediate 4 (Figure 2) was negligible.
Table 1.
Fluorinated Natural Product Derivativesa
| entry | Acid Chloride | NuH | product | % de | % yield |
|---|---|---|---|---|---|
| 1 | p-MeO-PACc | aminoglutethimide | 3a | 99 | 98 |
| 2 | phthPCd | artemisinin lactol | 3b | 81 | 75 |
| 3 | phthPCd | mitomycin C | 3c | 99 | 58 |
| 4 | p-MeO-PACc | taxol | 3d | 99 | 43 |
| 5 | p-MeO-PACc | cholesterol | 3e | 99 | 77 |
| 6 | p-MeO-PACc | vitamin D3 | 3f | 99 | 75 |
| 7 | phthPCd | glutathione diethyl ester | 3g | 99 | 40 |
| 8 | phthPCd | diacetylnormorphine | 3h | 99 | 32 |
| 9 | phthPCd | dpm-6-APEb | 3i | 99 | 74 |
All Reactions were run using standard reaction conditions.
dpm-6-APE = diphenylmethyl-6-aminopenicillanic ester
p-MeO-PAC = p-methoxyphenylacetyl chloride
phthPC = 3-phthalimidopropionyl chloride
Figure 2.

Reactivity using taxol and baccatin III
We then examined several other biologically relevant compounds in order to determine the scope and versatility of this method. Artemisinin was an attractive target due to its role in combating multiple drug-resistant strains of malaria.8 Quenching the fluorination of 3-phthalimidopropionyl chloride with artemisinin lactol affords 3b in 75% yield and 81% de.9 Mitomycin C, a drug possessing both antibiotic and chemotherapeutic properties, contains an aziridine ring that is essential for its mechanism of action.10 Studies have shown that N-acylaziridine derivatives of mitomycin C demonstrate decreased cytotoxicity in vivo while maintaining potency.11 Using our fluorination methodology, mitomycin C is acylated (3c, Table 1) exclusively on the aziridine nitrogen as opposed to its enamino (c, Figure 3), a result that can be rationalized by simple resonance arguments. The structural integrity of the acylaziridine is maintained under the mild conditions of our reaction, and is verified by IR spectroscopy.
Figure 3.

Natural Product Derivatives
Derived from: a) (R)-(+)-aminoglutethimide b) artemisinin lactol c) mitomycin C d) taxol e) cholesterol f) vitamin D3 g) glutathione h) diacetylnormorphine i) 6-aminopenicillanic acid
The next logical progression was to explore molecules containing multiple (and potentially competing) nucleophilic sites. Controlling site reactivity in natural product chemistry can be extremely difficult and often requires the use of protecting groups to ensure reaction at the desired site. Methods that bypass the need for such laborious and costly protections and deprotections would most certainly be a welcome addition to the synthetic repertoire.12 With this in mind, our attention turned to the chemotherapeutic drug taxol,13 which has three distinct and potentially nucleophilic hydroxyl groups. When p-methoxylphenylacetyl chloride was fluorinated and quenched with taxol, the sole product, resulting from attack by the secondary hydroxyl of the side chain, was obtained in 43% yield and excellent diastereoselectivity. When baccatin III (containing the basic taxol core) was used as a nucleophile, no product could be isolated, further supporting that under standard fluorination conditions, taxol reacts selectively from the secondary hydroxyl of its side chain (Figure 2). Another useful application of this fluorination method manifests in the modification of peptide-based therapeutic agents.14 By transacylation of the putative reactive fluorinateintermediate 4 with nucleophilic amino acid derivatives, a virtually limitless number of fluorinated peptides can be accessed. For example, 3-phthalimidopropionyl chloride was fluorinated and then quenched with glutathione diethyl ester affording 3g in 40% yield and with 99% de.
The solubility of the nucleophile used to quench the reaction proved critical during this investigation. Whereas nonpolar nucleophiles such as cholesterol and vitamin D3 dissolved readily in organic solvents and gave products in high yield, other substrates such as glutathione, morphine, and 6-aminopenicillanic acid displayed marginal solubility, and led to drastically decreased yields of the target compounds. However, simple modifications to these nucleophiles improved their solubility and yields increased (Table 1). Glutathione and 6-aminopenicillanic acid were esterified with ethanol and diphenyldiazomethane,15 respectively. Morphine was derivatized16 by acylation, and N-demethylation to provide a soluble nucleophile with an accessible secondary amine that was acylated in 32% yield.
In conclusion, we have demonstrated that our α-fluorination methodology pairs successfully with natural product derivatization to form biologically relevant molecules with consistently excellent diastereoselectivity. This new method should appeal to the synthetic chemist who seeks to utilize mild conditions for the asymmetric installation of fluorine. Future efforts will focus on a detailed mechanistic investigation of this method and an expansion to hybrid drug synthesis.
Experimental Section
General Procedure for the Syntheses of Fluorinated Products
To a dry 10 mL round bottom flask equipped with a stir bar was added trans-(PPh3)2PdCl2 (3.5 mg, 0.0083 mmol, 0.050 eq) and benzoylquinidine (BQd) (7.1 mg, 0.016 mmol, 0.10 eq), and kept under an atmosphere of nitrogen. THF (1.0 mL) was added, and the mixture was cooled to −78 °C. Hünig’s base (0.030 mL, 0.18 mmol, 1.1 eq) was then added neat to the mixture. Thereupon a solution of N-fluorobenzenesulfonimide (NFSi, 52 mg, 0.16 mmol, 1.0 eq) in 0.33 mL THF was added, followed by a solution of freshly distilled p-methoxyphenylacetyl chloride (0.021 mL, 0.16 mmol, 1.0 eq) in 0.66 mL THF. The reaction was maintained at −78 °C for 8 h. 4-Dimethylaminopyridine (DMAP, 2.0 mg, 0.016 mmol, 0.10 eq) and (R)-(+)-aminoglutethimide (12 mg, 0.055 mmol, 0.33 eq) were added as a solution in THF (0.50 mL), and the reaction warmed to room temperature overnight. The product was then subjected to column chromatography on silica gel with a mixture of ethyl acetate/hexanes as the eluent (unless otherwise noted).
(R)-2-fluoro-2-(4-methoxyphenyl)-N-(R)-(+)-aminoglutethimide (3a)
yellow solid: % yield = 98, % de = 99; mp = 55–60 °C; [α]20D = −3.42° (c = 0.012, CH2Cl2); 1H NMR (CDCl3) (@ 25 °C) δ 8.32 (br s, 1H), 8.20 (s, 1H), 7.65 (d, 2H), 7.40 (d, 2H), 7.25 (d, 2H), 6.95 (d, 2H), 5.85 (d, 1H, J = 49.8 Hz), 3.80 (s, 3H), 2.55 (d, 1H), 2.40-2.30 (m, 2H), 2.30-2.10 (m, 1H), 1.95-1.80 (m, 1H), 1.25 (t, 1H), 0.85 (t, 3H); 13C NMR (CDCl3) δ 175.3, 172.5, 161.0, 160.1, 136.5, 134.5, 129.5, 129.5, 127.8, 120.4, 114.5, 92.2 (d, J = 190.4 Hz), 56.0, 52.0, 34.0, 29.5, 27.0, 9.0; 19F NMR (CDCl3) δ −169.9 (ddd, J = 49.8, 15.5, 5.8 Hz); IR (cm−1, CaF2, CH2Cl2) 1706; HRMS (ESI+) calc for C22H23FN2O4Na+: 421.153407, found 421.152714.
Supplementary Material
Acknowledgments
T.L. thanks the NIH (Grant GM064559) for support. M.T.S. thanks Johns Hopkins University for a Marks Fellowship.
Footnotes
Supporting Information Available: General experimental procedures and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Taylor MS, Jacobsen EN. Proc Natl Acad Sci USA. 2004;101:5368–5373. doi: 10.1073/pnas.0307893101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mohr JT, Krout MR, Stoltz BM. Nature. 2008;455:323–332. doi: 10.1038/nature07370. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Clardy J, Walsh C. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]; (d) Carruthers W, Coldham I. Modern Methods of Organic Synthesis. 4. Cambridge University Press: New York; New York: 2006. [Google Scholar]; (e) Gershon H, Schulman SG, Spevack AD. J Med Chem. 1967;10:536–541. doi: 10.1021/jm00316a008. [DOI] [PubMed] [Google Scholar]; (f) Ismail FMD. J Fluorine Chem. 2002;118:27–33. [Google Scholar]; (g) Maienfisch P, Hall RG. Chimia. 2004;58:93–99. [Google Scholar]
- 2.For recent texts on the impact of fluorine on bioactivity, see: Kirk KL. J Fluorine Chem. 2006;127:1013–1029.Thomas CJ. Curr Topics in Med Chem. 2006;6:1529–1543. doi: 10.2174/156802606777951109.Liu P, Sharon A, Chu CK. J Fluorine Chem. 2008;129:743–766. doi: 10.1016/j.jfluchem.2008.06.007.Park BK, Kitteringham NR. Drug Metab Rev. 1994;26:605–643. doi: 10.3109/03602539408998319.Smart BE. J Fluorine Chem. 2001;109:3–11.Ojima I. Fluorine in Medicinal Chemistr and Chemical Biology. Wiley-Blackwell; Chichester, U.K: 2009. Chambers RD. Fluorine in Organic Chemistry. Wiley; New York: 1973.
- 3.(a) Hamashima Y, Takano H, Hotta D, Sodeoka M. Org Lett. 2003;5:3225–3228. doi: 10.1021/ol035053a. [DOI] [PubMed] [Google Scholar]; (b) Hamashima Y, Yagi K, Takano H, Tamas L, Sodeoka M. J Am Chem Soc. 2002;124:14530–14531. doi: 10.1021/ja028464f. [DOI] [PubMed] [Google Scholar]
- 4.Paull DH, Scerba MT, Alden-Danforth E, Widger LR, Lectka T. J Am Chem Soc. 2008;130:17260–17261. doi: 10.1021/ja807792c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.A generalized reaction scheme with specific reaction conditions is outlined in Figure 1.
- 6.It should be noted that stereochemical assignment of the products listed in Table 1 was inferred from prior investigations in our laboratory. No autocatalytic functionalization of any nucleophile was observed. Catalysis by the pseudoenantiomeric alkaloid benzoylquinine (BQ) gives direct access to the other diastereomeric product with identical yield and diastereomeric excess. Achiral alkaloid catalysts were tested and gave mixtures of diastereomers.
- 7.Siraki AG, Bonini MG, Jiang J, Ehrenshaft M, Mason RP. Chem Res Toxicol. 2007;20:1038–1045. doi: 10.1021/tx6003562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White NJ. J Clin Invest. 2004;113:1084–1092. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Posner GH, McRiner AJ, Paik I, Sur S, Borstnik K, Xie S, Shapiro TA, Alagbala A, Foster B. J Med Chem. 2004;47:1299–1301. doi: 10.1021/jm0303711. [DOI] [PubMed] [Google Scholar]
- 10.Tomasz M. Chemistry & Biology. 1995;2:575–579. doi: 10.1016/1074-5521(95)90120-5. [DOI] [PubMed] [Google Scholar]
- 11.Fishbein PL, Kohn H. J Med Chem. 1987;30:1767–1773. doi: 10.1021/jm00393a015. [DOI] [PubMed] [Google Scholar]
- 12.The pioneering work of Miller utilizes synthetic peptides to achieve site-selectivity in natural product derivatization. For recent examples, see: Lewis CA, Miller SJ. Angew Chem Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490.Sanchez-Roselló M, Puchlopek ALA, Morgan AJ, Miller SJ. J Org Chem. 2008;73:1774–1782. doi: 10.1021/jo702334z.Morgan AJ, Komiya S, Xu Y, Miller SJ. J Org Chem. 2006;71:6923–6931. doi: 10.1021/jo0610816.Lewis CA, Longcore KE, Miller SJ, Wender PA. J Nat Prod. 2009;72:1864–1869. doi: 10.1021/np9004932.
- 13.Mathew AE, Mejillano MR, Nath JP, Himes RH, Stella VJ. J Med Chem. 1992;35:145–151. doi: 10.1021/jm00079a019. [DOI] [PubMed] [Google Scholar]
- 14.Salwiczek M, Samsonov S, Vagt T, Nyakatura E, Fleige E, Numata J, Cölfen H, Pisabarro MT, Koksch B. Chem Eur J. 2009;15:7628–7636. doi: 10.1002/chem.200802136. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 15.(a) Smith LI, Howard KL. Org Synth, Coll. 1955;3:351. [Google Scholar]; (b) Jensen SE, Westlake DWS, Bowers RJ, Wolfe S. J Antibiot. 1982;35:1351–1360. doi: 10.7164/antibiotics.35.1351. [DOI] [PubMed] [Google Scholar]
- 16.Cooper GK, Rapoport H. J Label Compd Radiopharm. 1985;22:1201–1207. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.