

Abstract
Asparagine-linked glycosylation is a major form of posttranslational modifications, which plays important roles in protein folding, intracellular signaling, and a number of other biological recognition events [1]. Glycoproteins are often characterized by their structural micro-heterogeneity where different glycoforms have the same polypeptide backbone but differ in the pendant oligosaccharides. Of particular interest are the findings that subtle difference in the attached glycans can have a significant impact on the biological functions of a given glycoprotein [2, 3]. The urgent need of pure glycoforms for functional studies and biomedical applications has stimulated a great interest in exploring new methods for making homogeneous glycoproteins [4]. Major advances include the application of native chemical ligation and expressed protein ligation for constructing full-size glycoproteins [5–7], chemoselective ligation to introduce homogeneous glycans [8], and the engineering of yeast glycosylation pathways to produce single glycoforms [9]. Yet another interesting advance in the field is the endoglycosidase-catalyzed transglycosylation for glycosylation engineering and glycoprotein synthesis [10–16].
Keywords: glycoprotein, glycopeptide, chemoenzymatic synthesis, transglycosylation, sugar oxazoline
Endo-β-N-acetylglucosaminidases (ENGases) of the glycosyl hydrolase family 85 (GH85) are endoglycosidases that release N-glycans from glycoproteins by hydrolyzing the glycosidic bond in the chitobiose (GlcNAcβ1-4GlcNAc) core. Two ENGases, the Endo-A from Arthrobacter protophormiae and the Endo-M from Mucor hiemalis, have been used for glycopeptide and glycoprotein synthesis because of their potent transglycosylation activity [10–16]. The two enzymes have distinct substrate specificity. Endo-M can hydrolyze both high-mannose type and complex type N-glycans, but Endo-A has been shown to act only on high-mannose type N-glycans. An interesting development in the field is the exploration of synthetic sugar oxazolines, the mimics of the oxazolinium ion intermediate generated in a substrate-assisted mechanism, as activated donor substrates for the enzymatic transglycosylation [11–14]. To address the problem of product hydrolysis, glycosynthase mutants of Endo-M and Endo-A were generated that lack the hydrolytic activity but can still use the activated sugar oxazolines for transglycosylation [15, 16]. In particular, we and others have demonstrated that wild type Endo-A was able to take some truncated and selectively modified sugar oxazolines for transglycosylation with diminished product hydrolysis [11, 12, 14]. These results suggest that Endo-A could tolerate selected modifications at the outer mannose moieties of the highly activated Man3GlcNAc-oxazoline substrate. We report in this communication our new findings that wild-type Endo-A possesses low but promiscuous activity on full-size complex-type glycan oxazolines for transglycosylation without product hydrolysis. The transglycosylation activity of Endo-A on complex sialoglycan oxazoline, coupled with its high hydrolytic activity on high-mannose type N-glycans, enables a one-pot conversion of ribonuclease B to sialylated ribonuclease C. In addition, we also demonstrate that a combined use of Endo-A and an Endo-M mutant provides an efficient chemoenzymatic synthesis of homogeneous sialylated N-glycoproteins, which are hitherto difficult to obtain for functional studies [3, 6, 17].
Despite the observations that wild type Endo-A could take some truncated and modified sugar oxazolines for transglycosylation [11, 12, 14], initial study under a common enzymatic reaction condition did not reveal transglycosylation activity of Endo-A on a complex-type glycan oxazoline (CT-oxazoline, 1) [16]. This data suggested that the substitution at the C-2 positions of the 3- and/or 6-branch mannoses of the Man3GlcNAc-oxazoline with a Galβ1→4GlcNAcβ1 moiety, as found in the complex type N-glycan, conferred detrimental effects on substrate recognition by Endo-A. Nevertheless, the recently reported structures of Endo-A and its complex with a substrate analogue Man3GlcNAc-thiazoline [18] inspired us to re-investigate the activity of Endo-A toward various complex type glycan oxazolines. The structure of the Endo-A in complex with Man3GlcNAc-thiazoline indicates that the C-2 position at the α-1,3-branch mannose are protruding outside of the catalytic site without confrontation with Endo-A, but the C-2 position of the α-1,6-branch mannose has only a narrow space, implying a potential conflict of the C-2 substituent with Endo-A. Interestingly, when we docked the CT-oxazoline (1) into the substrate-binding pocket of Endo-A, we found that the CT-oxazoline could take an alternative conformation to avoid a direct conflict of the C-2 substituent. That is, the Man3GlcNAc core still reached deeply in the center of the binding pocket, while the α-1,6-branch mannose could rotate to protrude outside the cavity (Figure 1). While some extra activation energy would be required to compensate this conformational twist, these preliminary modelling studies did imply potential substrate promiscuity of Endo-A toward complex type N-glycan oxazolines. To test this idea, we sought to evaluate Endo-A activity on full-size complex type N-glycans oxazolines under a more forceful reaction condition.
Figure 1.
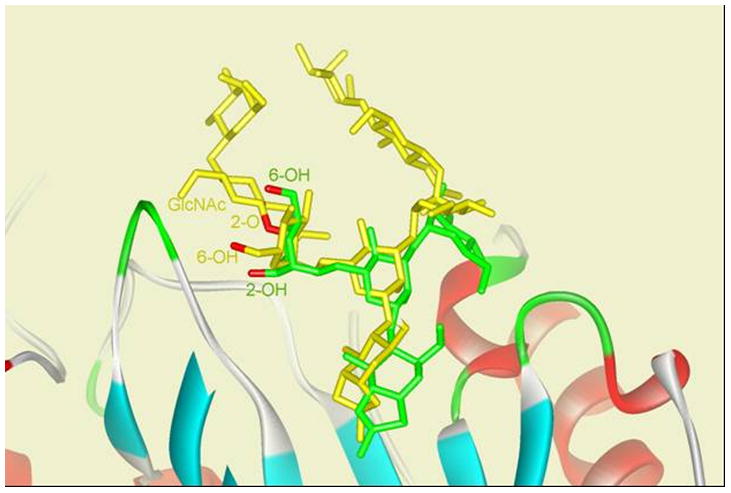
Conformational comparison of Man3GlcNAc-thiazoline (green) and CT-oxazoline (yellow) docked into substrate-binding site of Endo-A (macromolecule displayed in ribbon).
We have previously described a semi-synthesis of an asialoglycan oxazoline (1, C T-oxazoline) by a multiple-step approach including acetylation of the free glycan, oxazoline ring formation under Lewis acid catalysis, and final de-O-acetylation [16]. The synthesis of sialoglycan oxazoline could be more complex by this approach because of the presence of the sialic acid residues. Recently Shoda and co-workers reported an efficient method for synthesizing sugar oxazolines from free 2-acetamido sugars in aqueous solution, using 2-chloro-1,3-dimethylimidazolinium chloride (DMC) as the reagent for a single-step conversion [19]. They have shown that sialylated glycan oxazoline could be generated efficiently from the corresponding free glycan, although isolation and further characterization of the product were not described. We thus followed this procedure for preparing the sialoglycan oxazoline 2 (Scheme 1). The sialoglycopeptide (SGP) isolated from hen’s egg yolk [20] was hydrolyzed by Endo-M to give the free N-glycan (SCT), which was then treated with excess DMC (15 mol equiv) in the presence of TEA (42 mol equiv). Monitoring of the reaction by high-performance anion-exchange chromatography (HPAEC) indicated essentially 100% conversion of SCT to the sialoglycan oxazoline 2 within 15 min (Figure S1, Supporting Information). We also investigated the use of inorganic bases for the DMC-promoted transformation and found that NaOH and K2CO3 were also effective for the conversion to give a high yield of the sialoglycan oxazoline (2) (Table S1, Supporting Information,). The product was readily isolated from the reaction mixture by gel filtration (Sephadex G15; eluent, 0.05% aqueous TEA), giving the sialoglycan oxazoline 2 in 90% yield (Scheme 1). Similarly, treatment of the asialoglycan (CT) with an excess amount of DMC and Et3N in water gave the corresponding asialoglycan oxazoline (1) in excellent yield.
Scheme 1.

Semi-synthesis of sialylated N-glycan oxazoline.
First, we examined the Endo-A catalyzed transglycosylation of sialoglycan oxazoline 2 using a GlcNAc-pentapeptide (3) derived from erythropoietin (aa 37–41) as the acceptor substrate [12]. Incubation of the donor 2 (15 mM) and acceptor 3 (5 mM) with a catalytic amount of wild type Endo-A (10 mU/μl) at 23°C for 5 h gave only trace amount of the transglycosylation product (monitored by HPLC). This result is consistent with our previous observation that a desialylated complex-type N-glycan oxazoline was a poor substrate for wild type Endo-A [16]. However, when the enzymatic reaction was performed under high substrate concentrations (75 mM for the donor and 25 mM for the acceptor) with a significantly increased amount of Endo-A (200 mU/μl), a smooth transglycosylation reaction was observed, giving the transglycosylation product (4) in 68% yield after incubation for 8 h (Scheme 2). Encouraged by this result, we re-investigated the Endo-A catalyzed reaction between the asialoglycan oxazoline (1) and GlcNAc-peptide (3). We found that under the more drastic condition (high substrate concentration and large amount of enzyme), Endo-A also showed significant transglycosylation activity on the asialoglycan oxazoline (1) to give a 60% yield of the corresponding transglycosylation product (5) (Scheme 2). It was found that the complex-type N-glycopeptide products 4 and 5 were completely resistant to hydrolysis by Endo-A even for an extended incubation time, confirming the notion that Endo-A could act on the highly activated glycan oxazolines for transglycosylation but lack the activity to hydrolyze the “ground-state” products thus formed. The transglycosylation product 4 was characterized by ESI-MS analysis coupled with specific enzymatic transformations (See Supporting Information). Recently, Fairbanks and co-workers reported that Endo-A could act on a further truncated bi-antennary complex type N-glycan oxazoline, in which the C-2 positions of the outer mannoses of the core were substituted with only a GlcNAc moiety, for a slow transglycosylation to give a 11% yield of the corresponding product, but extended incubation somehow did not lead to increased yield [14]. The discovery of the substrate promiscuity of Endo-A toward full-size complex-type N-glycan oxazolines, together with its complete lack of hydrolytic activity toward the ground-state complex-type product, raises an exciting opportunity to improve and expand Endo-A’s potential for transglycosylation via a directed evolution strategy [21]. In comparison, we examined the transglycosylation of sialoglycan oxazoline (2) to GlcNAc-peptide (3) by wild-type Endo-M. It was found that the Endo-M catalyzed reaction proceeded rapidly to give a maximal yield of the glycopeptide product 4 within a short time. After that, however, the product yield decreased quickly with time, due to a rapid product hydrolysis catalyzed by Endo-M (Figure S2, Supporting Information). In contrast, Endo-A catalyzed transglycosylation was much slower, but the product was accumulated because of its lack of hydrolytic activity on the product.
Scheme 2.

Endo-A and EndoM-N175A act on sialylated N-glycan oxazoline for transglycosylation without product hydrolysis. Enzyme activity definition: 1 mU was defined as the amount of Endo-A or EndoM-N175A needed for transferring 1 nmol of Man3GlcNAc-oxazoline to GlcNAc-PNP in 1 min at 23 °C.
We also tested the sialoglycan oxazoline (2) with an Endo-M mutant, EndoM-N175A, which was previously shown to be able to transglycosylate the desialylated glycan oxazoline (1) without product hydrolysis [16]. It was found that the sialoglycan oxazoline (2) was a good substrate for EndoM-N175A, leading to formation of the transglycosylation product (4) in 88% yield (Scheme 2). The ability of wild type Endo-A and the mutant EndoM-N175A to use the sialoglycan oxazoline for transglycosylation without product hydrolysis now provides a quick access to homogeneous sialylated complex N-glycopeptides.
The transglycosylation activity of Endo-A on sialoglycan oxazoline, together with its known hydrolytic activity on high-mannose type N-glycans, prompted us to test the possibility to switch the nature of N-glycans in a glycoprotein in a one-pot manner. We have chosen bovine ribonuclease (RNase) as a model system. It is known that bovine pancreatic RNase exists in different glycoforms: RNase A is unglycosylated; RNase B carries a heterogeneous high-mannose type N-glycan (Man5–9GlcNAc2) at the Asn-34; and RNase C bears a heterogeneous complex-type N-glycan [22]. Recently a desialylated glycoform of RNase C was synthesized by an elegant combination of native chemical ligation and expressed protein ligation [7]. To test a one-pot chemoenzymatic approach, a mixture of RNase B (6 mM), sialoglycan oxazoline 2 (72 mM), and Endo-A (700 mU/μl) in a phosphate buffer (pH 7.0) was incubated at 23 °C. It was expected that RNase B would be first hydrolyzed by Endo-A to give the GlcNAc-RNase, which would then serve as an acceptor for the subsequent Endo-A catalyzed transglycosylation, leading to the ligation of the complex type N-glycan to the protein. An aliquot was taken from the reaction mixture at intervals and analyzed by SDS-PAGE (Figure 2). It was found that the cleavage of the high-mannose N-glycans of RNase B by Endo-A was very quick under the high enzyme concentration and the glycan cleaving reaction was complete within 5 min to give the GlcNAc-RNase (Figure 2, lane 2). The transglycosylation was relatively slow, but a new protein band corresponding to the product sialylated RNase C, which was about 2 kDa larger than the GlcNAc-RNase, was clearly formed at 3 h (Figure 2, lane 3). Thus, Endo-A permits a one-pot conversion of the RNase B to the sialylated RNase C in the presence of the sialoglycan oxazoline (Scheme 3). The sialylated RNase C product was isolated by RP-HPLC and its identity was characterized by a combination of several analytic means. First, the purified product appeared as a single band (Figure 2, lane 4) that was about 2 kDa larger in size than the GlcNAc-RNase B (Figure 2, lane 2), suggesting an attachment of a typical N-glycan; second, treatment of the RNase C product with Endo-M converted the RNase C back to GlcNAc-RNase B as a single band (Figure 2, lane 5), indicating that the glycan in the product was attached to the inner most GlcNAc rather than other parts of the protein; third, deconvolution of the ESI-MS spectrum (Figure 3) of the product gave a molecular mass of 15888 Da, which matches well with the molecular mass of the expected sialylated RNase C (calculated, M = 15886 Da); finally, treatment of the RNase C product by PNGase F released a single N-glycan that was confirmed to be the sialylated, bi-antennary complex-type N-glycan (MALDI-TOF MS: calculated, M = 2222.78; found (m/z), 2246.17 [M+Na+]). These studies indicate that a one-pot protocol is feasible for converting heterogeneous RNase B to a homogeneous RNase C. Nevertheless, the low transglycosylation activity of Endo-A on the complex type glycan oxazoline resulted in a relatively low yield. A prolonged incubation led to a moderate increase of the transglycosylation yield of the product, but we were aware that an overnight incubation also resulted in some non-enzymatic side reactions between the oxazoline and the ribonuclease when both were present at high concentrations. The by-products are yet to be further characterized, but a preliminary MS analysis of the mixture suggested that they were likely generated by slow chemical reactions of the oxazoline with some side chains in the proteins. Thus, generation of more active Endo-A mutants, e.g., via directed evolution, would be a logical next step for enhancing the overall efficiency of the one-pot protein glycosylation transformation.
Figure 2.
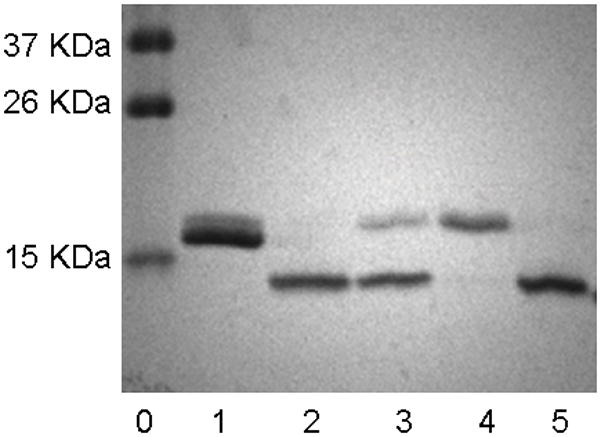
SDS-PAGE analysis of the one-pot transglycosylation reaction and the product. lane 0: marker; lane 1: RNase B; lane 2: an aliquot of the one-pot reaction mixture at 5min; lane 3: an aliquot of the one-pot reaction mixture at 3 h; lane 4: purified sialylated RNase C; lane 5: sialylated RNase C treated with Endo-M.
Scheme 3.

Endo-A and EndoM-N175A catalyzed one-pot conversion of RNase B to sialylated RNase C.
Figure 3.

ESI-MS of the sialylated RNase C.
As an alternative approach, we also tested a combined use of Endo-A and EndoM-N175A for a one-pot conversion. EndoM-N175A was already shown to be efficient to catalyze the transglycosylation of a desialylated glycan oxazoline, but it does not hydrolyze high-mannose type N-glycans in RNase B. In addition, both the Endo-M mutant and Endo-A would not hydrolyze the “ground-state” complex-type glycoprotein product. Indeed, incubation of a mixture of RNase B, oxazoline (2), Endo-A, and EndoM-N175A together in a phosphate buffer (pH 7.0) resulted in a substantial increase of the transglycosylation efficiency, giving a 70% isolated yield of the sialylated RNase C (Scheme 3). In this case, only a catalytic amount of Endo-A was required. Taken together, the combined use of the two enzymes with distinct substrate specificity offers a useful method for glycosylation engineering of glycoproteins by readily switching the personality of the attached N-glycans.
In summary, we have found that wild-type Endo-A possesses a low but promiscuous activity on sialylated and desialylated complex-type N-glycan oxazolines for transglycosylation, which enables a one-pot conversion of heterogeneous RNase B to homogeneous RNase C. We have also shown that a combined use of the distinct substrate specificity of Endo-A and an Endo-M mutant permits an efficient synthesis of homogeneous sialylated N-glycoproteins that are usually difficult to obtain for functional studies. The discovery of the promiscuous substrate activity of Endo-A on complex-type N-glycan oxazolines raises an exciting opportunity to improve and expand Endo-A’s transglycosylation activity by a directed evolution strategy.
Supplementary Material
Acknowledgments
The work was supported by the National Institutes of Health (R01 GM080374). We thank Prof. Kaoru Takegawa for providing the pGEX-2T/Endo-A plasmid for expressing the recombinant Endo-A enzyme.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.a) Dwek RA. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]; b) Helenius A, Aebi M. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]; c) Haltiwanger RS, Lowe JB. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]; d) Dube DH, Bertozzi CR. Nat Rev Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]; e) Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. Annu Rev Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- 2.a) Jefferis R. Biotechnol Prog. 2005;21:11–16. doi: 10.1021/bp040016j. [DOI] [PubMed] [Google Scholar]; b) Walsh G, Jefferis R. Nat Biotechnol. 2006;24:1241–1252. doi: 10.1038/nbt1252. [DOI] [PubMed] [Google Scholar]
- 3.a) Kaneko Y, Nimmerjahn F, Ravetch JV. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]; b) Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Science. 2008;320:373–376. doi: 10.1126/science.1154315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Grogan MJ, Pratt MR, Marcaurelle LA, Bertozzi CR. Annu Rev Biochem. 2002;71:593–634. doi: 10.1146/annurev.biochem.71.110601.135334. [DOI] [PubMed] [Google Scholar]; b) Buskas T, Ingale S, Boons GJ. Glycobiology. 2006;16:113R–136. doi: 10.1093/glycob/cwj125. [DOI] [PubMed] [Google Scholar]; c) Bennett RCS, Wong CH. Chem Soc Rev. 2007;36:1227–1238. doi: 10.1039/b617709c. [DOI] [PubMed] [Google Scholar]; d) Gamblin DP, Scanlan EM, Davis BG. Chem Rev. 2009;109:131–163. doi: 10.1021/cr078291i. [DOI] [PubMed] [Google Scholar]; e) Bernardes GJ, Castagner B, Seeberger PH. ACS Chem Biol. 2009;4:703–713. doi: 10.1021/cb900014n. [DOI] [PubMed] [Google Scholar]; f) Rich JR, Withers SG. Nat Chem Biol. 2009;5:206–215. doi: 10.1038/nchembio.148. [DOI] [PubMed] [Google Scholar]
- 5.Macmillan D, Bertozzi CR. Angew Chem. 2004;116:1379–1383. doi: 10.1002/anie.200352673. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:1355–1359. doi: 10.1002/anie.200352673. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto N, Tanabe Y, Okamoto R, Dawson PE, Kajihara Y. J Am Chem Soc. 2008;130:501–510. doi: 10.1021/ja072543f. [DOI] [PubMed] [Google Scholar]
- 7.a) Piontek C, Ring P, Harjes O, Heinlein C, Mezzato S, Lombana N, Pohner C, Puttner M, Varon Silva D, Martin A, Schmid FX, Unverzagt C. Angew Chem. 2009;121:1968–1973. doi: 10.1002/anie.200804734. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:1936–1940. doi: 10.1002/anie.200804734. [DOI] [PubMed] [Google Scholar]; b) Piontek C, Varon Silva D, Heinlein C, Pohner C, Mezzato S, Ring P, Martin A, Schmid FX, Unverzagt C. Angew Chem. 2009;121:1974–1978. doi: 10.1002/anie.200804735. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:1941–1945. doi: 10.1002/anie.200804735. [DOI] [PubMed] [Google Scholar]
- 8.a) van Kasteren SI, Kramer HB, Jensen HH, Campbell SJ, Kirkpatrick J, Oldham NJ, Anthony DC, Davis BG. Nature. 2007;446:1105–1109. doi: 10.1038/nature05757. [DOI] [PubMed] [Google Scholar]; b) Hirano K, Macmillan D, Tezuka K, Tsuji T, Kajihara Y. Angew Chem. 2009;121:9721–9724. doi: 10.1002/anie.200904376. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:9557–9560. doi: 10.1002/anie.200904376. [DOI] [PubMed] [Google Scholar]
- 9.a) Hamilton SR, Bobrowicz P, Bobrowicz B, Davidson RC, Li H, Mitchell T, Nett JH, Rausch S, Stadheim TA, Wischnewski H, Wildt S, Gerngross TU. Science. 2003;301:1244–1246. doi: 10.1126/science.1088166. [DOI] [PubMed] [Google Scholar]; b) Wildt S, Gerngross TU. Nat Rev Microbiol. 2005;3:119–128. doi: 10.1038/nrmicro1087. [DOI] [PubMed] [Google Scholar]; c) Hamilton SR, Davidson RC, Sethuraman N, Nett JH, Jiang Y, Rios S, Bobrowicz P, Stadheim TA, Li H, Choi BK, Hopkins D, Wischnewski H, Roser J, Mitchell T, Strawbridge RR, Hoopes J, Wildt S, Gerngross TU. Science. 2006;313:1441–1443. doi: 10.1126/science.1130256. [DOI] [PubMed] [Google Scholar]
- 10.a) Yamamoto K. J Biosci Bioeng. 2001;92:493–501. doi: 10.1263/jbb.92.493. [DOI] [PubMed] [Google Scholar]; b) Wang LX. Carbohydr Res. 2008;343:1509–1522. doi: 10.1016/j.carres.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang LX, Huang W. Curr Opin Chem Biol. 2009;13:592–600. doi: 10.1016/j.cbpa.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Li B, Zeng Y, Hauser S, Song H, Wang LX. J Am Chem Soc. 2005;127:9692–9693. doi: 10.1021/ja051715a. [DOI] [PubMed] [Google Scholar]; b) Li H, Li B, Song H, Breydo L, Baskakov IV, Wang LX. J Org Chem. 2005;70:9990–9996. doi: 10.1021/jo051729z. [DOI] [PubMed] [Google Scholar]; c) Li B, Song H, Hauser S, Wang LX. Org Lett. 2006;8:3081–3084. doi: 10.1021/ol061056m. [DOI] [PubMed] [Google Scholar]; d) Ochiai H, Huang W, Wang LX. J Am Chem Soc. 2008;130:13790–13803. doi: 10.1021/ja805044x. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Rising TW, Claridge TD, Moir JW, Fairbanks AJ. Chem Bio Chem. 2006;7:1177–1180. doi: 10.1002/cbic.200600183. [DOI] [PubMed] [Google Scholar]; f) Rising TW, Claridge TD, Davies N, Gamblin DP, Moir JW, Fairbanks AJ. Carbohydr Res. 2006;341:1574–1596. doi: 10.1016/j.carres.2006.03.007. [DOI] [PubMed] [Google Scholar]; g) Rising TW, Heidecke CD, Moir JW, Ling Z, Fairbanks AJ. Chem Eur J. 2008;14:6444–6464. doi: 10.1002/chem.200800365. [DOI] [PubMed] [Google Scholar]
- 12.Zeng Y, Wang J, Li B, Hauser S, Li H, Wang LX. Chem Eur J. 2006;12:3355–3364. doi: 10.1002/chem.200501196. [DOI] [PubMed] [Google Scholar]
- 13.Wei Y, Li C, Huang W, Li B, Strome S, Wang LX. Biochemistry. 2008;47:10294–10304. doi: 10.1021/bi800874y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsons TB, Moir JW, Fairbanks AJ. Org Biomol Chem. 2009;7:3128–3140. [Google Scholar]
- 15.a) Umekawa M, Huang W, Li B, Fujita K, Ashida H, Wang LX, Yamamoto K. J Biol Chem. 2008;283:4469–4479. doi: 10.1074/jbc.M707137200. [DOI] [PubMed] [Google Scholar]; b) Heidecke CD, Ling Z, Bruce NC, Moir JW, Parsons TB, Fairbanks AJ. Chembiochem. 2008;9:2045–2051. doi: 10.1002/cbic.200800214. [DOI] [PubMed] [Google Scholar]; c) Umekawa M, Li C, Higashiyama T, Huang W, Ashida H, Yamamoto K, Wang LX. J Biol Chem. 2010;285:511–521. doi: 10.1074/jbc.M109.059832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang W, Li C, Li B, Umekawa M, Yamamoto K, Zhang X, Wang LX. J Am Chem Soc. 2009;131:2214–2223. doi: 10.1021/ja8074677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barb AW, Brady EK, Prestegard JH. Biochemistry. 2009;48:9705–9707. doi: 10.1021/bi901430h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Ling Z, Suits MD, Bingham RJ, Bruce NC, Davies GJ, Fairbanks AJ, Moir JW, Taylor EJ. J Mol Biol. 2009;389:1–9. doi: 10.1016/j.jmb.2009.03.050. [DOI] [PubMed] [Google Scholar]; b) Yin J, Li L, Shaw N, Li Y, Song JK, Zhang W, Xia C, Zhang R, Joachimiak A, Zhang HC, Wang LX, Liu ZJ, Wang P. PLoS One. 2009;4:e4658. doi: 10.1371/journal.pone.0004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noguchi M, Tanaka T, Gyakushi H, Kobayashi A, Shoda SI. J Org Chem. 2009 doi: 10.1021/jo8024708. [DOI] [PubMed] [Google Scholar]
- 20.a) Seko A, Koketsu M, Nishizono M, Enoki Y, Ibrahim HR, Juneja LR, Kim M, Yamamoto T. Biochim Biophys Acta. 1997;1335:23–32. doi: 10.1016/s0304-4165(96)00118-3. [DOI] [PubMed] [Google Scholar]; b) Li H, Singh S, Zeng Y, Song H, Wang LX. Bioorg Med Chem Lett. 2005;15:895–898. doi: 10.1016/j.bmcl.2004.12.066. [DOI] [PubMed] [Google Scholar]
- 21.Hancock SM, Rich JR, Caines ME, Strynadka NC, Withers SG. Nat Chem Biol. 2009;5:508–514. doi: 10.1038/nchembio.191. [DOI] [PubMed] [Google Scholar]
- 22.a) Baynes JW, Wold F. J Biol Chem. 1976;251:6016–6024. [PubMed] [Google Scholar]; b) Becker RR, Halbrook JL, Hirs CH. J Biol Chem. 1973;248:7826–7832. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.