

Abstract
Cigarette smoking is a risk for Alzheimer’s disease (AD), the pathological hallmark of which is amyloid-β (Aβ) brain deposits. We found the adjusted risk of AD was significantly increased among medium level smokers (RR = 2.56; 95% CI = 1.65–5.52), with an even higher risk in the heavy smoking group (RR = 3.03; 95% CI = 1.25–4.02). This systematic review and original data further support this association. We searched Pubmed, Google scholar, and PsyINFO for original population study articles, meta-analyses, and reviews published between 1987 and 2011. Some studies were excluded due to design flaws including survivor bias. We performed analyses of: 1) amyloid precursor protein (APP) processing in N2a cells overexpressing Swedish mutant APP (SweAPP N2a) exposed to cigarette smoke condensate (CSC), 2) microglial inflammatory response to CSC, and 3) CSC exposed microglial phagocytosis of Aβ1–42. CSC significantly promotes neuronal Aβ generation, increases microglial IL-1β and TNF-α production, and decreases microglial Aβ1–42 phagocytosis. The mechanism underlying the epidemiological association of cigarette smoking with AD might involve the effect of cigarette smoke on APP processing, a reduction of Aβ clearance by microglia, and/or an increased microglial proinflammatory response. In vivo studies are required to fully elucidate how cigarette smoke promotes AD.
Keywords: Cigarette, Smoking, Alzheimer’s disease (AD), Dementia, Risk, Amyloid-β (Aβ)
CIGARETTE SMOKING AS A RISK FACTOR FOR PROMOTION OF ALZHEIMER’S DISEASE
In 2006, there were 26.6 million people with Alzheimer’s disease (AD), and this number is predicted to quadruple by the year 2050 (7). As such, an understanding of the mechanisms underlying this phenomenon is important. What follows is a brief background systematic review of the available literature that is appropriately designed without survivor bias, or tobacco company sponsorship. We will then move on to discuss a possible molecular mechanism underlying this epidemiology.
There is much evidence to indicate that cigarette smoking has a dose-dependent association with AD development (1,8,25,28,35,36,39,40). One of the first reports to identify cigarette smoking as a risk for the development of AD was published some 25 years ago. This was a case control study in which 98 men with clinical AD were compared to 162 controls matched by age and town of residence. AD cases were positively correlated with number of cigarettes smoked, although the result was not significant (47).
Ten years later, in a similar case-control study of 98 AD patients and 97 age- and sex-matched controls, daily smoking showed a trend toward an increase risk for AD [45.9% vs. 26.5% in non-smokers; odds ratio (OR) = 1.73, p = 0.08] while light smoking seemed to have a protective effect (56). A 1998 seminal report in the Lancet revealed that smoking was associated with a doubling of the risk of dementia and AD by Cox proportional hazards regression, after adjustment for age, sex, education, and alcohol intake [current smoker vs. never smoker, relative risk (RR) 2.3, 95% CI 1.3–4.1] (35). They first found which association between smoking and AD development was modified by apolipoprotein ε4 (apoE4) allele, which was a well-known genetic risk factor for incident AD (45).
Interestingly, current smokers without an apoE4 allele had significantly increased AD risk of 4.6 (95% CI 1.5–14.2). Those carrying the apoE4 alleles had a nonsignificantly increased risk of AD (RR 0.6, 95% CI 0.1–4.8). This may indicate that the risk conferred by the ApoE4 gene is so strong that cigarette smoking only has a negligible effect. The following year Merchant and colleagues (32) found that relative risk of AD among current smokers was almost double the control (RR 1.95, 95% CI 1.2–3.0). Consistently it was found that smokers had the highest risk of AD (RR 2.1, 95% CI 1.2–3.7) compared to never smokers in absent apoE4 allele participants; however, among carriers of apoE4 current smoker decreased in relative risk (RR 1.4, 95% CI 0.6–3.3). Interestingly, they found a trend among former smokers, who had a slight nonsignificant reduction in AD incidence as the relative risk was 0.7 (95% CI 0.5–1.1). This may be explained by the notion that those surviving former smokers may have more effective DNA repair mechanisms (5,33). As a consequence, the accumulation of aging-associated defects in DNA and DNA repair, which are presumed to be associated with AD, may be altered among surviving former smokers, reducing their susceptibility to AD (32).
In further support, in a pooled analysis of four prospective studies with 28,768 person-years of follow-up, it was found that current smoking significantly increases the risk of AD (28). Specifically, current smokers had a significantly increased relative risk of AD of 1.74 (95% CI 1.21–2.50) compared with never smokers. Tyas and colleagues (52) studied the association between smoking and AD and other types of dementia in the Honolulu Heart Program (1965–1971) and follow-up assessment for dementia (1991–1996) of 3,734 Japanese-American men (29). Neuropathologic data were available from 218 men. There was no significance increased in risk of AD incidence in current smokers (RR 1.0, 95% CI 0.61–1.63). However, the risk of AD in smokers increased with pack-years of smoking at medium (OR 2.18, 95% CI 1.07–4.69) and heavy levels (OR 2.40, 95% CI 1.16–5.17). This study found a lack of association between very heavy smoking and AD. This is probably explained by the hardy survivor bias effect, which will be expanded upon in the next section of this article (52). Upon autopsy in this study it was found that the number of amyloid plaques increased with amount smoked (52). Indeed, deposition of these amyloid-β peptide (Aβ) as β-amyloid plaques is a pathological hallmark of AD. They are accompanied by increased activation of microglia-mediated inflammatory responses (44).
We also conducted a prospective 2-year study of 2,820 persons aged 60 or older and found current smokers had significantly increased risk of AD (RR 2.71, CI 95% 1.63–5.42). When compared to light smokers, the risk of AD was significantly higher among smokers with a medium level of exposure (RR 2.56, 95% CI 1.65–5.52), with even greater risk of AD in the heavy smokers (RR 3.03, 95% CI 1.25–4.02) (25), adjusted by age, sex, education, blood pressure, and alcohol intake. This study further suggests that both smoking status and its intensity are associated with AD (25). In 2007, Reitz and colleagues published results from the Rotterdam study, a prospective cohort study of 6,868 subjects, 55 years or older and free of dementia at baseline studied over an average of 7.1 years. It was found that current smoking increases the relative risk of AD by 1.56 (95% CI 1.21–2.02). Moreover, in non-apoE4 carriers, current smoker increased in risk of AD incidence as well (RR 1.95, 95% CI 1.29–2.95) (39).
One potential explanation for nonassociation between smoking and AD in apoE4 carriers is that smoking may be harmful through alternative mechanisms, but beneficial in apoE4 carriers. This hypothesis is supported by previous findings that apoE4 carriers with AD have fewer nicotinic receptor binding sites and lower activity of choline ace-tyltransferase compared with noncarriers (35,37). Finally, Peters and colleagues (36) did a meta-analysis from 1996 to 2007 of individuals aged 65 and over, and concluded that current smoking is a significant risk for AD (RR 1.59, 95% CI 1.15–2.20).
In 2010 and 2011, two reports further reinforced the positive association between cigarette smoking and AD. In a meta-analysis by Cataldo and colleagues (8), smoking was a significant risk factor for AD after accounting for bias introduced by author tobacco industry affiliation and survival bias phenomenon in case-control studies. After controlling for study design, quality, secular trend, and tobacco industry affiliation, a significant increase in AD risk was associated with cigarette smoking for cohort studies of average quality in 2007 (1.45, 95% CI 1.16–1.80). This review also indicated case-control studies tended to demonstrate lower risks than cohort studies and the majority of the tobacco industry affiliated studies used case-control design (RR 0.60, 95% CI 0.27–1.32) (8). After controlling for study design, tobacco industry affiliation was independently associated with lower risk estimates. In contrast to the significant increase in risk of AD development in non-tobacco industry-associated studies (1.72 ± 0.19, p < 0.0005), the combination of case-control design and tobacco industry affiliation yielded a protective odds ratio of 0.86 (95% CI 0.75–0.98). Again these putative “protective” effects of smoking cigarettes was generated by poor study design, not accounting for survivor bias, and possible conflicts of interest due to affiliation with the tobacco industry (8).
In a seminal report by Rusanen and colleagues (40), it was found in a cohort of over 21,000 individuals those smoking >2 packs/day had a significantly higher risk of AD than nonsmokers [adjusted hazard ratio (HR) 2.57, 95% CI 1.63–4.03]. They adjusted for age, sex, education, race, marital status, hypertension, hyperlipidemia, body mass index, diabetes type II, heart disease, stroke, and alcohol use (40). These most recent findings in this well-designed study are consistent with previous studies showing that heavier smoking is associated with greater AD risk (8,25,28,35,36,39,40).
THE CONTROVERSY OVER THE MYTH THAT CIGARETTE SMOKING IS PROTECTIVE FOR AD
Several publications have indicated that smoking might have no, or a protective, effect (14,30), particularly those of case-control design (26). However this conclusion is likely misleading due to selection or “survivor” bias and funding of the research by the cigarette industry (8,22,33).
Addressing the latter, empirical support for selection bias due to censoring by death in epidemiologic studies of the effect of cigarette smoking on risk of AD is provided in a recent review by Hernán and colleagues (22). The deleterious effect of smoking on mortality is most likely not reduced in the demented. Conversely, in the elderly, comorbidity can have an additive effect on mortality (20). If this phenomenon occurs with dementia and smoking, smokers who become demented are likely eliminated early from the population, thus resulting in an underrepresentation of demented smokers in cross-sectional samples as well (22,53). This is supported by observations that smoking is associated with an increased mortality among patients with dementia but not controls, suggesting that patients with dementia who have been smokers may be underrepresented in cross-sectional samples (55). This hypothesis is also consistent with findings of a much greater effect of smoking on mortality in demented (HR 3.4) than in nondemented (HR 0.8) subjects (55). For example, in a study of a cohort of 668 people (75–101 years) it was found that smoking was not associated with prevalent AD. However, over a 3-year period incidence of AD was higher in the smokers. Over a 5-year follow-up period, the mortality rate was greater in smoking demented (HR 3.4) than nondemented (HR 0.8) patients (55). Thus, this report suggested that smoking is not protective against AD, and the cross-sectional association is likely due, at least in large (10), to earlier mortality in the smoking group (26,55).
Indeed, a systematic review of 12 studies by Hernán and colleagues (600 subjects, adjusting for age and sex) revealed a wide range of relative risk of AD for smokers versus nonsmokers (0.27–2.72). They found that the minimum age at entry (55–75 years) accounted for much of the inconsistency between studies, and it was suggested that selection bias secondary to censoring by death was the main explanation for the reversal of smoking-induced risk of AD with increasing age (22).
Indeed, it is known that study subjects with complete follow-up are healthier and have better age-specific cognitive scores than those with incomplete follow-up, which is precluded by earlier death due to smoking. A well-known potential result of these differences is selection bias: when the analysis is restricted to individuals with complete follow-up (e.g., those not too ill to participate) (12). In this scenario, it is very possible to find an exposure–outcome association that is not due to the causal effect of the exposure on the outcome (21). An extreme example of “incomplete follow-up” for nonfatal outcomes such as AD is death; thus, censoring by death may introduce selection bias as in the study by Wang and colleagues (55). In studies of the elderly (those at highest risk of developing AD), this selection bias may be quite significant as the death rate is high and mortality is often affected by the exposure (19)—in this case, cigarette smoking.
In earlier meta-analyses of case-control and cohort studies by Almeida et al. (3), similar conclusions were drawn. When the authors restricted their meta-analysis to the two cohort studies that described the number of subjects who were smokers at baseline and later developed AD, they found smoking almost doubled the risk of AD (RR 1.99, 95% CI 1.33–2.98) (3). In accord with these data, this group also showed smoking was positively associated with decreased gray matter density in brain regions previously associated with incipient AD (2).
HOW SMOKING MAY PROMOTE AD AT THE MOLECULAR AND CELLULAR LEVEL
Unlike pure nicotine, which we and others have found to be neuroprotective due to its activation of α-7 nicotinic acetylcholine receptors and reduction of proinflammatory signaling (18,58), cigarette smoke is an admixture of 4,000 different compounds (16,49). Of these, 81 have been classified as pharmacologically active, toxic, mutagenic, and carcinogenic in nature (42). Thus, it is not surprising that inhalation of this smoke would be toxic to the brain as well. The importance of highlighting cigarette smoking as a risk factor for AD is twofold in that it is modifiable (13), and also because it points to a need to understand how cigarette smoke contents can mechanistically promotes this disease outcome, as cigarette smoking continues to be prevalent worldwide.
In this regard, we have attempted to begin to understand this phenomenon in vitro. First, a brief discussion of the molecular origin of Aβ, the central pathology of AD, will be reviewed. Aβ peptide generation and aggregation as plaques are key pathological events in the development of AD (9,15,18,24,41,45). Aβ peptides and plaques have been extensively studied and evidenced to be neurotoxic, as they are reported mediators of apoptosis (27,31), inflammation (6), and oxidative stress (23,34). For this reason, some of the earliest proposed therapeutic strategies focused on prevention or elimination of these Aβ peptides and subsequent deposits. Aβ peptides are produced via the amyloidogenic pathway of amyloid precursor protein (APP) proteolysis, which involves the actions of β-and γ-secretases (43,46). Initially, β-secretase (BACE) cleaves APP, creating an Aβ-containing carboxyl terminal fragment known as β-C-terminal fragment (β-CTF) (48). This proteolysis also generates an amino terminal, soluble APP-β (sAPP-β) fragment, which is released extracellularly. Intra-cellularly, β-CTF is then cleaved by a multiprotein γ-secretase complex that results in generation of the Aβ peptide and a smaller γ-CTF (11,50). Oligomeric Aβ species are thought to be a driving force in AD-type neurodegeneration (54). Aβ peptides are metastable and can exist as monomeric, dimeric, and higher molecular weight oligomeric forms both in vitro and in vivo (54). It is becoming clear that Aβ dimers and oligomers are likely the neurotoxic species, because direct in vivo administration of these Aβ conformers injures neurons (54).
As cigarette smoke is an admixture of 4,000 different compounds (16), we used cigarette smoke condensate (CSC) to measure its effects in vitro. Since cigarette smoke is known to promote oxidative stress (32), which itself is known to promote β-secretase APP cleavage (51,57), we hypothesized that smoking may increase AD risk at the molecular level by activating this enzyme. To examine the effect CSC has on APP proteolysis, we first treated SweAPP N2a cells (N2a cells transfected with the human “Swedish” mutant form of APP) with a range of concentrations. Following Western blot, and ELISA analyses, we found that CSC effectively increases both Aβ1–40, 42 production in a concentration-dependent manner (Fig. 1).
Figure 1.
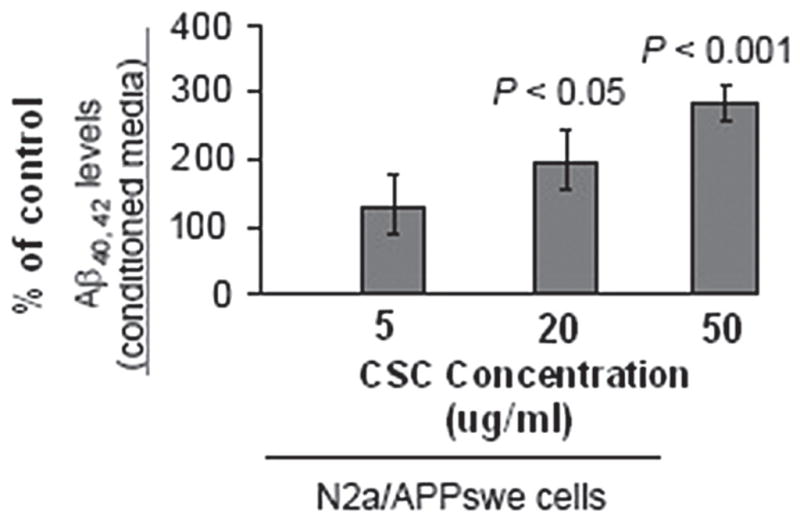
Cigarette smoke condensate (CSC) promotes Aβ generation in cultured neuronal cells. Aβ species were analyzed in cell lysates from CSC-treated SweAPP N2a cells by ELISA (n = 3 for each condition). Data are represented as the mean ± SD of a percentage of Aβ peptides secreted 24 h after CSC (Murty Pharmaceuticals, Lexington, KY; the batch of CSC is prepared from University of Kentucky reference cigarettes K1RF) (33,49) administration versus PBS. One-way ANOVA followed by post hoc comparison revealed significant differences between CSC-treated cells compared to control at 20 and 50 μg/ml treatment concentrations (p < 0.001 and p < 0.05, respectively).
Microglia are activated in close vicinity of Aβ plaques in AD patient brains and in transgenic mouse models of the disease (6). To determine the extent to which CSC promotes a proinflammatory phenotype of microglia, we exposed BV2 microglial cells to CSC at various concentrations for 24 h. Compared to control, CSC at doses of 20 μg/ml significantly promoted TNF-α and IL-1β release ELISA (Fig. 2).
Figure 2.

CSC increases proinflammatory cytokine release in BV2 microglia. Mouse microglial cells were plated in 12-well tissue culture plates (Nunclon™, Roskilde, Denmark) at 4 × 105 cells/well in DMEM media with 10% FBS and antibiotics. Cells were treated with CSC at various concentrations as indicated (from 5 to 50 μg/ml) for 24 h. Cell-free supernatants were then collected and assayed by TNF-α and IL-1β ELISA kits (eBioscience, San Diego, CA) in strict accordance with the manufacturer’s instructions. Compared with control, CSC treatment significantly increased TNF-α and IL-1β release at doses of 20 μg/ml and more. *p < 0.05; **p < 0.01 (see Fig. 1) by one-way ANOVA statistic analysis.
Next, since cigarette smoking has been positively associated with increased brain deposition of Aβ/β-amyloid and AD-like pathology (2,52) and microglial phagocytosis of Aβ has been considered a principle mechanism for the removal of Aβ from the brain parenchyma (4), we evaluated whether CSC could inhibit microglial uptake of Aβ. “Aged,” FITC-tagged Aβ1–42 was added to primary cultured microglia for 60 min in the presence of varying concentrations of CSC or PBS (negative control). As shown in Figure 2, addition of CSC significantly and dose-dependently decreased microglial uptake of Aβ1–42 compared with PBS (negative control; p < 0.05) for each of the doses of CSC tested (Fig. 3).
Figure 3.

CSC inhibits microglial phagocytosis of Aβ peptide. To determine whether CSC impairs microglia phagocytic capacity, “aged,” FITC-tagged Aβ1–42 was added to primary cultured BV2 microglia for 60 min in the presence of varying concentrations of CSC or PBS (negative control). As shown, addition of CSC significantly and dose-dependently decreased microglial uptake of Aβ1–42 compared to PBS (negative control; p < 0.05) for each of the doses of CSC tested except the 5 μg/ ml dose.
In sum, the results of previous studies indicating smoking may be protective for, or not inducing, AD are not supportable, especially in light of the most recent, well-designed, large clinical studies by ourselves and others which clearly indicate that moderate to heavy smoking is a significant direct risk factor for AD (8,25,28,35,36,39,40). Currently, Reitz and colleagues (38) are developing an algorithm for generating a summary risk score for prediction of AD development in the elderly. Interestingly, in the resulting scale, the risk factor of smoking aided in predicating development of late onset AD will be included (38). Taken together, moderate to heavy smoking is associated with increase of AD. The mechanism underlying this phenomenon may involve the effect of cigarette smoke on APP processing, a reduction of Aβ clearance by microglia, and/or an increased proinflammatory profile initiated or perpetuated by microglia. Future in vivo studies will most certainly be required to fully uncover the mechanisms by which cigarette smoke may promote AD at the cellular and molecular level.
Acknowledgments
This work is supported by NIH/NIMH 1K08MH082642-01A1 (B.G.) and a Veterans Affairs Merit grant (J.T.). The authors declare no financial or other relationship with information in this review article. Paul R. Sanberg was not involved in the peer review process of this article.
Biographies

Dr. Brian Giunta received his Bachelors in Microbiology at the University of South Florida in 1998. He earned his M.D. at the University Of South Florida College of Medicine in 2004. Dr. Giunta then completed his internship in Psychiatry at Duke University Medical Center in Durham, NC in 2005. Following, he earned his M.S. and Ph.D. in Medical Science from the University Of South Florida College Of Medicine in 2008 and 2010, respectively. Dr. Giunta became Assistant Professor of Psychiatry at USF in 2007 and is the recipient of an NIH Clinical Scientist Development award.

Dr. Juan Deng received her M.S. and M.D. from the Third Military Medical University of China, Chongqing in 2001 and 2009, respectively. She completed a research fellowship in Neurology from 2004 to 2010 also at the Third Military Medical University and has been an attending neurologist there since 2006.

Dr. Jingji Jin is a senior postdoctoral fellow in the Department of Psychiatry and Neurosciences College of Medicine at University of South Florida. She received her B.A. from Dalian University of Technology, and the M.S. from Dalian University of Technology in China. She earned her Ph.D. from the Ehime University School of Medicine in Japan, and pursued her postdoctoral research in the Departments of Cancer Biology and Pharmacology at University of Illinois College of Medicine prior to joining Dr. Giunta’s laboratory.

Mr. Edin Sadic is a third year undergraduate student at University of South Florida. He is currently studying BioMedical Sciences at USF and is following the Honors College 7-year Medical Program. Mr. Sadic be starting medical school at the University of South Florida College of Medicine in Fall 2012. He has spent the last 2 years gaining research experience in the lab of Dr. Giunta.

Ms. Saja Rum is an undergraduate student at the University of South Florida currently in her fourth year majoring in Biomedical Science. After graduating in the spring of 2012, Ms. Rum will obtain her Master’s in Physician Assistant studies.

Dr. Huadong Zhou received his M.D. and Ph.D. from the Third Military Medical University, Chongqing, China in 1981 and 1994, respectively. He completed clinical and research fellowships at Flinders University of South Australia in 2003 and at St Luke’s Brain and Stroke Institute in Kansas City, USA, in 2007. Currently, he is Chairman of the Department of Neurology at Third Military Medical University affiliated with Daping Hospital, Chongqing, China. Dr. Zhou specializes in diagnosis and treatment of Alzheimer’ disease, stroke, cerebrovascular interventional treatment, and sleep disorders. Dr. Zhou’s publications have focused heavily on epidemiological risk factors for stroke and dementia.

Dr. Paul R. Sanberg is Senior Associate Vice President for Research & Innovation, Distinguished University Professor, Executive Director of the Center of Excellence for Aging and Brain Repair, and Vice Chairman of Academics for the Department of Neurosurgery and Brain Repair in the College of Medicine, at the University of South Florida. Dr. Sanberg trained at York University, the University of British Columbia, the Australian National University, and Johns Hopkins University School of Medicine, among others. Dr. Sanberg is an inventor on approximately 30 health-related patents issued by the U.S. Patent and Trademark Office (USPTO), and numerous foreign patents. His research has focused on discovering innovative ways to repair damaged brain, and has helped lead the team that demonstrated that bone marrow and umbilical cord blood-derived stem cells can be transformed to neural cells that may be useful in stroke, spinal cord injury, and ALS. Dr. Sanberg is the founder of the National Academy of Inventors (at the University of South Florida) and currently serves as its president.

Dr. Jun Tan received his Bachelors of Medicine in China in 1983. He earned his M.S. at Fudan University in 1989 and Ph.D. in 1992, as well as postdoctoral studies at the University of Michigan in 1998. He became Assistant Professor of Psychiatry at USF, Associate Professor in 2004, and Professor in 2007. He is the author of 110 original scientific papers in prestigious journals such as Science, Nature Neuroscience, Nature Medicine, Nature Commutations, EMBO Journal, and Proceedings of the National Academy of Sciences. He is the recipient of multiple grants from the NIH totaling $10 million. In 2007, Dr. Tan was named the Robert A. Silver Endowed Chair in Developmental Neurobiology.
References
- 1.Aggarwal NT, Bienias JL, Bennett DA, Wilson RS, Morris MC, Schneider JA, Shah RC, Evans DA. The relation of cigarette smoking to incident Alzheimer’s disease in a biracial urban community population. Neuroepidemiology. 2006;26(3):140–146. doi: 10.1159/000091654. [DOI] [PubMed] [Google Scholar]
- 2.Almeida OP, Garrido GJ, Lautenschlager NT, Hulse GK, Jamrozik K, Flicker L. Smoking is associated with reduced cortical regional gray matter density in brain regions associated with incipient Alzheimer disease. Am J Geriatr Psychiatry. 2008;16(1):92–98. doi: 10.1097/JGP.0b013e318157cad2. [DOI] [PubMed] [Google Scholar]
- 3.Almeida OP, Hulse GK, Lawrence D, Flicker L. Smoking as a risk factor for Alzheimer’s disease: contrasting evidence from a systematic review of case-control and cohort studies. Addiction. 2002;97(1):15–28. doi: 10.1046/j.1360-0443.2002.00016.x. [DOI] [PubMed] [Google Scholar]
- 4.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko MK, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 5.Boerrigter ME, Wei JY, Vijg J. DNA repair and Alzheimer’s disease. J Gerontol. 1992;47(6):B177–B184. doi: 10.1093/geronj/47.6.b177. [DOI] [PubMed] [Google Scholar]
- 6.Bradt BM, Kolb WP, Cooper NR. Complement-dependent proinflammatory properties of the Alzheimer’s disease beta-peptide. J Exp Med. 1998;188(3):431–438. doi: 10.1084/jem.188.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007;3(3):186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 8.Cataldo JK, Prochaska JJ, Glantz SA. Cigarette smoking is a risk factor for Alzheimer’s disease: An analysis controlling for tobacco industry affiliation. J Alzheimers Dis. 2010;19:465–480. doi: 10.3233/JAD-2010-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Citron M, Diehl TS, Gordon G, Biere AL, Seubert P, Selkoe DJ. Evidence that the 42- and 40-amino acid forms of amyloid beta protein are generated from the beta-amyloid precursor protein by different protease activities. Proc Natl Acad Sci USA. 1996;93(23):13170–13175. doi: 10.1073/pnas.93.23.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Debanne SM, Bielefeld RA, Cheruvu VK, Fritsch T, Rowland DY. Alzheimer’s disease and smoking: Bias in cohort studies. J Alzheimers Dis. 2007;11(3):313–321. doi: 10.3233/jad-2007-11308. [DOI] [PubMed] [Google Scholar]
- 11.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 12.Euser SM, Schram MT, Hofman A, Westendorp RG, Breteler MM. Measuring cognitive function with age: The influence of selection by health and survival. Epidemiology. 2008;19(3):440–447. doi: 10.1097/EDE.0b013e31816a1d31. [DOI] [PubMed] [Google Scholar]
- 13.Flicker L. Modifiable lifestyle risk factors for Alzheimer’s disease. J Alzheimers Dis. 2010;20(3):803–811. doi: 10.3233/JAD-2010-091624. [DOI] [PubMed] [Google Scholar]
- 14.Fratiglioni L, Wang HX. Smoking and Parkinson’s and Alzheimer’s disease: Review of the epidemiological studies. Behav Brain Res. 2000;113(1–2):117–120. doi: 10.1016/s0166-4328(00)00206-0. [DOI] [PubMed] [Google Scholar]
- 15.Funamoto S, Morishima-Kawashima M, Tanimura Y, Hirotani N, Saido TC, Ihara Y. Truncated carboxyl-terminal fragments of beta-amyloid precursor protein are processed to amyloid beta-proteins 40 and 42. Biochemistry. 2004;43(42):13532–13540. doi: 10.1021/bi049399k. [DOI] [PubMed] [Google Scholar]
- 16.Gao S, Chen K, Zhao Y, Rich CB, Chen L, Li SJ, Toselli P, Stone P, Li W. Transcriptional and posttranscriptional inhibition of lysyl oxidase expression by cigarette smoke condensate in cultured rat fetal lung fibroblasts. Toxicol Sci. 2005;87(1):197–203. doi: 10.1093/toxsci/kfi212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giunta B, Ehrhart J, Townsend K, Sun N, Vendrame M, Shytle D, Tan J, Fernandez F. Galantamine and nicotine have a synergistic effect on inhibition of microglial activation induced by HIV-1 gp120. Brain Res Bull. 2004;64(2):165–170. doi: 10.1016/j.brainresbull.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Golde TE, Eckman CB, Younkin SG. Biochemical detection of Abeta isoforms: Implications for pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Biochim Biophys Acta. 2000;1502(1):172–187. doi: 10.1016/s0925-4439(00)00043-0. [DOI] [PubMed] [Google Scholar]
- 19.Greenland S. Quantifying biases in causal models: Classical confounding vs collider-stratification bias. Epidemiology. 2003;14(3):300–306. [PubMed] [Google Scholar]
- 20.Guralnik JM. Assessing the impact of comorbidity in the older population. Ann Epidemiol. 1996;6(5):376–380. doi: 10.1016/s1047-2797(96)00060-9. [DOI] [PubMed] [Google Scholar]
- 21.Hernán MA, Hernández-Díaz S, Robins JM. A structural approach to selection bias. Epidemiology. 2004;15(5):615–625. doi: 10.1097/01.ede.0000135174.63482.43. [DOI] [PubMed] [Google Scholar]
- 22.Hernán MA, Alonso A, Logroscino G. Cigarette smoking and dementia: Potential selection bias in the elderly. Epidemiology. 2008;19(3):448–450. doi: 10.1097/EDE.0b013e31816bbe14. [DOI] [PubMed] [Google Scholar]
- 23.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA. 1994;91(8):3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huse JT, Doms RW. Closing in on the amyloid cascade: Recent insights into the cell biology of Alzheimer’s disease. Mol Neurobiol. 2000;22(1–3):81–98. doi: 10.1385/MN:22:1-3:081. [DOI] [PubMed] [Google Scholar]
- 25.Juan D, Zhou DH, Li J, Wang JY, Gao C, Chen M. A 2-year follow-up study of cigarette smoking and risk of dementia. Eur J Neurol. 2004;11(4):277–282. doi: 10.1046/j.1468-1331.2003.00779.x. [DOI] [PubMed] [Google Scholar]
- 26.Kukull WA. The association between smoking and Alzheimer’s disease: Effects of study design and bias. Biol Psychiatry. 2001;49(3):194–199. doi: 10.1016/s0006-3223(00)01077-5. [DOI] [PubMed] [Google Scholar]
- 27.LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer’s A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet. 1995;9(1):21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- 28.Launer LJ, Andersen K, Dewey ME, Letenneur L, Ott A, Amaducci LA, Brayne C, Copeland JR, Dartigues JF, Kragh-Sorensen P, Lobo A, Martinez-Lage JM, Stijnen T, Hofman A. Rates and risk factors for dementia and Alzheimer’s disease: Results from EURODEM pooled analyses. EURODEM Incidence Research Group and Work Groups. European Studies of Dementia. Neurology. 1999;52(1):78–84. doi: 10.1212/wnl.52.1.78. [DOI] [PubMed] [Google Scholar]
- 29.Launer LJ, Masaki K, Petrovitch H, Foley D, Havlik RJ. The association between midlife blood pressure levels and late-life cognitive function. The Honolulu-Asia Aging Study. JAMA. 1995;274(23):1846–1851. [PubMed] [Google Scholar]
- 30.Letenneur L, Larrieu S, Barberger-Gateau P. Alcohol and tobacco consumption as risk factors of dementia: A review of epidemiological studies. Biomed Pharmacother. 2004;58(2):95–99. doi: 10.1016/j.biopha.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 31.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90(17):7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merchant C, Tang MX, Albert S, Manly J, Stern Y, Mayeux R. The influence of smoking on the risk of Alzheimer’s disease. Neurology. 1999;52(7):1408–1412. doi: 10.1212/wnl.52.7.1408. [DOI] [PubMed] [Google Scholar]
- 33.Moktar A, Singh R, Vadhanam MV, Ravoori S, Lillard JW, Gairola CG, Gupta RC. Cigarette smoke condensate-induced oxidative DNA damage and its removal in human cervical cancer cells. Int J Oncol. 2011;39(4):941–947. doi: 10.3892/ijo.2011.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murakami K, Irie K, Ohigashi H, Hara H, Nagao M, Shimizu T, Shirasawa T. Formation and stabilization model of the 42-mer Abeta radical: Implications for the long-lasting oxidative stress in Alzheimer’s disease. J Am Chem Soc. 2005;127(43):15168–15174. doi: 10.1021/ja054041c. [DOI] [PubMed] [Google Scholar]
- 35.Ott A, Slooter AJ, Hofman A, van Harskamp F, Witteman JC, Van Broeckhoven C, van Duijn CM, Breteler MM. Smoking and risk of dementia and Alzheimer’s disease in a population-based cohort study: The Rotterdam Study. Lancet. 1998;351(9119):1840–1843. doi: 10.1016/s0140-6736(97)07541-7. [DOI] [PubMed] [Google Scholar]
- 36.Peters R, Poulter R, Warner J, Beckett N, Burch L, Bulpitt C. Smoking, dementia and cognitive decline in the elderly, a systematic review. BMC Geriatr. 2008;8:36. doi: 10.1186/1471-2318-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poirier J, Delisle MC, Quirion R, Aubert I, Farlow M, Lahiri D, Hui S, Bertrand P, Nalbantoglu J, Gilfix BM, Gauthier S. Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Natl Acad Sci USA. 1995;92(26):12260–12264. doi: 10.1073/pnas.92.26.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reitz C, Tang MX, Schupf N, Manly JJ, Mayeux R, Luchsinger JA. A summary risk score for the prediction of Alzheimer disease in elderly persons. Arch Neurol. 2010;67(7):835–841. doi: 10.1001/archneurol.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reitz C, den Heijer T, van Duijn C, Hofman A, Breteler MM. Relation between smoking and risk of dementia and Alzheimer disease: The Rotterdam Study. Neurology. 2007;69(10):998–1005. doi: 10.1212/01.wnl.0000271395.29695.9a. [DOI] [PubMed] [Google Scholar]
- 40.Rusanen M, Kivipelto M, Quesenberry CP, Jr, Zhou J, Whitmer RA. Heavy smoking in midlife and long-term risk of Alzheimer disease and vascular dementia. Arch Intern Med. 2011;171(4):333–339. doi: 10.1001/archinternmed.2010.393. [DOI] [PubMed] [Google Scholar]
- 41.Sambamurti K, Greig NH, Lahiri DK. Advances in the cellular and molecular biology of the betaamyloid protein in Alzheimer’s disease. Neuromolec Med. 2002;1(1):1–31. doi: 10.1385/NMM:1:1:1. [DOI] [PubMed] [Google Scholar]
- 42.Sasco AJ, Secretan MB, Straif K. Tobacco smoking and cancer: A brief review of recent epidemiological evidence. Lung Cancer. 2004;45(Suppl 2):S3–S9. doi: 10.1016/j.lungcan.2004.07.998. [DOI] [PubMed] [Google Scholar]
- 43.Schenk DB, Rydel RE, May P, Little S, Panetta J, Lieberburg I, Sinha S. Therapeutic approaches related to amyloid-beta peptide and Alzheimer’s disease. J Med Chem. 1995;38(21):4141–4154. doi: 10.1021/jm00021a001. [DOI] [PubMed] [Google Scholar]
- 44.Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA. 1991;88(16):7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selkoe DJ. Alzheimer’s disease: Genotypes, phenotypes, and treatments. Science. 1997;275(5300):630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 46.Selkoe DL, Yamazaki T, Citron M, Podlisny MB, Koo EH, Teplow DB, Haass C. The role of APP processing and trafficking pathways in the formation of amyloid beta-protein. Ann NY Acad Sci. 1996;777:57–64. doi: 10.1111/j.1749-6632.1996.tb34401.x. [DOI] [PubMed] [Google Scholar]
- 47.Shalat SL, Seltzer B, Pidcock C, Baker EL., Jr Risk factors for Alzheimer’s disease: A case-control study. Neurology. 1987;37(10):1630–1633. doi: 10.1212/wnl.37.10.1630. [DOI] [PubMed] [Google Scholar]
- 48.Sinha S, Lieberburg I. Cellular mechanisms of beta-amyloid production and secretion. Proc Natl Acad Sci USA. 1999;96(20):11049–11053. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steele RH, Payne VM, Fulp CW, Rees DC, Lee CK, Doolittle DJ. A comparison of the mutagenicity of mainstream cigarette smoke condensates from a representative sample of the U.S. cigarette market with a Kentucky reference cigarette (K1R4F) Mutat Res. 1995;342(3–4):179–190. doi: 10.1016/0165-1218(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 50.Suo Z, Tan J, Placzek A, Crawford F, Fang C, Mullan M. Alzheimer’s beta-amyloid peptides induce inflammatory cascade in human vascular cells: The roles of cytokines and CD40. Brain Res. 1998;807(1–2):110–117. doi: 10.1016/s0006-8993(98)00780-x. [DOI] [PubMed] [Google Scholar]
- 51.Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo DG, Mattson MP, Tabaton M. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem. 2008;104(3):683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyas SL, White LR, Petrovitch H, Webster Ross G, Foley DJ, Heimovitz HK, Launer LJ. Mid-life smoking and late-life dementia: The Honolulu-Asia Aging Study. Neurobiol Aging. 2003;24(4):589–596. doi: 10.1016/s0197-4580(02)00156-2. [DOI] [PubMed] [Google Scholar]
- 53.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 54.Walsh DM, Klyubin I, Shankar GM, Townsend M, Fadeeva JV, Betts V, Podlisny MB, Cleary JP, Ashe KH, Rowan MJ, Selkoe DJ. The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33(Pt. 5):1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- 55.Wang HX, Fratiglioni L, Frisoni GB, Viitanen M, Winblad B. Smoking and the occurrence of Alzheimer’s disease: Cross-sectional and longitudinal data in a population-based study. Am J Epidemiol. 1999;149(7):640–644. doi: 10.1093/oxfordjournals.aje.a009864. [DOI] [PubMed] [Google Scholar]
- 56.Wang PN, Wang SJ, Hong CJ, Liu TT, Fuh JL, Chi CW, Liu CY, Liu HC. Risk factors for Alzheimer’s disease: A case-control study. Neuroepidemiology. 1997;16(5):234–240. doi: 10.1159/000109692. [DOI] [PubMed] [Google Scholar]
- 57.Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature. 1999;402(6761):533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- 58.Zamani MR, Allen YS. Nicotine and its interaction with beta-amyloid protein: A short review. Biol Psychiatry. 2001;49(3):221–232. doi: 10.1016/s0006-3223(00)01108-2. [DOI] [PubMed] [Google Scholar]