

Abstract
Transforming growth factor β1 (TGFβ1) promotes fibrosis by, among other mechanisms, activating quiescent fibroblasts into myofibroblasts and increasing the expression of extracellular matrices. Recent work suggests that peroxisome proliferator-activated receptor γ (PPARγ) is a negative regulator of TGFβ1-induced fibrotic events. We, however, hypothesized that antifibrotic pathways mediated by PPARγ are influenced by TGFβ1, causing an imbalance towards fibrogenesis. Consistent with this, primary murine primary lung fibroblasts responded to TGFβ1 with a sustained downregulation of PPARγ transcripts. This effect was dampened in lung fibroblasts deficient in Smad3, a transcription factor that mediates many of the effects of TGFβ1. Paradoxically, TGFβ1 stimulated the activation of the PPARγ gene promoter and induced the phosphorylation of PPARγ in primary lung fibroblasts. The ability of TGFβ1 to modulate the transcriptional activity of PPARγ was then tested in NIH/3T3 fibroblasts containing a PPARγ-responsive luciferase reporter. In these cells, stimulation of TGFβ1 signals with a constitutively active TGFβ1 receptor transgene blunted PPARγ-dependent reporter expression induced by troglitazone, a PPARγ activator. Overexpression of PPARγ prevented TGFβ1 repression of troglitazone-induced PPARγ-dependent gene transcription, whereas coexpression of PPARγ and Smad3 transgenes recapitulated the TGFβ1 effects. We conclude that modulation of PPARγ is controlled by TGFβ1, in part through Smad3 signals, involving regulation of PPARγ expression and transcriptional potential.
1. Introduction
Transforming growth factor β1 (TGFβ1) is a pleomorphic growth factor with anti-inflammatory and profibrotic properties that has been implicated in many forms of natural and experimental tissue fibrosis [1]. In lung, TGFβ1 is produced by epithelial cells, alveolar and tissue macrophages, and fibroblasts after exposure to injurious agents such as silica, bleomycin, hyperoxia, and paraquat among others [2]. A key role for TGFβ1 in lung fibrosis has been confirmed in studies showing the development of lung fibrosis in animals transfected with TGFβ1-producing adenovirus, and by work demonstrating inhibition of experimental lung fibrosis by interventions targeting TGFβ1 or its downstream signals [3, 4].
The profibrotic effects of TGFβ1 are mostly, but not entirely, mediated by intracellular signals triggered by the transcription factor Smad3 [5]. TGFβ1/Smad3 signaling stimulates connective tissue expression and epithelial-mesenchymal transition, events considered key to the development of lung fibrosis [6]. In fibroblasts, TGFβ1/Smad3 signaling stimulates their transdifferentiation into myofibroblasts and their expression of matrix genes like fibronectin and collagens [7]. Importantly, knockdown of the TGFβ1/Smad3 signaling pathway inhibits experimental lung fibrosis [8]. In a model of immune-mediated airway fibrosis, we demonstrated inhibition of myofibroblast transdifferentiation and matrix deposition in animals deficient in Smad3 [9].
Considering its importance in the development of lung fibrosis, research directed at investigating the factors that control TGFβ1/Smad3 signaling has intensified. This research has led to the exploration of peroxisome proliferator-activated receptor γ (PPARγ), a member of the ligand-activated nuclear hormone receptor superfamily of transcription factors that is known for its ability to regulate glucose and lipid metabolism and that has been implicated in insulin sensitivity, atherosclerosis, and inflammation [10, 11]. Preliminary studies in our laboratory suggested that PPARγ inhibits the effects of TGFβ1 through direct interactions with Smad3 (Ramirez et al., unpublished observations). Furthermore, others have demonstrated protection against TGFβ1-induced myofibroblast transdifferentiation in cells treated with PPARγ activators [12].
These observations suggest a promising role for PPARγ activators in the treatment of fibrotic lung disorders. However, we wondered if TGFβ1 influences PPARγ expression and/or activation in tissues. To test this idea, we examined the effects of TGFβ1 on PPARγ in murine primary lung fibroblasts and found that TGFβ1, in part through Smad3 signaling, differentially controls PPARγ expression levels, transcription, and activation. These observations suggest that TGFβ1/Smad3 signaling triggers profibrotic events, while concomitantly influencing the expression of PPARγ.
2. Materials and Methods
2.1. Cell Culture
NIH/3T3 fibroblasts were purchased from ATCC. Primary murine lung from Smad3-deficient mice and wildtype C57BL/6 were generated and maintained as previously described [7, 9]. Primary lung fibroblasts were used between passages 2–10 in all experimental conditions. Where indicated, serum-starved fibroblasts were first pretreated for 1 hour in the presence or absence of TGFβ1 (10 ng/mL) (R&D Systems, Minneapolis, MN, USA) followed by incubation with 10 μM of troglitazone (Cayman Chemical, Ann Arbor, MI, USA) for the specified time. The studies were approved by the institutional animal research review committee.
2.2. Western Blots
Whole cell extracts were processed and analyzed as described [7, 9] with antibodies to phospho-PPARγ (Millipore, Billerica, MA, USA) and PPARγ protein (Cell Signaling Technology). β-Actin was used as control (Sigma, St. Louis, MO, USA).
2.3. Reverse Transcription-Polymerase Chain Reaction
Total RNA was isolated and tested as previously described [7]. Murine forward and reverse primers for PCR reactions were based on GenBank published sequences and are as follows: PPARγ (5′-GAC CAC TCG CAT TCC TTT-3′; 5′-CCA CAG ACT CGG CAC TCA-3′) and 28s rRNA (5′-TTG AAA ATC CGG GGG AGA-3′; 5′-ACA TTG TTC CAA CAT GCC AG-3′). Amplicons were resolved on 1% agarose gels, stained with ethidium bromide, and visualized with a UV transilluminator.
2.4. Immunofluorescence Microscopy
A phospho-PPARγ antibody was applied to paraformaldehyde-fixed, Triton X-100-permeablized cells at 4°C overnight followed by an Alexa Fluor 555-labeled anti-rabbit secondary antibody (Invitrogen). Slides were cover-slipped with ProLong Gold mounting medium (Invitrogen) and viewed under epifluorescence microscopy (Olympus BX41, Melville, MY, USA). Images were captured using MagnaFire 2.1 digital image acquisition software (Goleta, CA, USA).
2.5. Plasmids
Murine pCMX-PPARγ [13] and the human PPARγ promoter [14] were generous gifts from L. Jameson, (Northwestern University, Chicago, IL, USA) and C.M. Hart (Emory University, Atlanta, GA, USA), respectively. Flag-Smad3, AP2 (PPRE)-Luc, and TβRI (CA) were purchased from Addgene (numbers 14827, 8858, and 14833, resp., Cambridge, MA, USA).
2.6. Transfection and Reporter Studies
A calcium-phosphate transient transfection protocol was followed [15]. Briefly, at 50% confluence, cells were exposed to fresh growth media for 1 hour. DNA precipitate was applied for 24 hours. Cells were then washed, cultured for 6 hours in complete serum-free media, and treated as indicated. For reporter assays, treated cells were lysed using 5x Passive Lysis Buffer (Promega, Madison, WI, USA) and exposed to luciferase reagent, which was prepared according to Dyer et al. [16]. Luminescence was measured with a Thermo Luminoskan Ascent luminometer (Waltham, MA, USA) and normalized to Renilla activity [16].
2.7. Data Analysis
Western blotting and RT-PCR experiments were performed in duplicates and repeated at least three times to ensure consistency. Reporter and proliferation data were also repeated thrice each with 3-4 replicates per experiment. All results are presented as mean ± SE. GraphPad Prism v3.0 was used to analyze data by one-way ANOVA computation with Tukey's multiple comparisons test. A P value of 0.05 was considered significant.
3. Results
3.1. Effects of TGFβ1 on PPARγ Expression
To begin to evaluate the effects of TGFβ1 on PPARγ expression, we first tested for PPARγ gene transcription in lung fibroblasts. After a small and nonsignificant increase in PPARγ expression, primary lung fibroblasts displayed a dramatic downregulation of PPARγ mRNA expression beginning after an hour of exposure to TGFβ1 and persisting for at least 48 hours (Figure 1(a)). To determine a mechanism by which TGFβ1 might regulate PPARγ expression, we examined the role of the transcription factor and TGFβ1 intracellular transducer, Smad3. For this, primary lung fibroblasts were harvested from the lungs of Smad3-deficient mice and wildtype mice from the same genetic background and were cultured in the presence or absence of TGFβ1 (Figure 1(b)). In wildtype primary lung fibroblasts, TGFβ1 downregulated PPARγ mRNA expression as previously demonstrated. However, this effect was greatly blunted in cells lacking Smad3, suggesting that Smad3 signaling is responsible for much of the inhibition of PPARγ gene expression observed in TGFβ1-treated fibroblasts.
Figure 1.
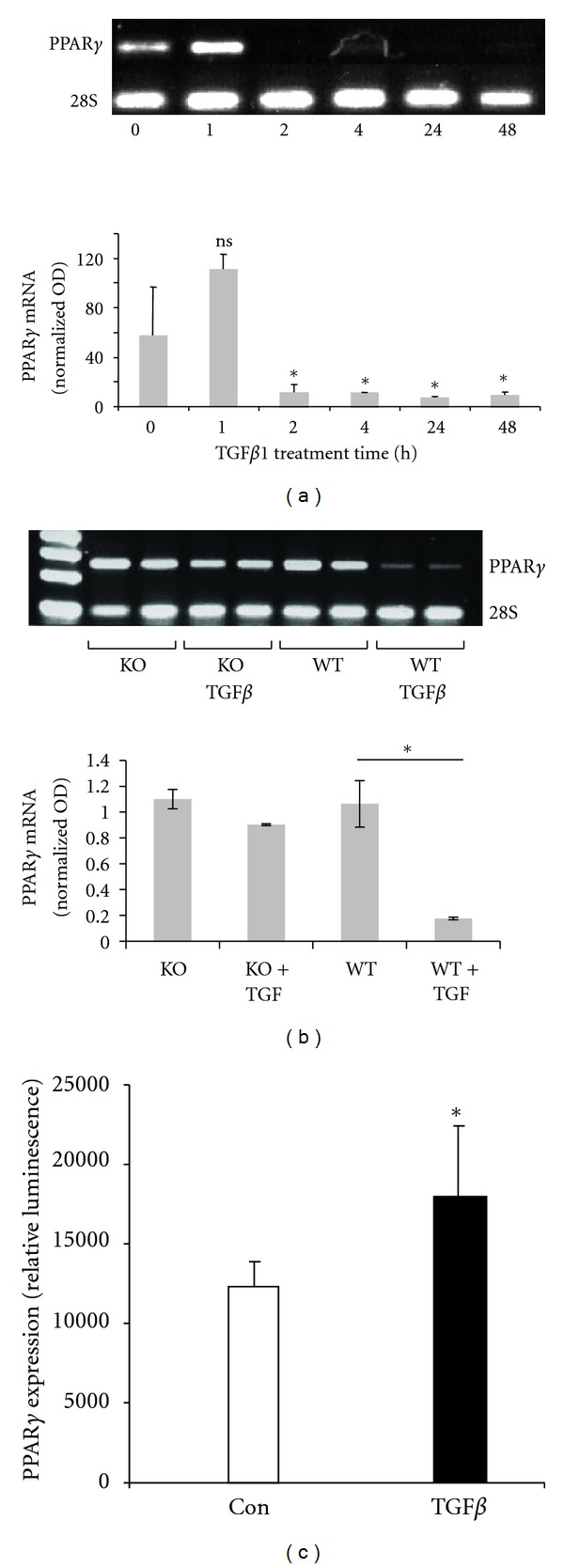
TGFβ1 modulates PPARγ expression, in part, via Smad3 signaling. (a) TGFβ1 and PPARγ mRNA expression. Murine primary lung fibroblasts were stimulated with TGFβ1 (10 ng/mL) for the indicated times. The treated cells were then submitted for analysis by RT-PCR for PPARγ. (b) Role of Smad3. Primary lung fibroblasts were isolated from the lungs of Smad3-deficient and wildtype mice and incubated with TGFβ1 (10 ng/mL) for 24 hours. Transcripts of PPARγ mRNA were detected using RT-PCR. *P < 0.05. (c) Regulation of PPARγ gene transcription. NIH/3T3 fibroblasts were transiently transfected with a luciferase reporter driven by the full-length PPARγ promoter and incubated with TGFβ1 (10 ng/mL). Cellular extracts were analyzed after 24 hours for luminescence. Luciferase activity was normalized to Renilla with data shown as fold change ± SE relative to control. *P < 0.05.
To test if the inhibition of PPARγ expression by TGFβ1/Smad3 signaling occurred at the level of the gene promoter, NIH/3T3 fibroblasts were transfected with full-length human PPARγ promoter ligated to a luciferase reporter (Figure 1(c)). However, instead of inhibiting PPARγ expression, we observed that TGFβ1 stimulated activity of the PPARγ gene promoter.
3.2. Effect of TGFβ1 on PPARγ Phosphorylation
Next, we determined if TGFβ1 could induce PPARγ posttranslational modifications, namely, phosphorylation, given TGFβ1's known ability to activate intracellular signaling molecules. To this end, primary lung fibroblasts were treated with TGFβ1 at several different time points and examined for phospho-PPARγ with an antibody that specifically detects phosphorylation at serine 82 of PPARγ1 (corresponding to serine 112 of PPARγ2). By Western blot, phosphorylation of PPARγ was detectable within 3 hours of TGFβ1 exposure, with maximal effects at 6 hours, but persistent up to 24 hours after stimulation when the experiment ended (Figure 2(a)). In parallel experiments, phospho-PPARγ was visualized by immunofluorescence in the nuclear compartment one hour after initiation of TGFβ1 treatment, peaked three hours later, but was mostly absent by six hours (Figure 2(b)).
Figure 2.
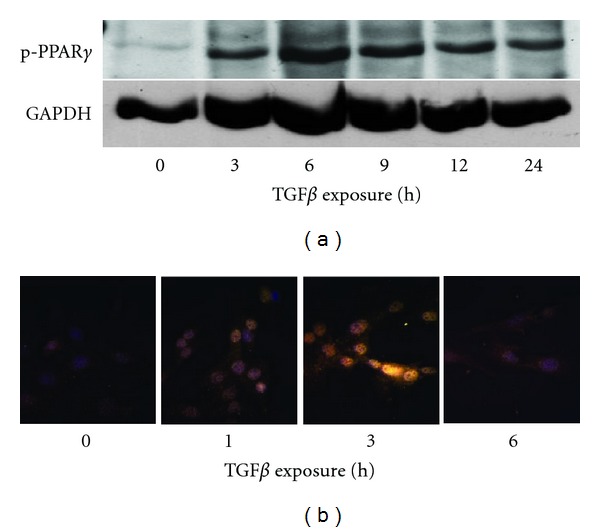
TGFβ1 induces the phosphorylation of PPARγ. Primary lung fibroblasts were treated with TGFβ1 (10 ng/mL) for the indicated times. Phosphorylation of PPARγ was detected by Western blot (a) and immunofluorescence microscopy (b) using a phospho-specific antibody.
3.3. Effects of TGFβ1 on PPARγ-Dependent Gene Expression
We then examined the functional effects of TGFβ1 signaling on the transcriptional capability of PPARγ after activation by the PPARγ ligand, troglitazone. These experiments were conducted in NIH/3T3 fibroblasts containing a luciferase reporter driven by a PPARγ response element. In other words, luciferase induction indicates stimulation of PPARγ-dependent gene expression. As shown in Figure 3(a), troglitazone stimulated PPARγ activation as demonstrated by the ability of troglitazone to induce PPARγ-dependent gene transcription. This effect was blunted by the overexpression of a constitutively-active type I TGFβ receptor, that is, TβRI or activin-like kinase 5 (ALK5), suggesting a role for TGFβ1 receptor activation in the TGFβ1-dependent repression of gene transcription by PPARγ. However, the inhibition of transcriptional ability of PPARγ by TGFβ1 signaling was overcome by the forced overexpression of the full-length PPARγ gene (Figure 3(b)). As expected, when a Smad3 transgene was cotransfected with the PPARγ transgene in stoichiometrically equivalent amounts, TGFβ1 was again able to partially offset activation of a PPARγ-responsive gene promoter induced by troglitazone (Figure 3(c)). These data indicate that stimulation of TGFβ1-dependent pathways, via activation of TGFβ1 receptors and Smad3 signaling, regulates the activity of PPARγ on gene transcription.
Figure 3.
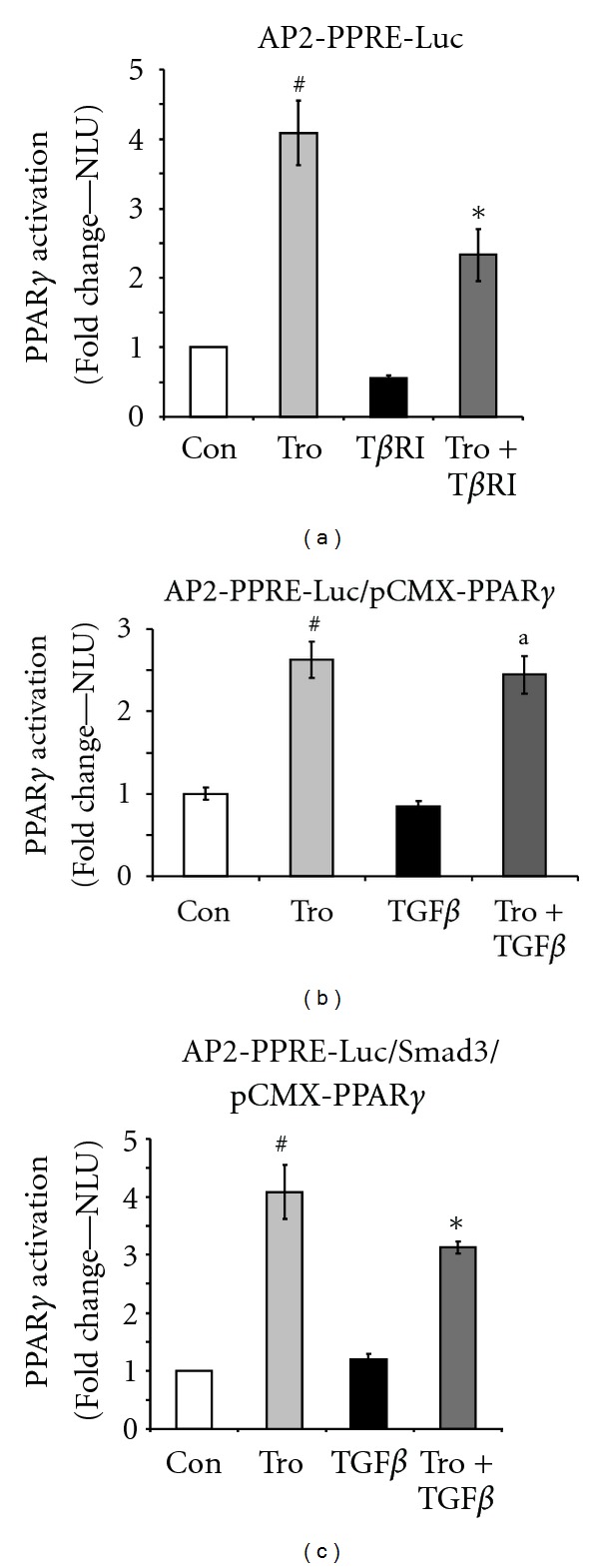
TGFβ1 signaling inhibits the transcriptional activity of PPARγ. PPARγ transcriptional activity was determined with luciferase assays in NIH/3T3 fibroblasts bearing a PPARγ response element-luciferase reporter (AP2-PPRE-Luc). Luciferase activity was normalized to Renilla with data shown as fold change ± SE relative to control. # P < 0.001 compared to control. *P < 0.001 compared to troglitazone. a P: ns compared to troglitazone. (a) Effects of troglitazone and TGFβRI. PPRE-containing NIH/3T3 fibroblasts were treated with troglitazone (Tro 10 μM) and/or cotransfected with a constitutively active type I TGFβ receptor (TGFβRI). At 24 hours, cells were analyzed for luciferase activity. (b) Overexpression of PPARγ gene. The full-length PPARγ gene and a PPRE were cotransfected into NIH/3T3 fibroblasts, which were then pretreated with TGFβ1 (10 ng/mL) for 1 hour, followed by troglitazone (10 μM) for 24 hours. (c) Role of Smad3. Smad3 and PPARγ were coexpressed in NIH3T3 fibroblasts with a PPRE. Cells were exposed to TGFβ1 (10 ng/mL) for 1 hour and then stimulated with troglitazone (10 μM) for 24 hours.
4. Discussion
Our studies suggest that TGFβ1 influences the expression and activity of PPARγ with potential implications to lung inflammation and fibrosis. PPARs are recognized as versatile members of the ligand-activated nuclear hormone receptor superfamily of transcription factors that includes receptors for steroids, thyroid hormone, retinoic acid, and vitamin D among others [10, 11]. PPARs are considered to play key roles in diverse physiological processes ranging from lipid metabolism to inflammation and have been implicated in diseases such as cancer, atherosclerosis, and lung injury [17]. Three subtypes of PPARs have been identified and cloned: PPARα, PPARβ/δ, and PPARγ. Of the three PPARs identified to date, PPARγ represents the most promising PPAR target in lung diseases in view of emerging reports implicating this molecule in various pulmonary processes [17]. Importantly, PPARγ has been described as a negative regulator of macrophage function since its activation suppresses the production of inflammatory cytokines, chemokines, metalloproteinases, and nitric oxide [18]. These PPARγ-mediated anti-inflammatory effects are not restricted to monocytic cells as treatment with PPARγ agonists results in inhibition of cytokine and chemokine production in other cells [18–20]. More recently, it has been reported that PPARγ activators inhibit TGFβ1-induced myofibroblast transdifferentiation [12].
In diseased tissues, PPARγ expression has been shown to relate inversely with that of TGFβ1 [21]. Thus, it appears that the balance between TGFβ1 and PPARγ may determine, among other factors, whether fibrogenesis predominates after tissue injury. However, in many patients and in experimental models, endogenous PPARγ is unable to counter the effects of TGFβ1. We reasoned that tissue injury results in the expression of factors capable of inhibiting PPARγ expression or of blunting its antifibrotic effects. We further hypothesized that TGFβ1 itself could directly influence PPARγ expression. The observations reported here suggest that this is indeed the case. We observed that TGFβ1 has an early, but transient, inductive effect on PPARγ mRNA expression in primary lung fibroblasts; this effect was likely caused by the ability of TGFβ1 to induce PPARγ gene transcription (Figure 1). This early effect was associated with PPARγ mRNA translation into protein and phosphorylation of the PPARγ protein. However, this effect was later associated with profound inhibition of PPARγ mRNA accumulation. The exact mechanisms responsible for this late effect are unknown, but increased mRNA degradation is likely. Based on these observations, we postulate that TGFβ1 expression and/or activation in injured tissues is associated with early induction of a counterregulatory factor, PPARγ. However, persistence of TGFβ1 expression/activation results in late inhibition of PPARγ mRNA accumulation. It should be highlighted that this “biphasic” effect of TGFβ1 on PPARγ (early stimulation and late repression) has been observed by others when studying the actions of TGFβ1 on PPARγ expression in vascular smooth muscle cells [22]. When studying these effects, one must consider cell type and other factors that could potentially affect the responses observed. For example, others have demonstrated that TGFβ increases PPARγ expression in H460 cells, but not in Ch27 cells, whereas nuclear accumulation of p-Smad3 was only observed in CH27 cells [23].
Interestingly, despite late inhibition of PPARγ mRNA accumulation, we observed persistent PPARγ phosphorylation at least 24 hours after TGFβ stimulation (Figure 2). This, however, did not correlate with persistent nuclear localization of PPARγ as demonstrated by cytochemistry (Figure 2(b)). The mechanisms responsible for these events remain unclear.
We also studied the role of Smad3 signaling in the effects observed. We found that Smad3 appears to mediate, at least in part, the effects of TGFβ1 since cells deficient in Smad3 showed blunting of the TGFβ1 downregulatory effect on PPARγ. This was not surprising considering that Smad3 mediates many of the effects of TGFβ1 in tissues. These data are consistent with observations made in dermal fibroblasts [21]. In other work, we reported that activation of P38 MAPK plays a role in TGFβ1-induced myofibroblast transdifferentiation [7], and others have documented the interplay between TGFβ1 and MAPKs in other systems [24]. Lin et al. (2011) reported that TGFβ-induced expression of PPARγ was related to activation of p38, but this was tested in H460 carcinoma cells [23]. Interestingly, they also showed that PPARγ1 can bind Smad3 and p-Smad3. Zheng and Chen, on the other hand, reported that exogenous TGFβ inhibits PPARγ expression in activated hepatic stellate cells and this appeared mediated through Smads [25]. Furthermore, and consistent with our data, blocking TGFβ signaling by dominant negative type II TGFβ receptor increased PPARγ. Choy and Derynck (2003) reported that TGFβ inhibits adipogenesis by signaling through Smad3 which, in turn, interacts with C/EBPs leading to transcriptional inhibition of the PPARγ2 promoter [26].
The studies described above suggest that TGFβ1 controls PPARγ transcription, mRNA accumulation, and protein phosphorylation in differential ways. Considering the many points at which TGFβ1 could influence PPARγ expression and activation, these studies did not allow us to predict the overall effects of TGFβ1 on PPARγ function. This critical issue was addressed by testing cells transfected with a PPARγ-responsive element that is transcribed only when activated PPARγ enters the nucleus and interacts with target genes. To test the system, we first showed that troglitazone, a PPARγ activator, stimulated PPARγ-dependent gene expression (Figure 3(a)). Interestingly, expression of a constitutively active TGFβ1 receptor, TGFβ1RI, reduced the effect of troglitazone implicating this receptor in mediating the effects of TGFβ1. Together, TGFβRII and TGFβRI form an activated ligand-receptor complex capable of stimulating downstream signals like Smads [27].
As expected, the inhibition of transcriptional ability of PPARγ by TGFβ1 was overcome by the forced expression of the full-length PPARγ gene. This is an important observation because it suggests that targeting the TGFβ1/PPARγ balance by enhancing PPARγ activation may have potential therapeutic relevance. Several natural and synthetic compounds have been identified as activators of PPARγ. The insulin-sensitizing antidiabetic drugs known as thiazolidinediones were the first compounds identified as PPARγ agonists [28]; troglitazone, used in this report, is a thiazolidinedione, but it is no longer available commercially due to liver toxicity [29]. However, other thiazolidinediones are available and are being tested in clinical trials in different areas.
Finally, we tested the role of Smad3 by cotransfecting cells with the Smad3 and PPARγ transgenes in stoichiometrically equivalent amounts. As before, TGFβ1 was able to partially offset activation of a PPARγ-responsive gene promoter induced by troglitazone. These data indicate that stimulation of TGFβ1-dependent pathways, via activation of TGFβ1 receptors and Smad3 signaling, regulates the activity of PPARγ on gene transcription.
In conclusion, modulation of PPARγ is intricately controlled by TGFβ1, in part through Smad3 signals, involving tight regulation of PPARγ expression levels and transcriptional potential. These mechanisms begin to explain how TGFβ1 is able to overcome the anti-inflammatory and antifibrotic effects of PPARγ and may have implications in vivo. The commercial availability of PPARγ activators capable of tilting the TGFβ1/PPARγ balance to reduce, delay, and even reverse fibrosis raises the possibility of targeting PPARγ in humans with fibrotic lung disease.
Conflict of Interests
The authors declare that they have no conflicts of interests.
Acknowledgment
This work was supported by NIH NHLBI Grant 1K08HL077533 (A. Ramirez).
References
- 1.Bonniaud P, Margetts PJ, Ask K, Flanders K, Gauldie J, Kolb M. TGF-β and Smad3 signaling link inflammation to chronic fibrogenesis. Journal of Immunology. 2005;175(8):5390–5395. doi: 10.4049/jimmunol.175.8.5390. [DOI] [PubMed] [Google Scholar]
- 2.Keane MP, Belperio JA, Strieter RM. Cytokine biology and the pathogenesis of interstitial lung disease. In: Schwarz MI, King TE, editors. Interstitial Lung Disease. 5th edition. chapter 16. Shelton, Conn, USA: People's Medical Publishing House; 2011. pp. 365–386. [Google Scholar]
- 3.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor- β1 induces prolonged severe fibrosis in rat lung. Journal of Clinical Investigation. 1997;100(4):768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCormick LL, Zhang Y, Tootell E, Gilliam AC. Anti-TGF-β treatment prevents skin and lung fibrosis in murine sclerodermatous graft-versus-host disease: a model for human scleroderma. Journal of Immunology. 1999;163(10):5693–5699. [PubMed] [Google Scholar]
- 5.Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-β signaling in fibrosis. Growth Factors. 2011;29:196–202. doi: 10.3109/08977194.2011.595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willis BC, Borok Z. TGF-β-induced EMT: mechanisms and implications for fibrotic lung disease. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2007;293(3):L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 7.Ramirez AM, Shen Z, Ritzenthaler JD, Roman J. Myofibroblast transdifferentiation in obliterative bronchiolitis: TGF-β signaling through Smad3-dependent and -independent pathways. American Journal of Transplantation. 2006;6(9):2080–2088. doi: 10.1111/j.1600-6143.2006.01430.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhao J, Shi W, Wang YL, et al. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2002;282(3):L585–L593. doi: 10.1152/ajplung.00151.2001. [DOI] [PubMed] [Google Scholar]
- 9.Ramirez AM, Takagawa S, Sekosan M, Jaffe HA, Varga J, Roman J. Smad3 deficiency ameliorates experimental obliterative bronchiolitis in a heterotopic tracheal transplantation model. American Journal of Pathology. 2004;165(4):1223–1232. doi: 10.1016/S0002-9440(10)63382-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giaginis C, Tsourouflis G, Theocharis S. Peroxisome proliferator-activated receptor-γ (PPAR-γ) ligands: novel pharmacological agents in the treatment of ischemia reperfusion injury. Current Molecular Medicine. 2008;8(6):562–579. doi: 10.2174/156652408785748022. [DOI] [PubMed] [Google Scholar]
- 11.Rabinovitch M. PPARg and the pathobiology of pulmonary arterial hypertension. Advances in Experimental Medicine and Biology. 2010;661:447–458. doi: 10.1007/978-1-60761-500-2_29. [DOI] [PubMed] [Google Scholar]
- 12.Kulkarni AA, Thatcher TH, Olsen KC, Maggirwar SB, Phipps RP, Sime PJ. PPAR-γ ligands repress TGFβ-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PLoS One. 2011;6(1) doi: 10.1371/journal.pone.0015909.e15909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park Y, Freedman BD, Lee EJ, Park S, Jameson JL. A dominant negative PPARγ mutant shows altered cofactor recruitment and inhibits adipogenesis in 3T3-L1 cells. Diabetologia. 2003;46(3):365–377. doi: 10.1007/s00125-003-1037-4. [DOI] [PubMed] [Google Scholar]
- 14.Blanquicett C, Kang BY, Ritzenthaler JD, Jones DP, Hart CM. Oxidative stress modulates PPARγ in vascular endothelial cells. Free Radical Biology and Medicine. 2010;48(12):1618–1625. doi: 10.1016/j.freeradbiomed.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kingston RE, Chen CA, Rose JK. Calcium phosphate transfection. Current Protocols in Cell Biology, chapter 9, unit 9. 1, 2003. [DOI] [PubMed]
- 16.Dyer BW, Ferrer FA, Klinedinst DK, Rodriguez R. A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Analytical Biochemistry. 2000;282(1):158–161. doi: 10.1006/abio.2000.4605. [DOI] [PubMed] [Google Scholar]
- 17.Standiford TJ, Keshamouni VC, Reddy RC. Peroxisome proliferator-activated receptor-γ as a regulator of lung inflammation and repair. Proceedings of the American Thoracic Society. 2005;2(3):226–231. doi: 10.1513/pats.200501-010AC. [DOI] [PubMed] [Google Scholar]
- 18.Sime PJ. The antifibrogenic potential of PPARγ ligands in pulmonary fibrosis. Journal of Investigative Medicine. 2009;56(2):534–538. doi: 10.2310/JIM.0b013e31816464e9. [DOI] [PubMed] [Google Scholar]
- 19.Reddy RC. Immunomodulatory role of PPAR-γ in alveolar macrophages. Journal of Investigative Medicine. 2009;56(2):522–527. doi: 10.2310/JIM.0b013e3181659972. [DOI] [PubMed] [Google Scholar]
- 20.Becker J, Delayre-Orthez C, Frossard N, Pons F. Regulation of inflammation by PPARs: a future approach to treat lung inflammatory diseases? Fundamental and Clinical Pharmacology. 2006;20(5):429–447. doi: 10.1111/j.1472-8206.2006.00425.x. [DOI] [PubMed] [Google Scholar]
- 21.Wei J, Ghosh AK, Sargent JL, et al. PPARγ downregulation by tgf in fibroblast and impaired expression and function in systemic sclerosis: a novel mechanism for progressive fibrogenesis. PLoS One. 2010;5(11) doi: 10.1371/journal.pone.0013778.e13778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu M, Zhang J, Lin Y, et al. Early stimulation and late inhibition of peroxisome proliferator-activated receptor γ (PPARγ) gene expression by transformino growth factor β in human aortic smooth muscle cells: role of early growth-response factor-1 (Egr-1), activator protein 1 (AP1) and Smads. Biochemical Journal. 2003;370(3):1019–1025. doi: 10.1042/BJ20021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin LC, Hsu SL, Wu CL, Liu WC, Hsueh CM. Peroxisome proliferator-activated receptor γ (PPARγ) plays a critical role in the development of TGFβ resistance of H460 cell. Cellular Signalling. 2011;23(10):1640–1650. doi: 10.1016/j.cellsig.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 24.Terai K, Call MK, Saika S, et al. Crosstalk between TGFb and MAPK signaling during corneal wound healing. Investigative Ophthalmology & Visual Science. 2011;52:8208–8215. doi: 10.1167/iovs.11-8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng S, Chen A. Disruption of transforming growth factor-β signaling by curcumin induces gene expression of peroxisome proliferator-activated receptor-γ in rat hepatic stellate cells. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2007;292(1):G113–G123. doi: 10.1152/ajpgi.00200.2006. [DOI] [PubMed] [Google Scholar]
- 26.Choy L, Derynck R. Transforming growth factor-β inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. Journal of Biological Chemistry. 2003;278(11):9609–9619. doi: 10.1074/jbc.M212259200. [DOI] [PubMed] [Google Scholar]
- 27.Santibañez JFS, Quintanilla M, Bernabeu C. TGF-β/TGF-β receptor system and its role in physiological and pathological conditions. Clinical Science. 2011;121(6):233–251. doi: 10.1042/CS20110086. [DOI] [PubMed] [Google Scholar]
- 28.Yki-Jarvinen H. Thiazolidinediones. The New England Journal of Medicine. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 29.Scheen AJ. Thiazolidinediones and liver toxicity. Diabetes and Metabolism. 2001;27(3):305–313. [PubMed] [Google Scholar]