

Abstract
DNA replication origin activity changes during development. Chromatin modifications are known to influence the genomic location of origins and the time during S phase that they initiate replication in different cells. However, how chromatin regulates origins in concert with cell differentiation remains poorly understood. Here, we use developmental gene amplification in Drosophila ovarian follicle cells as a model to investigate how chromatin modifiers regulate origins in a developmental context. We find that the histone acetyltransferase (HAT) Chameau (Chm) binds to amplicon origins and is partially required for their function. Depletion of Chm had relatively mild effects on origins during gene amplification and genomic replication compared with previous knockdown of its ortholog HBO1 in human cells, which has severe effects on origin function. We show that another HAT, CBP (Nejire), also binds amplicon origins and is partially required for amplification. Knockdown of Chm and CBP together had a more severe effect on nucleosome acetylation and amplicon origin activity than knockdown of either HAT alone, suggesting that these HATs collaborate in origin regulation. In addition to their local function at the origin, we show that Chm and CBP also globally regulate the developmental transition of follicle cells into the amplification stages of oogenesis. Our results reveal a complexity of origin epigenetic regulation by multiple HATs during development and suggest that chromatin modifiers are a nexus that integrates differentiation and DNA replication programs.
Keywords: DNA replication, Histone acetylation, Gene amplification
INTRODUCTION
A central unanswered question in DNA replication is the mechanism by which multicellular eukaryotes select sites in their genome to act as origins (Mechali, 2010). A strict DNA consensus for origins has not been identified. Moreover, which loci are selected to be origins and when they initiate during S phase can change during development. In humans, origin misregulation is the basis for specific heritable developmental syndromes and contributes to genome instability and cancer (Bicknell et al., 2011a; Bicknell et al., 2011b; Guernsey et al., 2011; Hook et al., 2007; Shima et al., 2007). Evidence suggests that chromatin modification plays an important role in origin developmental plasticity, but the mechanism for origin specification and integration with development remains little understood. In this study, we investigate these questions using as a model the origins that mediate developmental gene amplification in the Drosophila ovary.
Origin DNA is bound by a pre-replicative complex (pre-RC) that is then activated to initiate replication during S phase (Remus and Diffley, 2009). Analysis of origins in S. cerevisiae identified a DNA consensus sequence for the binding site of the origin recognition complex (ORC), a component of the pre-RC. In multicellular eukaryotes, sites of pre-RC binding and replication initiation have been mapped genome-wide in a number of organisms, yet a strict DNA consensus for origins has not emerged (Bell et al., 2010; Cadoret, 2008; Cayrou et al., 2011; Eaton et al., 2011; Hiratani et al., 2008; MacAlpine et al., 2010; Schwaiger et al., 2009). Moreover, metazoan ORC has little binding specificity in vitro, except for a bias for poly(A)-poly(T) tracts and superhelical DNA (Bielinsky et al., 2001; Remus et al., 2004; Vashee et al., 2003). Despite this apparent lack of sequence specificity, replication initiation occurs at preferred genomic sites in vivo. Which sites are selected to be origins and the time that they initiate replication during S phase can both change during development (Hiratani et al., 2008; Mechali, 2010; Nordman and Orr-Weaver, 2012; Sasaki et al., 1999; Shinomiya and Ina, 1991). Despite recent advances, the mechanisms that determine differential origin usage during development remain largely undefined.
The developmental plasticity of origins provided early evidence that epigenetic mechanisms might play an important role in origin regulation in eukaryotes (Edenberg and Huberman, 1975; Hyrien et al., 1995; Shinomiya and Ina, 1991). Recent genomic analyses have shown a correlation between active origin loci and chromatin status, including nucleosome position, histone modification and histone variants (Bell et al., 2010; Cadoret, 2008; Cayrou et al., 2011; Eaton et al., 2011; Hiratani et al., 2008; MacAlpine et al., 2010; Muller et al., 2010; Schwaiger et al., 2009). Several studies have demonstrated that the acetylation of nucleosomes promotes ORC binding, active origin selection and early replication initiation during S phase (Aggarwal and Calvi, 2004; Danis et al., 2004; Hartl et al., 2007; Kim et al., 2011; Pappas et al., 2004; Schwaiger et al., 2009; Vogelauer et al., 2002). Moreover, a number of specific histone acetyltransferases (HATs) and histone deacetylases (HDACs) have been shown to influence origin activity (Aggarwal and Calvi, 2004; Doyon et al., 2006; Espinosa et al., 2010; Iizuka et al., 2006; Iizuka and Stillman, 1999; Karmakar et al., 2010; Miotto and Struhl, 2008; Miotto and Struhl, 2010; Pappas et al., 2004; Vogelauer et al., 2002; Wong et al., 2010). Nevertheless, how different HATs and HDACS regulate origins in concert with development remains poorly understood.
Early evidence for a role of histone acetylation in origin regulation came from analysis of developmental gene amplification in the Drosophila ovary (Aggarwal and Calvi, 2004; Hartl et al., 2007). Late in oogenesis, the somatic follicle cells surrounding the oocyte cease genomic replication and begin site-specific replication from origins at only six loci (Calvi, 2006; Kim et al., 2011). The reinitiation of replication from these origins results in the amplification of DNA copy number for genes involved in eggshell synthesis (Spradling, 1981). Similar to other origins, these amplicon origins are bound by the pre-RC and regulated by the cell cycle kinases CDK2 and CDC7 [Cdc2c and l(1)G0148 – FlyBase] (Calvi, 2006; Calvi et al., 1998; Claycomb and Orr-Weaver, 2005; Landis and Tower, 1999). Precisely at the onset of stage 10B, nucleosomes at amplicon origins become hyperacetylated, ORC binds and the origin becomes active (Aggarwal and Calvi, 2004; Austin et al., 1999). At the best-characterized origin, DAFC-66D, acetylation rapidly declines in stage 11/12, ORC departs and replication initiation ceases, but replication forks continue to migrate outwards (Aggarwal and Calvi, 2004; Austin et al., 1999). Evidence suggested that nucleosome acetylation contributes to amplicon origin locus specificity and the efficiency with which they initiate replication (Aggarwal and Calvi, 2004). We recently showed that multiple lysines on histones H3 and H4 are hyperacetylated at the amplicon origins, and that acetylation of certain lysines is dependent on different steps of pre-RC assembly (Liu et al., 2012). The acetylation on multiple lysines suggested that several HATs might regulate amplicon origins, but the identity of these HATs is not known. Moreover, it is not known how developmental signals integrate with chromatin modifiers to ensure amplicon activity at the proper time in oogenesis.
Here, we use gene amplification in the Drosophila ovary to investigate the epigenetic regulation of origins in a developmental context. We show that the HAT Chameau (Chm) is required for normal levels of amplification, but, unlike its human ortholog HBO1, Chm is not absolutely required for gene amplification or genomic replication. We further show that the HAT CBP (Nejire) collaborates with Chm to promote wild-type levels of gene amplification. Importantly, our data suggest that Chm and CBP might function both locally at the amplicon origins and globally in the developmental transition of follicle cells into the amplification stage of oogenesis. Our results imply that epigenetic mechanisms might have a more general role for the coordination of DNA replication programs with cell development.
MATERIALS AND METHODS
Drosophila strains and genetics
Standard techniques were used for culture of Drosophila melanogaster at 25°C. UAS:Flag-Mcm6 (Schwed et al., 2002) and c323:Gal4 (Manseau et al., 1997) and OregonRmodencode (Roy et al., 2010) were described previously. UAS:Myc-chm and chm14 strains were a gift from Y. Graba and J. Pradel (Grienenberger et al., 2002). Orc2dsRNA and chmdsRNA-2 were obtained from the Vienna Drosophila RNAi Center (Dietzl et al., 2007). chmdsRNA-1, chmshRNA, CBPdsRNA-3 and RNAi against mof, Taf1 and enok were obtained from the Transgenic RNAi Project (TRiP) via the Bloomington Drosophila Stock Center (BDSC) (Ni et al., 2008; Ni et al., 2011). RNAi against Pcaf (GCN5) was also from the BDSC (Carre et al., 2005). RNAi against Tip60 was a gift from F. Elefant (Zhu et al., 2007). CBPdsRNA-1, CBPdsRNA-2 and CBP mutant flies were gifts from J. Kumar (Kumar et al., 2004).
Immunolabeling and microscopy
Antibody, BrdU and DAPI labeling were performed as previously described (Calvi and Lilly, 2004). We used the following antibodies and concentrations: mouse anti-BrdU 1:20 (Becton Dickinson), rabbit anti-CBP 1:200 [gift from M. Mannervik (Lilja et al., 2003)], rabbit anti-H4K12ac 1:200 (Upstate Technologies), mouse anti-Cut 1:15 [Developmental Studies Hybridoma Bank (Blochlinger et al., 1990)]. Secondary antibodies Alexa 488 anti-rabbit, Alexa 488 anti-mouse, Alexa 568 anti-mouse and Alexa 633 anti-mouse were used at 1:500 (Invitrogen). Images were taken with a Leica DMRA2 widefield epifluorescence microscope, except for Fig. 5 and Fig. 6A, which were taken with a Leica SP5 laser scanning confocal microscope.
Fig. 5.

CBP associates with the amplification origin at DAFC-66D. (A-C) Stage 10B egg chamber containing CBPdsRNA-1-expressing clones co-labeled with anti-CBP (A green) and BrdU (B red). (C) Merged image of A and B shows co-localization of CBP and BrdU foci. Insets show higher magnification image of a single nucleus. (D) ChIP with anti-CBP from Oregon-R whole ovaries followed by qPCR for DAFC-66D as described in Fig. 1E. Error bars indicate represent the range of two biological replicates. Scale bars: 10 μm.
Fig. 6.
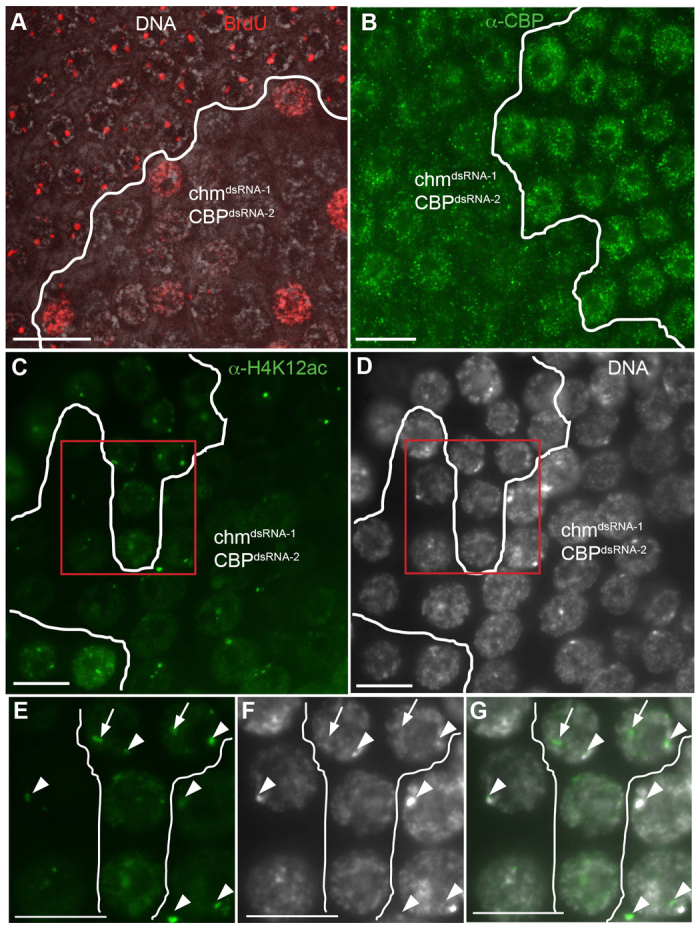
Combined knockdown of Chm and CBP causes a severe reduction in amplification. (A) chmdsRNA-1 and CBPdsRNA-2 were both expressed in clones using the Flp-Out system and labeled with BrdU. (B) chmdsRNA-1; CBPdsRNA-2 double-RNAi clones labeled with anti-CBP. (C) chmdsRNA-1; CBPdsRNA-2 double-RNAi clone labeled with anti-H4K12Ac. (D) The same clone from C labeled with DAPI. (E-G) Magnified images of nuclei from C and D (boxed regions). (E) Anti-H4K12Ac, (F) DAPI, (G) merged image. H4K12Ac foci corresponding to amplicons are indicated by arrows and the islands of H4K12Ac near the DAPI-bright heterochromatin are indicated by arrowheads. Scale bars: 10 μm.
Quantification and statistical analysis
BrdU and DAPI fluorescence were quantified using Openlab v5.0.2 (Improvision). For quantification of BrdU fluorescence, the brightest amplification focus in each nucleus was measured, which corresponds to DAFC-66D. Only nuclei containing visible BrdU foci were included. For both BrdU and DAPI fluorescence, 20 wild-type nuclei in each egg chamber were measured to determine the wild-type average fluorescence. Fluorescence intensity of nuclei in each mutant clone was normalized to the average wild-type fluorescence within the same egg chamber. Statistical significance was determined using an unpaired t-test, except for the quantification of chm14 clone size compared with wild-type twin spot in Fig. 3B, which used a paired t-test.
Fig. 3.

Compromised Chm function has mild effects on cell proliferation and genomic replication. (A) The number of nuclei in Flp-Out clones. n=9 stage 7-9 egg chambers representing more than 700 cells per genotype. (B) The number of nuclei in homozygous chm14 clones compared with their homozygous wild-type ‘twin spot’ (GFP/GFP). n=21 egg chambers and more than 400 cells per genotype. (C) To assess polyploid DNA content after endocycles, total DAPI fluorescence was measured within stage 10B follicle cells with reduced Chm (red bars) relative to wild-type cells (blue bars) within the same egg chamber. Error bars indicate s.e.m. (D) BrdU labeling of endocycling chm14 cells from a stage 9 egg chamber. Scale bar: 25 μm.
Generation of Chm antibody
The unique N-terminal 114 amino acids of Chm were cloned into pGEX-6P-1 (GE Healthcare) and expressed in competent E. coli BL-21 cells (Stratagene). GST-Chm1-114 was purified with Glutathione Sepharose 4B beads (GE Healthcare), then eluted off the beads and sent to Covance Research Products for injection into rabbits. Chm1-114 was then removed from GST by PreScission Protease (GE Healthcare) cleavage, conjugated to Affigel-10 (BioRad), and used for affinity purification of anti-Chm antibody from serum.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described (Negre et al., 2006). Whole ovaries were isolated by blending well-conditioned females in cold PBS/0.02% Tween 20. Ovaries were allowed to settle for 2-3 minutes, then separated from remaining fly tissue using wire mesh filters. Ovaries were then fixed for 15 minutes in 1.8% paraformaldehyde, during which cells were lysed using a Potter homogenizer and dounce homogenizer (Kontes). This was followed by quenching with 225 mM glycine. Lysates were sonicated to a modal length of 500 bp. Immunoprecipitations were performed overnight at 4°C with primary antibody, then incubated for 2 hours with Protein A agarose beads (Invitrogen). Beads were centrifuged and washed four times with lysis buffer, twice with Tris/EDTA, and eluted overnight at 65°C to reverse crosslinks. Eluted material and input were treated with 1 μg RNaseA (Roche 11119915001) and 50 μg proteinase K (New England Biolabs). DNA was purified by phenol/chloroform extraction and ethanol precipitated. qPCR analysis was performed as described (Liu et al., 2012) using the following primers (5′-3′, forward and reverse): ACE3, GCAGTGGCCTGAAAATTCTGCT and AGCTTAGTGCGGCAGTTTGGAA; Cp18, TCCCTCATACGGTGGTGGATA and CCGGAGTACTGAGATCCCACAT; Ori-β, GACTCTTCCAAATGGGAACCAC and CAACCGACCTGGAAACCATTAC; ACE3+10kb, GAGACAAGAGGACCAGCCCATC and GAAAGGGAACCCAAAGCAAACC; Rh2, AGGACCTCAATGGGTTGAGAGA and CCATGGTTGGAGTAAATGACCA.
RESULTS
The Drosophila ortholog of HBO1, Chm, interacts with the MCM complex in ovarian follicle cells
The unit of egg production during Drosophila oogenesis is the egg chamber (Fig. 1A) (Bastock and St Johnston, 2008; Spradling, 1993). Egg chambers consist of one oocyte and 15 sister germline nurse cells surrounded by an epithelial sheet of somatic follicle cells. Egg chambers proceed through 14 defined stages of development as they migrate down a structure called the ovariole (King, 1970). The somatic follicle cells undergo transitions in the cell cycle program in coordination with egg chamber development. Two follicle cell stem cells (FSCs) reside in the anterior of the ovariole in a structure called the germarium (Margolis and Spradling, 1995; Nystul and Spradling, 2007). FSC daughter cells proliferate and then form an epithelial sheet around the germline cyst, which together bud off as a stage 1 egg chamber. Follicle cells increase in number by a canonical mitotic division cycle before stage 7, and then switch into an endocycle comprising G and S phases without mitosis (Deng et al., 2001; Lopez-Schier and St Johnston, 2001; Mahowald et al., 1979; Shcherbata et al., 2004). Follicle cells undergo three endocycles during stage 7-10A and then in stage 10B enter the amplification stage during which only select loci reinitiate DNA replication (Calvi, 2006; Calvi et al., 1998; Claycomb and Orr-Weaver, 2005; Spradling and Mahowald, 1980).
Fig. 1.
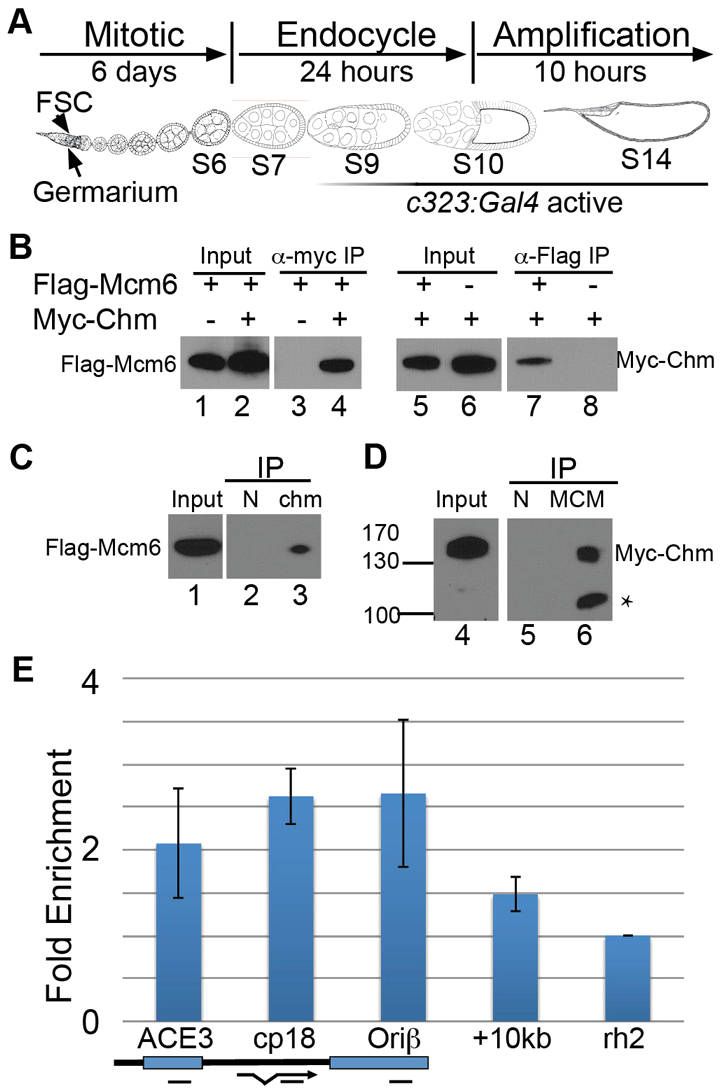
Chm interacts with the MCM complex and binds the DAFC-66D origin in amplification stage follicle cells. (A) Drosophila egg chamber development during oogenesis. Development proceeds from left to right as shown. The somatic follicle cells surrounding each egg chamber originate from follicle stem cells (FSCs) in the germarium, proliferate via the mitotic cycle in stages 1-6, proceed through three endocycles in stages 7-10A, and then amplify specific loci in stages 10B-14. The stages during which c323:Gal4 becomes increasingly active are indicated beneath by a graded line. (B) Myc-Chm interacts with Flag-Mcm6. UAS:Myc-chm and/or UAS:Flag-Mcm6 were expressed in stage 9-14 follicle cells using c323:Gal4. Ovary lysates were immunoprecipitated (IP) with antibodies against Myc (lanes 3 and 4) or Flag (lanes 7 and 8) and analyzed by western blot against either Flag-Mcm6 (lanes 1-4) or Myc-Chm (lanes 5-8). The presence (+) or absence (−) of Flag-Mcm6 and Myc-Chm in the different fly strains is indicated above each lane. (C) Endogenous Chm interacts with Flag-Mcm6. c323:Gal4; UAS:Flag-Mcm6 ovary lysates were immunoprecipitated with anti-Chm (lane 3) or normal rabbit serum (N, lane 2) and western blots incubated with anti-Flag. (D) The endogenous MCM complex interacts with Myc-Chm. c323:Gal4; UAS:Myc-chm ovaries were immunoprecipitated with anti-pan-MCM (lane 6) or normal mouse serum (N, lane 5) and western blots incubated with anti-Myc. Asterisk indicates myc-Chm degradation product. (E) Myc-Chm binds the DAFC-66D origin in vivo. ChIP of c323:Gal4; UAS:Myc-chm whole-ovary lysates with anti-Myc. Enriched DNA was analyzed by qPCR using primers to ACE3, Ori-β and Cp18 regions of the DAFC-66D origin (map below), a region 10 kb distal from the core origin, and the negative control locus Rhodopsin 2 (Rh2). Fold enrichment in the pellet was normalized to input and to the Rh2 locus. Plotted values represent the average of two biological replicates and error bars represent the range. Enrichment was not observed with pre-immune serum (data not shown).
Nucleosome acetylation plays an important role in the developmental activation of the amplicon origins. To determine which HATs are responsible for this acetylation, we initially focused on the gene chameau (chm), the Drosophila ortholog of human HBO1 (KAT7 – Human Genome Nomenclature Committee). In human cells, HBO1 associates with CDT1 and other pre-RC subunits and acetylates nucleosomes at origins, which is required for subsequent binding of the hexameric minichromosome maintenance (MCM) helicase complex to origin DNA during pre-RC assembly (Doyon et al., 2006; Iizuka et al., 2006; Iizuka and Stillman, 1999; Miotto and Struhl, 2008; Miotto and Struhl, 2010). Human HBO1 protein also physically interacts with the Drosophila ortholog of the MCM subunit Mcm2 in a yeast two-hybrid assay (Burke et al., 2001). We had previously shown that Chm can stimulate gene amplification in Drosophila when tethered to an amplicon origin in vivo, suggesting that the function of HBO1/Chm in origin regulation might be conserved from flies to humans (Aggarwal and Calvi, 2004). It is not known, however, whether Chm normally regulates Drosophila origins during developmental gene amplification or genomic replication. Previous characterization of chm mutants in Drosophila showed that it is required as a transcriptional co-activator during metamorphosis and also influences heterochromatic silencing, but a role in DNA replication has not been explored (Grienenberger et al., 2002; Miotto et al., 2006).
To determine whether Chm regulates amplicon origins, we first tested whether Chm physically interacts with the MCM complex in Drosophila follicle cells during gene amplification. The UAS:Myc-chm and UAS:Flag-Mcm6 epitope-tagged transgenes were expressed in stage 9-14 follicle cells using the c323:Gal4 driver (Fig. 1A) (Grienenberger et al., 2002; Manseau et al., 1997). We had previously used c323:Gal4 and UAS:Flag-Mcm6 to show that Drosophila Mcm6 associates with the MCM complex and is required for developmental gene amplification (Schwed et al., 2002). Immunoprecipitation (IP) of Myc-Chm resulted in co-IP of Flag-Mcm6 protein (Fig. 1B, lane 4), and the reciprocal IP of Flag-Mcm6 resulted in co-IP of Myc-Chm (Fig. 1B, lane 7). In controls, anti-Myc did not result in IP of Flag-Mcm6 from fly strains lacking Myc-Chm, nor did anti-Flag result in IP of Myc-Chm without Flag-Mcm6 (Fig. 1B, lanes 3 and 8).
To examine endogenous Chm protein, we generated a polyclonal rabbit antibody against the unique N-terminal 114 amino acids of Chm. IP of endogenous Chm protein with this anti-Chm antibody resulted in co-IP of Flag-Mcm6 (Fig. 1C). IP of endogenous MCM proteins with a pan-MCM antibody that recognizes all six subunits of the MCM complex resulted in co-IP of Myc-Chm (Fig. 1D) (Klemm and Bell, 2001). We were unable to detect co-IP of Chm with the MCM complex in wild-type cells without overexpression. Together, these results suggest that Chm physically interacts with the MCM complex in follicle cells when amplification origins are active and nucleosomes at the origins are acetylated.
Myc-Chm associates with the DAFC-66D origin
We next examined whether Chm associates with amplicon origins in vivo. We used Myc-Chm for ChIP followed by qPCR for DAFC-66D amplicon origin DNA, normalized to input and the Rhodopsin 2 (Rh2) control locus. To achieve specificity for follicle cells late in oogenesis during amplification, we again used c323:Gal4 to drive expression of UAS:Myc-chm. ChIP with anti-Myc antibodies showed that Myc-Chm was enriched at DAFC-66D, including the ACE3 and Ori-β regions, which are both required in cis for origin function (Fig. 1E) (Heck and Spradling, 1990; Orr-Weaver and Spradling, 1986). Myc-Chm was also enriched within the body of the Cp18 gene, but was less enriched at a region 10 kb from ACE3 (Fig. 1E). This pattern of enrichment is consistent with the broad domain of histone acetylation that was previously defined at DAFC-66D (Kim et al., 2011; Liu et al., 2012). Together, these results suggest that Chm protein associates with the MCM complex and the DAFC-66D amplicon origin during gene amplification in follicle cells late in oogenesis.
Chm HAT activity is required for wild-type amplification levels
Our results suggested that Chm protein associates with and might directly regulate amplicon origins. To genetically test whether chm is required in the adult ovary, it was necessary to use conditional knockdown because chm homozygous null mutants are pupal lethal due to reduced expression of genes regulated by the JNK pathway (Grienenberger et al., 2002; Miotto et al., 2006). We therefore induced RNAi against chm by expressing UAS:hairpin RNA transgenes in follicle cell clones using a Flp recombinase-inducible Act:Gal4 (hereafter referred to as Flp-Out) (Pignoni and Zipursky, 1997). In this system, heat shock induction of hsp70:Flp recombinase results in activation of Act:Gal4 in a single founder cell, which after several days of cell proliferation results in a clone of cells in stage 10B egg chambers that expresses UAS:dsRed and the UAS:hairpin RNA. We then assayed developmental amplification in these cells by measuring BrdU incorporation into amplification foci using fluorescence microscopy, and compared it with control cells not expressing the UAS:hairpin RNA in the same egg chamber (Calvi and Lilly, 2004). As proof of principle, we created clones expressing dsRNA against the origin-binding protein Orc2, and analyzed amplification 5 days later. Similar to loss-of-function mutants of Orc2, knockdown of Orc2 by RNAi abolished BrdU incorporation into amplification foci (Fig. 2B) (Landis et al., 1997). RNAi knockdown of the pre-RC protein Double parked (Dup), which is the fly ortholog of Cdt1, also abolished amplification (data not shown), whereas clones expressing Gal4 alone had no effect (Fig. 2A).
Fig. 2.

Compromised Chm function reduces, but does not eliminate, amplification. (A-E) UAS:RNA hairpin transgenes corresponding to the indicated gene were expressed in follicle cell clones using a Flp-inducible Act:Gal4 (Flp-Out), and assayed for gene amplification by incorporation of BrdU, which labels sites of active amplification as foci. Gray, DAPI; red, anti-BrdU labeling. Clones expressing different hairpin RNA transgenes are outlined in each panel. (A) Act:Gal4 alone, (B) Orc2dsRNA, (C) chmdsRNA-1, (D) chmdsRNA-2, (E) chmshRNA. (F) Gene amplification in permanent chm14/chm14 stem cell clones was analyzed by BrdU labeling (red) and compared with control heterozygous chm14/GFP cells in the same egg chamber. (A-F) Follicle cells from stage 10B egg chambers are shown. Scale bars: 10 μm. (G) Quantification of BrdU fluorescence intensity at DAFC-66D in cells with reduced chm (red bars) relative to wild-type cells (blue bars) within the same egg chamber. n>40 nuclei from at least three separate egg chambers. **P<0.01, ***P<0.0001. Error bars indicate s.e.m.
We used three different Drosophila strains that contain Gal4-inducible hairpin RNA for knockdown of Chm. These included two separate dsRNA hairpins (dsRNA-1 and dsRNA-2) and one short hairpin microRNA (shRNA) (Dietzl et al., 2007; Ni et al., 2008; Ni et al., 2011). Clones expressing either chmdsRNA-1 or chmdsRNA-2 transgenes had prominent amplicon BrdU foci beginning in stage 10B (Fig. 2C,D). Quantification revealed, however, that there was a small but significant reduction in the intensity of BrdU foci within the chmdsRNA-expressing cells compared with control cells in the same egg chambers (Fig. 2G). By contrast, expression of the chmshRNA transgene resulted in a greater reduction in BrdU focal intensity, including some cells with no detectable BrdU foci (Fig. 2E,G). In addition, some cells within each clone had BrdU incorporation throughout the nucleus, indicating that the normal site-specific replication in these cells was disrupted (Fig. 2E). The RNAi results suggested that Chm is required for wild-type levels of developmental gene amplification.
RNAi knockdown of Chm with different hairpin transgenes resulted in different severities of phenotype. Although this could be due to greater knockdown by chmshRNA, it was possible that the more severe phenotype of chmshRNA was caused by the off-target knockdown of other genes. We could not distinguish between these possibilities with our Chm polyclonal antibody because it did not reliably detect endogenous Chm protein in fixed tissues (data not shown). Therefore, to further test whether Chm is required for developmental gene amplification, we analyzed a chm14 null allele, which is a ~8 kb deletion that removes the last three exons of the gene and disrupts the conserved Myst family HAT domain (Grienenberger et al., 2002). Since animals homozygous for this allele die during metamorphosis, we created an FRT chm14 chromosome and generated homozygous mutant clones in heterozygous chm14/chm+ Ubi:GFP animals. To maximize depletion of Chm protein, stage 10 follicle cells were assayed for amplification 10 days after heat induction of hsp70:Flp recombinase, at which point permanent GFP-negative clones are derived from a single homozygous chm14 FSC in the germarium (Margolis and Spradling, 1995). These chm14 homozygous mutant follicle cells had BrdU amplification foci of normal appearance beginning in stage 10B (Fig. 2F). Quantification of fluorescence intensity, however, showed that BrdU incorporation was slightly but significantly reduced in the chm14 homozygous cells compared with chm14 heterozygous cells in the same egg chamber (Fig. 2G). This mild phenotype for the chm14 null cells, which are derived from null stem cells after 10 days and ~8 cell divisions, was similar to that of chmdsRNA-1 and chmdsRNA-2. It is likely, therefore, that the more severe phenotype of chmshRNA is due to off-target effects, and thus this hairpin construct was not analyzed further. The combined results of this genetic mosaic analysis suggested that Chm is at least partially required for developmental gene amplification.
Chm knockdown has mild effects on cell proliferation and genomic DNA replication
Knockdown of HBO1 in human cells in culture results in severe defects in MCM loading at origins, which impairs DNA replication and cell proliferation (Doyon et al., 2006; Iizuka et al., 2006; Iizuka and Stillman, 1999; Miotto and Struhl, 2008; Miotto and Struhl, 2010). Therefore, we were surprised to observe large clones of follicle cells after RNAi knockdown and from the mutant chm14 FSC. This suggests that depletion of Chm does not significantly affect FSC divisions or the mitotic proliferation of follicle cells that occurs before stage 7 of oogenesis. To quantify this, we counted the number of cells in the Flp-Out dsRed+ transient clones expressing chmdsRNA-2, and compared them with control clones generated in animals that do not have a UAS:RNAi transgene. Although chmdsRNA-2-expressing clones had a wide range of cell numbers, this range was comparable to that of control clones, suggesting that depletion of Chm does not affect cell proliferation (Fig. 3A). However, the RNAi and FSC clones do not have a wild-type ‘twin spot’ to facilitate direct comparison of proliferation rates between Chm-depleted and wild-type cells in the same egg chambers. Therefore, we generated transient chm14 homozygous mutant clones, and compared the number of cells in these GFP-negative clones with the corresponding wild-type GFP+/GFP+ twin spots derived from the same recombination event. The number of cells in chm14 mutant clones was not significantly different from the twin spot controls (Fig. 3B). Together, the chm14 and RNAi results suggest that depletion of Chm does not have a detectable effect on the proliferation of follicle cells that occurs before stage 7 of oogenesis.
We also determined whether depletion of Chm compromises endoreplication of DNA during the three follicle cell endocycles that occur during stages 7-10A. During each endocycle, DNA endoreplication results in an approximate doubling of cellular DNA content and total DAPI fluorescence (Calvi et al., 1998; Mahowald et al., 1979; Maqbool et al., 2010). To assay final DNA content, we measured DAPI fluorescence in cells of stage 10B mutant clones and compared it with wild-type cells within the same egg chambers. Follicle cells within the permanent chm14 clones had similar DAPI fluorescence to wild-type cells (Fig. 3C). DAPI fluorescence was also similar between chmdsRNA-2 and control cells (Fig. 3C). To directly analyze endoreplication, we labeled ovaries with BrdU. Examination of endocycling follicle cells during stages 7-10A indicated that chm14 clones are indistinguishable from wild type in the appearance and fraction of BrdU-labeled cells (Fig. 3D). These data suggest that depletion of Chm has, at most, only a mild effect on endoreplication.
Knockdown of the Drosophila CBP/p300 ortholog Nejire impairs gene amplification
The biochemical results suggested that Chm associates with the MCM complex and amplicon origins, but genetic depletion did not completely eliminate amplification. We therefore considered the possibility that the function of Chm at the amplicon origins is partially redundant with other HATs. In support of this hypothesis, we recently found that numerous lysines on histones H3 and H4 are hyperacetylated during gene amplification, suggesting that multiple HATs might regulate origins in follicle cells (Liu et al., 2012). To determine whether other HATs regulate gene amplification, we expressed UAS hairpin RNAs against different HATs using the Flp-Out system. These included RNAi transgenes against Pcaf (GCN5), mof, enok, Taf1, Tip60 and CBP (see Materials and methods). From this candidate screen, we found that knockdown of Nejire (Nej), which is the Drosophila ortholog of the HAT CBP/p300, impaired amplification (Akimaru et al., 1997).
We used two strains that have the same UAS:CBP hairpin RNA inserted in different genomic positions and have been shown to disrupt normal eye development (Kumar et al., 2004). Expression of UAS:CBPdsRNA-1 severely reduced incorporation of BrdU into amplification foci, whereas UAS:CBPdsRNA-2 did not, the likely result of a genomic position effect on hairpin RNA expression (Fig. 4A,B,I). Consistent with the severity of phenotypes, UAS:CBPdsRNA-2 clones had reduced but detectable CBP protein, whereas CBP protein was undetectable in UAS:CBPdsRNA-1 clones (Fig. 4E,F). Expression of a third CBP hairpin RNA, UAS:CBPdsRNA-3, also severely disrupted amplification and greatly reduced CBP protein levels (Fig. 4C,G) (Ni et al., 2008). In addition, some cells in the UAS:CBPdsRNA-3 and UAS:CBPdsRNA-1 clones had BrdU labeling throughout the nucleus (Fig. 4A,C). We attempted to validate these RNAi results by clonal analysis of CBP loss-of-function alleles using the Flp/FRT system. Clones of the mild alleles CBP131 and CBPS342 did not have an effect on BrdU incorporation at amplicon loci (Fig. 4D,I; data not shown). CBP131 clones also had no effect on CBP protein levels (Fig. 4H). We could not recover follicle cell clones of more severe CBP alleles, indicating that they are cell lethal and precluding our ability to test their effect on gene amplification (data not shown). Nonetheless, the RNAi results suggested that CBP is required for normal developmental gene amplification, a result that we pursue further below.
Fig. 4.

RNAi knockdown of CBP reduces amplification. (A-C,E-G) Three separate hairpin RNA transgenes against CBP were expressed in clones using the Flp-Out system and labeled with BrdU (red, A-C) or anti-CBP (green, E-G). (D) CBP131 clones were generated by Flp/FRT and labeled with BrdU. (H) CBP131 clones labeled with anti-CBP. (I) Quantification of BrdU fluorescence intensity in UAS:hairpin RNA-expressing clones. n>40 nuclei from at least three egg chambers. ***P<0.0001. Error bars indicate s.e.m. Scale bars: 10 μm.
CBP associates with the DAFC-66D origin
To test whether CBP functions locally at amplicon origins, we determined whether CBP co-localizes with BrdU amplification foci in follicle cells. Ovaries clonally expressing CBPdsRNA-1 were labeled for BrdU and CBP (Lilja et al., 2003). Wild-type stage 10B follicle cells had anti-CBP labeling throughout the nucleus, with a significantly brighter focus of CBP labeling. This focus co-localized with the brightest BrdU amplification focus, which corresponds to DAFC-66D (Fig. 5A-C) (Calvi et al., 1998). In the neighboring CBPdsRNA-1-expressing cells, both the overall levels and focal labeling of CBP at DAFC-66D were reduced or eliminated, indicating that focal anti-CBP labeling in wild-type cells reflects enrichment of CBP protein at DAFC-66D.
To quantify the binding of CBP to DAFC-66D with higher resolution, we used anti-CBP antibodies for ChIP on whole ovaries from wild-type flies. This showed that CBP is 2-fold enriched at ACE3 relative to the Rh2 control locus (Fig. 5D). CBP enrichment was lower at Ori-β and within the body of the Cp18 gene, and was not enriched 10 kb from ACE3 (Fig. 5D). Since these experiments used chromatin from whole ovaries, this is a conservative estimate for CBP occupancy at DAFC-66D in amplification stage follicle cells. The combined imaging and ChIP results suggest that CBP associates with, and might act locally at, DAFC-66D.
Combined knockdown of chm and CBP compromises nucleosome acetylation and severely reduces amplification
Our results suggested that both Chm and CBP associate with the DAFC-66D amplicon origin and contribute to normal levels of gene amplification. To further explore whether both HATs contribute to amplification, we co-expressed hairpin RNA against Chm and CBP using the Flp-Out method. To examine possible synergism between these knockdowns, we chose to co-express chmdsRNA-1 and CBPdsRNA-2 because each of these hairpins on their own had little to no effect on gene amplification (Fig. 2C and Fig. 4B). By contrast, co-expression of both these RNAi hairpins together resulted in severe reductions in BrdU incorporation at amplicon foci. Foci were undetectable in most cells, and some cells had incorporation of BrdU throughout the nucleus (Fig. 6A). CBP protein levels were similar between the chmdsRNA-1; CBPdsRNA-2 double- and CBPdsRNA-2 single-knockdown cells, suggesting that the severe phenotype of the double knockdown was not caused by further reductions in CBP protein (Fig. 6B and Fig. 4F). The synergistic phenotype of Chm and CBP double RNAi further suggests that these HATs are both important for the regulation of developmental gene amplification.
To evaluate whether Chm and CBP are required for histone acetylation in follicle cells, we labeled clones expressing UAS:hairpin RNA transgenes with antibodies against histone H4 acetylated on lysine 12 (anti-H4K12Ac), a modification that we showed is greatly enriched at amplicon origins when they are active (Liu et al., 2012). Anti-H4K12Ac antibodies labels throughout follicle cell nuclei, with more prominent labeling at foci that correspond to the hyperacetylated and active amplicon origins (Fig. 6C-G) (Liu et al., 2012). In addition, anti-H4K12Ac brightly labels a focus that corresponds to a chromatin domain near the heterochromatic DAPI bright spot (Fig. 6C-G). Although knockdown of either HAT alone did not have a detectable effect on H4K12 acetylation (data not shown), co-expressing chmdsRNA-1; CBPdsRNA-2 reduced both total nuclear and focal labeling of H4K12ac at the amplicons, but did not affect the H4K12Ac focus near the DAPI-bright spot (Fig. 6C-G). Together, these results suggest that Chm and CBP collaborate to acetylate H4K12 and regulate amplification origins in follicle cells.
Knockdown of chm and CBP compromises the developmental transition to amplification
The imaging and ChIP results suggested that Chm and CBP proteins might act locally at the amplicon origins. However, knockdown of CBP resulted in nucleus-wide incorporation of BrdU in some cells, a cellular phenotype that was also frequent in the chm; CBP double knockdown (Fig. 4A,C and Fig. 6A). To explain this result, we considered the possibility that Chm and CBP might be globally required for the transition of follicle cells from endocycles to amplification during stage 10A/B. To address this, we used an antibody against the transcription factor Cut, which is normally undetectable during endocycles but increases to higher levels at the onset of amplification in stage 10B (Blochlinger et al., 1990; Sun and Deng, 2005). Cut expression in stage 10B egg chambers was slightly reduced by expression of chmdsRNA-1 or chmdsRNA-2, and not detectably reduced after expression of CBPdsRNA-2, all of which had only mild effects on amplification (Fig. 7A-C). By contrast, CBPdsRNA-1 single knockdown, which had a more severe amplification phenotype, reduced Cut labeling further (Fig. 7D). Importantly, the double knockdown with chmdsRNA-1; CBPdsRNA-2, each of which had mild effects on amplification on its own, had much lower levels of Cut, consistent with the strong effect of this double knockdown on amplification (Fig. 7E). chmdsRNA-1; CBPdsRNA-2 double-knockdown clones also showed an increase in the levels of Hindsight (Hnt; Pebbled – FlyBase), which is normally downregulated during the transition from endocycles to amplification (data not shown) (Sun and Deng, 2007).
Fig. 7.
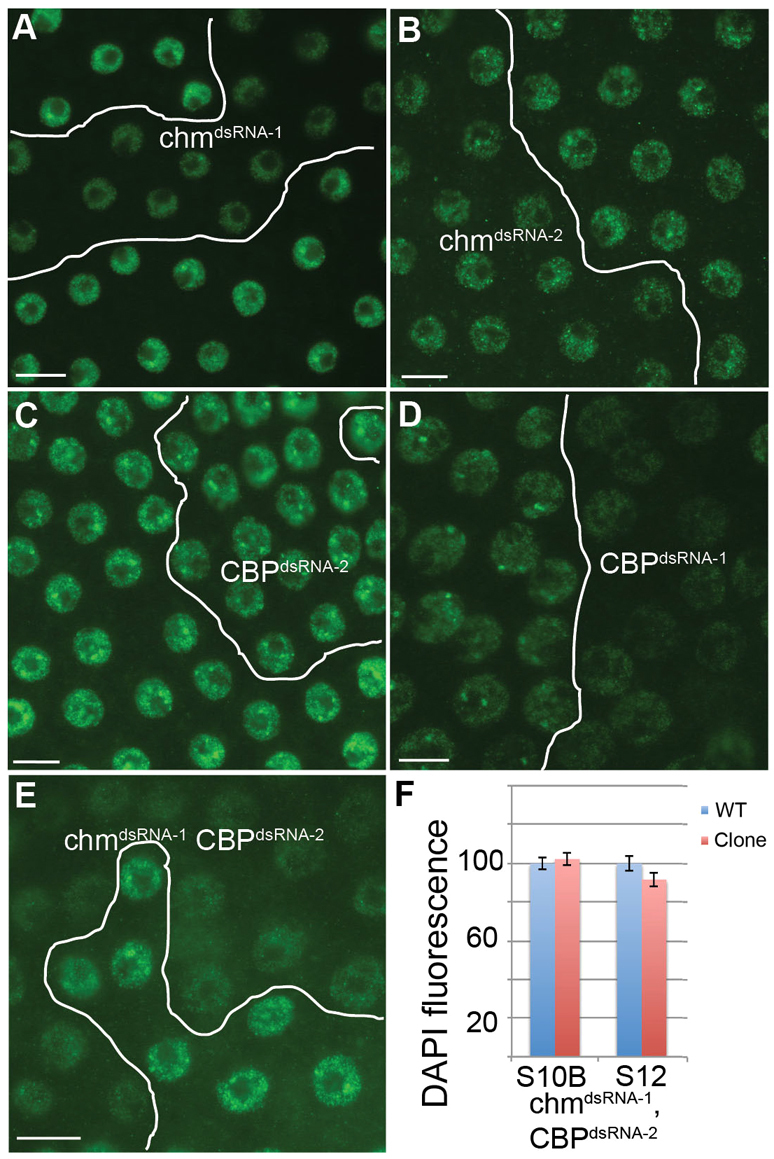
Knockdown of Chm and CBP perturbs the developmental transition to amplification. (A-E) Stage 10B follicle cell Flp-Out clones were labeled with anti-Cut (green). (A) chmdsRNA-1, (B) chmdsRNA-2, (C) CBPdsRNA-2, (D) CBPdsRNA-1, or (E) chmdsRNA-1; CBPdsRNA-2. (F) Quantification of total DAPI fluorescence in stage 10B and stage 12 follicle cells expressing chmdsRNA-1; CBPdsRNA-2 (red) compared with wild-type cells in the same egg chamber (blue). n=60 nuclei from three egg chambers. Error bars indicate s.e.m. Scale bars: 10 μm.
There are two possible explanations for persistent genomic replication in stage 10B. Either the normal three follicle cell endocycles are delayed relative to egg chamber development, or follicle cells are undergoing an additional fourth endocycle. DAPI fluorescence intensity was not significantly different in the chmdsRNA-1; CBPdsRNA-2 double-knockdown clones in stage 10B or stage 12 (Fig. 7F). Therefore, we could not distinguish whether follicle cells are just finishing a delayed third, or just beginning a new fourth, genomic endoreplication. In summary, the results suggest that Chm and CBP, in addition to acting locally at amplicons, might act globally to regulate the developmental transition of follicle cells from endocycles to gene amplification.
DISCUSSION
We investigated which HAT enzymes participate in the epigenetic regulation of gene amplification origins in the Drosophila ovary. We found that the HAT Chm, which is the Drosophila ortholog of human HBO1, is important for DNA replication programs in ovarian follicle cells. However, in stark contrast to the absolute requirement of HBO1 at mammalian origins, knockdown of chm did not completely eliminate gene amplification in follicle cells and had only mild effects on endoreplication and cell proliferation. Our results suggest that a second HAT, CBP, collaborates with Chm, and that both HATs act locally to acetylate nucleosomes at the origin to stimulate gene amplification. In addition to this local function, our data suggest that these HATs might also be globally required for follicle cells to undergo the developmental transition from endocycles into the amplification stage of oogenesis. These data imply that Chm and CBP integrate follicle cell development with DNA replication programs, and raise the possibility that the coordination of origin activity with cell differentiation by chromatin modifiers might be a general theme in development.
Acetylation at amplicon origins
We and others had shown that amplicon origins become hyperacetylated when they are active beginning in stage 10B, and that this acetylation is important for origin specification and efficiency (Aggarwal and Calvi, 2004; Hartl et al., 2007). However, which HATs are responsible for the developmental regulation of amplicon origins remained unknown. Our data suggest that the HAT Chm binds the MCM complex, associates with amplicon DNA, and is required for normal amplicon origin activity in follicle cells late in oogenesis (Fig. 8). These data are consistent with the requirement of HBO1 for loading the MCM complex during pre-RC formation in human cells and frog extracts (Doyon et al., 2006; Iizuka et al., 2006; Iizuka and Stillman, 1999; Miotto and Struhl, 2008; Miotto and Struhl, 2010). Furthermore, we have previously shown that tethering Chm directly to DAFC-66D promotes amplification (Aggarwal and Calvi, 2004). However, whereas depletion of HBO1 has severe effects on origin activity and cellular proliferation, the depletion of Chm did not completely eliminate amplification, had only mild effects on endoreplication, and no demonstrable effects on cell proliferation. One possibility is that we have not completely eliminated Chm activity. We were unable to directly assess the degree of knockdown among the different genotypes because our Chm antibody recognizes overexpressed protein but does not detect endogenous levels of Chm. Moreover, because the knockdown was only in a subset of follicle cells, we were unable to assess the relative levels of chm mRNA by qPCR. Importantly, the chm14 allele, which deletes essential regions of the Myst family HAT domain, had mild effects on DNA replication in follicle cell clones, and imaginal discs of homozygous chm14 larvae also did not have defects in cell proliferation (data not shown). Together, our data suggest that, although Chm associates with and regulates origins similar to its mammalian ortholog HBO1, Chm might not be absolutely essential for the function of all origins.
Fig. 8.
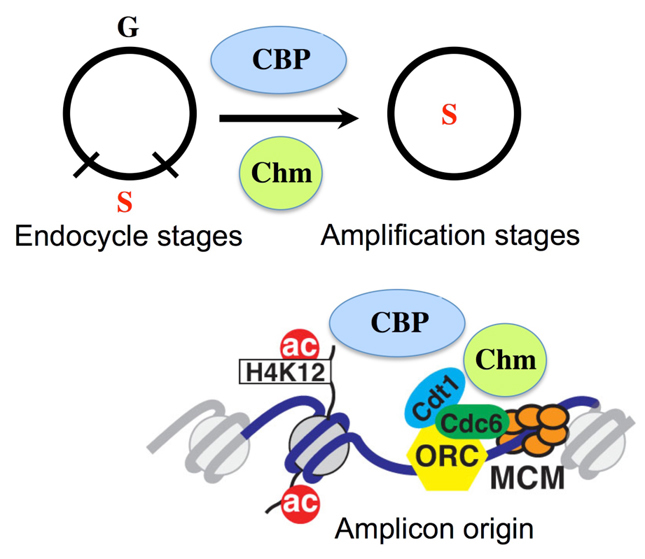
Model for regulation of developmental gene amplification by Chm and CBP. Chm and CBP are globally required for the developmental transition of follicle cells from endocycles to amplification stages (above). In addition, Chm and CBP are locally required during amplification stages for acetylation of H4K12 and for activity of the amplicon origins. See text for details.
The relatively mild phenotype of chm mutants raised the possibility that there is a developmental complexity of origin regulation by multiple HATs that has not been revealed by the analysis of cells in culture. In agreement with this interpretation, we found that the HAT CBP also associated with amplicon origin DAFC-66D, and that CBP knockdown partially compromised gene amplification (Fig. 8). While some of the hairpin RNAs against Chm and CBP had mild phenotypes, using these hairpin RNAs to knockdown both HATs together resulted in a synergistic, more severe replication phenotype and also reduced acetylation of H4K12. One interpretation is that these HATs are partially redundant in function or contribute in parallel in a non-additive way to origin activity. This idea is consistent with recent evidence that the level of nucleosome hyperacetylation quantitatively correlates with the number of initiation events at different amplicon origins (Kim et al., 2011; Liu et al., 2012). Our recent finding that a multitude of lysines are hyperacetylated on nucleosomes when these origins are active also raises the possibility that other HATs regulate amplicon origins and await discovery. An important future goal is to identify the cadre of HATs and HDACS at these origins and determine how the balance of their activity regulates origin specificity and developmental timing.
Although our data suggest that both Chm and CBP have a local function at amplicon origins, the mechanism by which acetylation promotes origin activity remains unclear. Our previous evidence suggested that nucleosome acetylation correlates with the locus specificity of ORC binding, but recent evidence also suggests that acetylation on some lysines occurs during pre-RC assembly and is downstream of, and depends on, ORC binding (Aggarwal and Calvi, 2004; Liu et al., 2012). It is also possible that Chm or CBP regulates the origin by acetylating pre-RC or other non-histone proteins, consistent with recent evidence from other systems (Glozak and Seto, 2009; Iizuka et al., 2006). Another important question is how these HATs are recruited specifically to the amplicon origins. Two transcription factor complexes have been shown to bind to the amplicons and to be required for normal levels of amplification: the Myeloblastosis-Multivulva B complex (Myb-MuvB) and the steroid hormone Ecdysone receptor-Ultraspiracle (EcR-Usp) (Beall et al., 2002; Hackney et al., 2007; Shea et al., 1990). It might be that one or both of these complexes mediates chromatin regulation of the origins by recruiting HATs and other chromatin modifiers. In fact, CBP is known to be a transcriptional co-activator of Myb in both Drosophila and mammalian cells (Dai et al., 1996; Hou et al., 1997).
Integration of DNA replication programs with development
Our data suggest that CBP and Chm also contribute indirectly to amplicon origin activity by globally regulating the development of follicle cells (Fig. 8). The most severe knockdown of these HATs resulted in reduced expression of Cut and persistent Hnt, two developmental markers for the endocycle-to-amplification stage transition (Sun and Deng, 2005). One possibility is that Chm and CBP control the expression of genes that are important for this developmental transition, consistent with their known activity as transcriptional regulators from flies to humans (Chan and La Thangue, 2001; Doyon et al., 2006; Miotto et al., 2006; Miotto and Struhl, 2006). The evidence for both local and global activity of these HATs suggest that they integrate the activity of amplicon origins with follicle cell development.
Our results using the model amplicon origins might have wider implications for understanding how plastic origin activity is coordinated with development. DNA replication programs can vary widely in development, with both the genomic location of origins and the time that they initiate during S phase differing among cells (Mechali, 2010; Nordman and Orr-Weaver, 2012). These variations in DNA replication mirror changes in the modification of the epigenome during development and underscore the important contribution of acetylation and other chromatin modifications to origin activity. Highly relevant to our results, mammalian CBP, as part of the DARRT complex, is required for transcription of the β-globin genes and for the initiation of DNA replication at the β-globin locus early during S phase in erythroid cells (Aladjem, 2007; Dhar et al., 1988; Karmakar et al., 2010). Disruption of the DARRT complex not only prevents transcription and origin function locally, but also affects cell differentiation globally (Karmakar et al., 2010). In light of this and other emerging evidence in the field, our results raise the possibility that epigenetic regulators actively coordinate DNA replication programs with cell differentiation as a general theme during development.
Acknowledgements
We thank Y. Graba, J. Pradel, J. Kumar, F. Elefant, the Bloomington Drosophila Stock Center and the Vienna Drosophila RNAi Center for fly stocks; M. Mannervik for the generous gift of anti-CBP antibody; Jim Powers and the Indiana Light Microscopy and Imaging Center (LMIC) for assistance with confocal microscopy; and members of the B.R.C. laboratory for critical reading of the manuscript and helpful discussion.
Footnotes
Funding
This work was supported by the National Institutes of Health (NIH) [postdoctoral fellowship F32 GM080089 to K.H.M., R01 GM61290-11 to B.R.C.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
References
- Aggarwal B. D., Calvi B. R. (2004). Chromatin regulates origin activity in Drosophila follicle cells. Nature 430, 372-376 [DOI] [PubMed] [Google Scholar]
- Akimaru H., Chen Y., Dai P., Hou D. X., Nonaka M., Smolik S. M., Armstrong S., Goodman R. H., Ishii S. (1997). Drosophila CBP is a co-activator of cubitus interruptus in hedgehog signalling. Nature 386, 735-738 [DOI] [PubMed] [Google Scholar]
- Aladjem M. I. (2007). Replication in context: dynamic regulation of DNA replication patterns in metazoans. Nat. Rev. Genet. 8, 588-600 [DOI] [PubMed] [Google Scholar]
- Austin R. J., Orr-Weaver T. L., Bell S. P. (1999). Drosophila ORC specifically binds to ACE3, an origin of DNA replication control element. Genes Dev. 13, 2639-2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastock R., St Johnston D. (2008). Drosophila oogenesis. Curr. Biol. 18, R1082-R1087 [DOI] [PubMed] [Google Scholar]
- Beall E. L., Manak J. R., Zhou S., Bell M., Lipsick J. S., Botchan M. R. (2002). Role for a Drosophila Myb-containing protein complex in site-specific DNA replication. Nature 420, 833-837 [DOI] [PubMed] [Google Scholar]
- Bell O., Schwaiger M., Oakeley E. J., Lienert F., Beisel C., Stadler M. B., Schubeler D. (2010). Accessibility of the Drosophila genome discriminates PcG repression, H4K16 acetylation and replication timing. Nat. Struct. Mol. Biol. 17, 894-900 [DOI] [PubMed] [Google Scholar]
- Bicknell L. S., Bongers E. M., Leitch A., Brown S., Schoots J., Harley M. E., Aftimos S., Al-Aama J. Y., Bober M., Brown P. A., et al. (2011a). Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat. Genet. 43, 356-359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicknell L. S., Walker S., Klingseisen A., Stiff T., Leitch A., Kerzendorfer C., Martin C. A., Yeyati P., Al Sanna N., Bober M., et al. (2011b). Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat. Genet. 43, 350-355 [DOI] [PubMed] [Google Scholar]
- Bielinsky A. K., Blitzblau H., Beall E. L., Ezrokhi M., Smith H. S., Botchan M. R., Gerbi S. A. (2001). Origin recognition complex binding to a metazoan replication origin. Curr. Biol. 11, 1427-1431 [DOI] [PubMed] [Google Scholar]
- Blochlinger K., Bodmer R., Jan L. Y., Jan Y. N. (1990). Patterns of expression of cut, a protein required for external sensory organ development in wild-type and cut mutant Drosophila embryos. Genes Dev. 4, 1322-1331 [DOI] [PubMed] [Google Scholar]
- Burke T. W., Cook J. G., Asano M., Nevins J. R. (2001). Replication factors MCM2 and ORC1 interact with the histone acetyltransferase HBO1. J. Biol. Chem. 276, 15397-15408 [DOI] [PubMed] [Google Scholar]
- Cadoret J. C. (2008). Genome-wide studies highlight indirect links between human replication origins and gene regulation. Proc. Natl. Acad. Sci. USA 105, 15837-15842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi B. R. (2006). Developmental DNA amplification. In DNA Replication and Human Disease (ed. DePamphilis M. L.), pp. 233-255 Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Calvi B. R., Lilly M. A. (2004). BrdU labeling and nuclear flow sorting of the Drosophila ovary. In Drosophila Cytogenetics Protocols (ed. Henderson D.), pp. 203-213 Totowa: Humana Press; [DOI] [PubMed] [Google Scholar]
- Calvi B. R., Lilly M. A., Spradling A. C. (1998). Cell cycle control of chorion gene amplification. Genes Dev. 12, 734-744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carre C., Szymczak D., Pidoux J., Antoniewski C. (2005). The histone H3 acetylase dGcn5 is a key player in Drosophila melanogaster metamorphosis. Mol. Cell. Biol. 25, 8228-8238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C., Coulombe P., Vigneron A., Stanojcic S., Ganier O., Peiffer I., Rivals E., Puy A., Laurent-Chabalier S., Desprat R., et al. (2011). Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 21, 1438-1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H. M., La Thangue N. B. (2001). p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 114, 2363-2373 [DOI] [PubMed] [Google Scholar]
- Claycomb J. M., Orr-Weaver T. L. (2005). Developmental gene amplification: insights into DNA replication and gene expression. Trends Genet. 21, 149-162 [DOI] [PubMed] [Google Scholar]
- Dai P., Akimaru H., Tanaka Y., Hou D. X., Yasukawa T., Kanei-Ishii C., Takahashi T., Ishii S. (1996). CBP as a transcriptional coactivator of c-Myb. Genes Dev. 10, 528-540 [DOI] [PubMed] [Google Scholar]
- Danis E., Brodolin K., Menut S., Maiorano D., Girard-Reydet C., Mechali M. (2004). Specification of a DNA replication origin by a transcription complex. Nat. Cell Biol. 6, 721-730 [DOI] [PubMed] [Google Scholar]
- Deng W. M., Althauser C., Ruohola-Baker H. (2001). Notch-Delta signaling induces a transition from mitotic cell cycle to endocycle in Drosophila follicle cells. Development 128, 4737-4746 [DOI] [PubMed] [Google Scholar]
- Dhar V., Mager D., Iqbal A., Schildkraut C. L. (1988). The coordinate replication of the human beta-globin gene domain reflects its transcriptional activity and nuclease hypersensitivity. Mol. Cell. Biol. 8, 4958-4965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzl G., Chen D., Schnorrer F., Su K. C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S., et al. (2007). A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151-156 [DOI] [PubMed] [Google Scholar]
- Doyon Y., Cayrou C., Ullah M., Landry A. J., Cote V., Selleck W., Lane W. S., Tan S., Yang X. J., Cote J. (2006). ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell 21, 51-64 [DOI] [PubMed] [Google Scholar]
- Eaton M. L., Prinz J. A., MacAlpine H. K., Tretyakov G., Kharchenko P. V., MacAlpine D. M. (2011). Chromatin signatures of the Drosophila replication program. Genome Res. 21, 164-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg H. J., Huberman J. A. (1975). Eukaryotic chromosome replication. Annu. Rev. Genet. 9, 245-284 [DOI] [PubMed] [Google Scholar]
- Espinosa M. C., Rehman M. A., Chisamore-Robert P., Jeffery D., Yankulov K. (2010). GCN5 is a positive regulator of origins of DNA replication in Saccharomyces cerevisiae. PLoS ONE 5, e8964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glozak M. A., Seto E. (2009). Acetylation/deacetylation modulates the stability of DNA replication licensing factor Cdt1. J. Biol. Chem. 284, 11446-11453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grienenberger A., Miotto B., Sagnier T., Cavalli G., Schramke V., Geli V., Mariol M. C., Berenger H., Graba Y., Pradel J. (2002). The MYST domain acetyltransferase Chameau functions in epigenetic mechanisms of transcriptional repression. Curr. Biol. 12, 762-766 [DOI] [PubMed] [Google Scholar]
- Guernsey D. L., Matsuoka M., Jiang H., Evans S., Macgillivray C., Nightingale M., Perry S., Ferguson M., LeBlanc M., Paquette J., et al. (2011). Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat. Genet. 43, 360-364 [DOI] [PubMed] [Google Scholar]
- Hackney J. F., Pucci C., Naes E., Dobens L. (2007). Ras signaling modulates activity of the ecdysone receptor EcR during cell migration in the Drosophila ovary. Dev. Dyn. 236, 1213-1226 [DOI] [PubMed] [Google Scholar]
- Hartl T., Boswell C., Orr-Weaver T. L., Bosco G. (2007). Developmentally regulated histone modifications in Drosophila follicle cells: initiation of gene amplification is associated with histone H3 and H4 hyperacetylation and H1 phosphorylation. Chromosoma 116, 197-214 [DOI] [PubMed] [Google Scholar]
- Heck M., Spradling A. (1990). Multiple replication origins are used during Drosophila chorion gene amplification. J. Cell Biol. 110, 903-914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratani I., Ryba T., Itoh M., Yokochi T., Schwaiger M., Chang C. W., Lyou Y., Townes T. M., Schubeler D., Gilbert D. M. (2008). Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6, e245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook S. S., Lin J. J., Dutta A. (2007). Mechanisms to control rereplication and implications for cancer. Curr. Opin. Cell Biol. 19, 663-671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou D. X., Akimaru H., Ishii S. (1997). Trans-activation by the Drosophila myb gene product requires a Drosophila homologue of CBP. FEBS Lett. 413, 60-64 [DOI] [PubMed] [Google Scholar]
- Hyrien O., Maric C., Mechali M. (1995). Transition in specification of embryonic metazoan DNA replication origins. Science 270, 994-997 [DOI] [PubMed] [Google Scholar]
- Iizuka M., Stillman B. (1999). Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J. Biol. Chem. 274, 23027-23034 [DOI] [PubMed] [Google Scholar]
- Iizuka M., Matsui T., Takisawa H., Smith M. M. (2006). Regulation of replication licensing by acetyltransferase Hbo1. Mol. Cell. Biol. 26, 1098-1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar S., Mahajan M. C., Schulz V., Boyapaty G., Weissman S. M. (2010). A multiprotein complex necessary for both transcription and DNA replication at the beta-globin locus. EMBO J. 29, 3260-3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. C., Nordman J., Xie F., Kashevsky H., Eng T., Li S., MacAlpine D. M., Orr-Weaver T. L. (2011). Integrative analysis of gene amplification in Drosophila follicle cells: parameters of origin activation and repression. Genes Dev. 25, 1384-1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King R. C. (1970). Ovarian Development in Drosophila melanogaster. New York: Academic Press; [Google Scholar]
- Klemm R. D., Bell S. P. (2001). ATP bound to the origin recognition complex is important for preRC formation. Proc. Natl. Acad. Sci. USA 98, 8361-8367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J. P., Jamal T., Doetsch A., Turner F. R., Duffy J. B. (2004). CREB binding protein functions during successive stages of eye development in Drosophila. Genetics 168, 877-893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis G., Tower J. (1999). The Drosophila chiffon gene is required for chorion gene amplification, and is related to the yeast Dbf4 regulator of DNA replication and cell cycle. Development 126, 4281-4293 [DOI] [PubMed] [Google Scholar]
- Landis G., Kelley R., Spradling A., Tower J. (1997). The k43 gene, required for chorion gene amplification and diploid cell chromosome replication, encodes the Drosophila homolog of yeast origin recognition complex subunit 2. Proc. Natl. Acad. Sci. USA 94, 3888-3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilja T., Qi D., Stabell M., Mannervik M. (2003). The CBP coactivator functions both upstream and downstream of Dpp/Screw signaling in the early Drosophila embryo. Dev. Biol. 262, 294-302 [DOI] [PubMed] [Google Scholar]
- Liu J., McConnell K., Dixon M., Calvi B. R. (2012). Analysis of model replication origins in Drosophila reveals new aspects of the chromatin landscape and its relationship to origin activity and the prereplicative complex. Mol. Biol. Cell 23, 200-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Schier H., St Johnston D. (2001). Delta signaling from the germ line controls the proliferation and differentiation of the somatic follicle cells during Drosophila oogenesis. Genes Dev. 15, 1393-1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine H. K., Gordan R., Powell S. K., Hartemink A. J., MacAlpine D. M. (2010). Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res. 20, 201-211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahowald A., Caulton J., Edwards M., Floyd A. (1979). Loss of centrioles and polyploidization in follicle cells of Drosophila melanogaster. Exp. Cell Res. 118, 404-410 [DOI] [PubMed] [Google Scholar]
- Manseau L., Baradaran A., Brower D., Budhu A., Elefant F., Phan H., Philp A. V., Yang M., Glover D., Kaiser K., et al. (1997). GAL4 enhancer traps expressed in the embryo, larval brain, imaginal discs, and ovary of Drosophila. Dev. Dyn. 209, 310-322 [DOI] [PubMed] [Google Scholar]
- Maqbool S. B., Mehrotra S., Kolpakas A., Durden C., Zhang B., Zhong H., Calvi B. R. (2010). Dampened activity of E2F1-DP and Myb-MuvB transcription factors in Drosophila endocycling cells. J. Cell Sci. 123, 4095-4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis J., Spradling A. (1995). Identification and behavior of epithelial stem cells in the Drosophila ovary. Development 121, 3797-3807 [DOI] [PubMed] [Google Scholar]
- Mechali M. (2010). Eukaryotic DNA replication origins: many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 11, 728-738 [DOI] [PubMed] [Google Scholar]
- Miotto B., Struhl K. (2006). Differential gene regulation by selective association of transcriptional coactivators and bZIP DNA-binding domains. Mol. Cell. Biol. 26, 5969-5982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B., Struhl K. (2008). HBO1 histone acetylase is a coactivator of the replication licensing factor Cdt1. Genes Dev. 22, 2633-2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B., Struhl K. (2010). HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol. Cell 37, 57-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B., Sagnier T., Berenger H., Bohmann D., Pradel J., Graba Y. (2006). Chameau HAT and DRpd3 HDAC function as antagonistic cofactors of JNK/AP-1-dependent transcription during Drosophila metamorphosis. Genes Dev. 20, 101-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P., Park S., Shor E., Huebert D. J., Warren C. L., Ansari A. Z., Weinreich M., Eaton M. L., MacAlpine D. M., Fox C. A. (2010). The conserved bromo-adjacent homology domain of yeast Orc1 functions in the selection of DNA replication origins within chromatin. Genes Dev. 24, 1418-1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre N., Lavrov S., Hennetin J., Bellis M., Cavalli G. (2006). Mapping the distribution of chromatin proteins by ChIP on chip. Methods Enzymol. 410, 316-341 [DOI] [PubMed] [Google Scholar]
- Ni J. Q., Markstein M., Binari R., Pfeiffer B., Liu L. P., Villalta C., Booker M., Perkins L., Perrimon N. (2008). Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster. Nat. Methods 5, 49-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J. Q., Zhou R., Czech B., Liu L. P., Holderbaum L., Yang-Zhou D., Shim H. S., Tao R., Handler D., Karpowicz P., et al. (2011). A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat. Methods 8, 405-407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordman J., Orr-Weaver T. L. (2012). Regulation of DNA replication during development. Development 139, 455-464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystul T., Spradling A. (2007). An epithelial niche in the Drosophila ovary undergoes long-range stem cell replacement. Cell Stem Cell 1, 277-285 [DOI] [PubMed] [Google Scholar]
- Orr-Weaver T., Spradling A. (1986). Drosophila chorion gene amplification requires an upstream region regulating s18 transcription. Mol. Cell. Biol. 6, 4624-4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas D. L., Jr, Frisch R., Weinreich M. (2004). The NAD(+)-dependent Sir2p histone deacetylase is a negative regulator of chromosomal DNA replication. Genes Dev. 18, 769-781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignoni F., Zipursky S. L. (1997). Induction of Drosophila eye development by decapentaplegic. Development 124, 271-278 [DOI] [PubMed] [Google Scholar]
- Remus D., Diffley J. F. (2009). Eukaryotic DNA replication control: Lock and load, then fire. Curr. Opin. Cell Biol. 21, 771-777 [DOI] [PubMed] [Google Scholar]
- Remus D., Beall E. L., Botchan M. R. (2004). DNA topology, not DNA sequence, is a critical determinant for Drosophila ORC-DNA binding. EMBO J. 23, 897-907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Ernst J., Kharchenko P. V., Kheradpour P., Negre N., Eaton M. L., Landolin J. M., Bristow C. A., Ma L., Lin M. F., et al. (2010). Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330, 1787-1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T., Sawado T., Yamaguchi M., Shinomiya T. (1999). Specification of regions of DNA replication initiation during embryogenesis in the 65-kilobase DNApolalpha-dE2F locus of Drosophila melanogaster. Mol. Cell. Biol. 19, 547-555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaiger M., Stadler M. B., Bell O., Kohler H., Oakeley E. J., Schubeler D. (2009). Chromatin state marks cell-type- and gender-specific replication of the Drosophila genome. Genes Dev. 23, 589-601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwed G., May N., Pechersky Y., Calvi B. R. (2002). Drosophila minichromosome maintenance 6 is required for chorion gene amplification and genomic replication. Mol. Biol. Cell 13, 607-620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbata H. R., Althauser C., Findley S. D., Ruohola-Baker H. (2004). The mitotic-to-endocycle switch in Drosophila follicle cells is executed by Notch-dependent regulation of G1/S, G2/M and M/G1 cell-cycle transitions. Development 131, 3169-3181 [DOI] [PubMed] [Google Scholar]
- Shea M., King D., Conboy M., Mariani B., Kafatos F. (1990). Proteins that bind to chorion cis-regulatory elements: A new C2H2 protein and a C2C2 steroid receptor-like component. Genes Dev. 4, 1128-1140 [DOI] [PubMed] [Google Scholar]
- Shima N., Alcaraz A., Liachko I., Buske T. R., Andrews C. A., Munroe R. J., Hartford S. A., Tye B. K., Schimenti J. C. (2007). A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 39, 93-98 [DOI] [PubMed] [Google Scholar]
- Shinomiya T., Ina S. (1991). Analysis of chromosomal replicons in early embryos of Drosophila melanogaster by two-dimensional gel electrophoresis. Nucleic Acids Res. 19, 3935-3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling A. (1981). The organization and amplification of two chromosomal domains containing Drosophila chorion genes. Cell 27, 193-201 [DOI] [PubMed] [Google Scholar]
- Spradling A. (1993). Developmental genetics of oogenesis. In The Development of Drosophila melanogaster (ed. Bate M., Martinez-Arias A.), pp. 1-70 Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Spradling A., Mahowald A. (1980). Amplification of genes for chorion proteins during oogenesis in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 77, 1096-1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Deng W. M. (2005). Notch-dependent downregulation of the homeodomain gene cut is required for the mitotic cycle/endocycle switch and cell differentiation in Drosophila follicle cells. Development 132, 4299-4308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Deng W. M. (2007). Hindsight mediates the role of notch in suppressing hedgehog signaling and cell proliferation. Dev. Cell 12, 431-442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashee S., Cvetic C., Lu W., Simancek P., Kelly T. J., Walter J. C. (2003). Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev. 17, 1894-1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelauer M., Rubbi L., Lucas I., Brewer B. J., Grunstein M. (2002). Histone acetylation regulates the time of replication origin firing. Mol. Cell 10, 1223-1233 [DOI] [PubMed] [Google Scholar]
- Wong P. G., Glozak M. A., Cao T. V., Vaziri C., Seto E., Alexandrow M. (2010). Chromatin unfolding by Cdt1 regulates MCM loading via opposing functions of HBO1 and HDAC11-geminin. Cell Cycle 9, 4351-4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Singh N., Donnelly C., Boimel P., Elefant F. (2007). The cloning and characterization of the histone acetyltransferase human homolog Dmel\TIP60 in Drosophila melanogaster: Dmel\TIP60 is essential for multicellular development. Genetics 175, 1229-1240 [DOI] [PMC free article] [PubMed] [Google Scholar]