

Abstract
The transcription factors NsdC and NsdD are required for sexual development in Aspergillus nidulans. We now show these proteins also play a role in asexual development in the agriculturally important aflatoxin (AF)-producing fungus Aspergillus flavus. We found that both NsdC and NsdD are required for production of asexual sclerotia, normal aflatoxin biosynthesis, and conidiophore development. Conidiophores in nsdC and nsdD deletion mutants had shortened stipes and altered conidial heads compared to those of wild-type A. flavus. Our results suggest that NsdC and NsdD regulate transcription of genes required for early processes in conidiophore development preceding conidium formation. As the cultures aged, the ΔnsdC and ΔnsdD mutants produced a dark pigment that was not observed in the wild type. Gene expression data showed that although AflR is expressed at normal levels, a number of aflatoxin biosynthesis genes are expressed at reduced levels in both nsd mutants. Expression of aflD, aflM, and aflP was greatly reduced in nsdC mutants, and neither aflatoxin nor the proteins for these genes could be detected. Our results support previous studies showing that there is a strong association between conidiophore and sclerotium development and aflatoxin production in A. flavus.
INTRODUCTION
The filamentous fungus Aspergillus flavus produces a number of secondary metabolites, including the toxic and carcinogenic aflatoxins (AFs). AFs are polyketide-derived compounds produced by A. flavus during growth on crops such as corn, peanuts, cottonseed, and tree nuts (5). Ingestion of foods contaminated with AFs has been implicated in acute toxicoses, while chronic, low-level exposure can lead to immune suppression and liver cancer (34, 41). In addition to the health risks associated with AFs, there are also significant adverse economic impacts to producers due to market rejection of contaminated crops and livestock losses as well as costs associated with monitoring for AF contamination (36, 45).
A. flavus occurs as a saprophyte in soils and normally reproduces clonally by means of conidia (asexual spores), although recent evidence suggests that sexual recombination is possible in A. flavus (25). Infestations of A. flavus in crops are sustained by production and dissemination of airborne conidia or sclerotia (reproductive bodies formed from mycelia that are capable of resisting unfavorable environmental conditions). Sclerotia can remain dormant for long periods of time until favorable conditions allow germination and production of more conidial inoculum (17, 19). High concentrations of AFs may occur in both conidia and sclerotia of A. flavus. Increased animal toxicity has been attributed to the combined activity of AFs and other metabolites present in sclerotia (42). Sclerotia are proposed by some researchers to be a vestige of the sexual structures termed cleistothecia produced by Aspergillus spp. such as A. nidulans. Cleistothecia are compact mycelial structures that harbor the ascospores (20). The ability to reproduce sexually has now been demonstrated in both A. flavus and A. parasiticus (25, 26, 33). These studies demonstrated the presence of ascospore-containing ascocarps within the matrix of sclerotia, supporting the role of these structures in sexual as well as asexual reproduction.
Fungal development and secondary metabolite production are known to be coregulated in many filamentous fungi, including A. flavus (9, 14, 38). Numerous environmental factors that affect fungal growth, such as nutritional status, pH, temperature, stress, and light, are involved in regulation of developmental processes, including sexual or asexual reproduction (12, 17, 21). A number of common regulatory factors that govern morphological differentiation in fungi, especially in A. nidulans, have been identified, and some of these factors have been found to control genes involved in secondary metabolite production as well (8, 32, 43, 46). For example, the Velvet complex proteins, LaeA, VeA, and VelB, regulate A. nidulans development as well as secondary metabolite production in a light-responsive manner (6, 35, 37, 39). The VeA-LaeA-VelB heterotrimeric complex also appears to have a similar function in A. flavus, because inactivation of either VeA or LaeA results in loss of AF and sclerotium production (2, 18, 29).
Previous work identified a group of A. nidulans mutants termed NSD (never in sexual development) because they failed to produce cleistothecia (22). NsdD is a GATA-type zinc finger transcription factor (23, 24), while NsdC is a C2H2 zinc finger-type DNA-binding protein that has been shown to negatively regulate asexual sporulation (30). We found homologs of these genes in A. flavus and A. parasiticus. Because of the relationship between development of cleistothecia and sclerotia, we postulated that in A. flavus NsdC and NsdD could be required for transcriptional regulation of genes necessary for the related developmental processes of sclerotium morphogenesis and conidiation. In addition, based on the existence of genetic links between morphogenesis and secondary metabolism (9), we considered it possible that Nsd proteins could regulate AF production. To test this hypothesis, we examined sclerotium formation, conidiophore morphology, and AF production in A. flavus nsdC and nsdD gene knockout mutants.
MATERIALS AND METHODS
Strains, media, and growth conditions.
An Aspergillus flavus CA14 Δku70 ΔniaD ΔpyrG parental strain (referred to as CA14; obtained from P.-K. Chang, SRRC) was used as the host for transformation. Three ΔnsdC (CA14 Δku70 ΔniaD ΔnsdC) and ΔnsdD (CA14 Δku70 ΔpyrG ΔnsdD) mutants were obtained, and initial characterization of these strains showed that all had a consistent phenotype. Cultures were point inoculated onto YGT agar (0.5% yeast extract, 2% glucose, and 1 ml of trace element solution per liter of medium) (27) supplemented with 1 mg/ml uracil and 1 mg/ml uridine (YGT-U) and incubated at 30°C in the light, a condition that promotes conidiation. Conidia were collected from plates in 0.01% Triton X-100 and stored at 4°C.
Vector construction and fungal transformation.
The A. flavus nsdC and nsdD gene orthologs were identified by a gene index search of the Aspergillus Comparative Database (http://www.broadinstitute.org/annotation/genome/aspergillus_group/MultiHome.html). A PCR-based method was used to construct an nsdD-niaD knockout plasmid, following essentially the same protocol as described in reference 11 and as depicted in Fig. S1A in the supplemental material. Briefly, 5′ and 3′ regions of the nsdD gene were amplified using oligonucleotide primers. The locations of the primers (contig 3; in parentheses following the primer sequence) within and flanking nsdD are based on the Broad Institute Aspergillus Comparative Database nucleotide sequence data for the A. flavus genome. The primers used were 5′ nsdD KpnI-NotI (5′-GGTACCGCGGCCGCACGGTACACTATTAGATCATAGCCGA-3′) (nucleotide [nt] 2009099) and 5′ nsdD XhoI (5′-CTCGAGATGCTTCTGCTTGTAGTTGGGA-3′) (nt 2009840). The 3′ end of nsdD was amplified using the primers 3′ nsdD XbaI (5′-TCTAGATTCGAAGAAGCGACGAGGCGTAAG-3′) (nt 2010653) and 3′ nsdD NotI (5′-GCGGCCGCTCGGAACATTTCATGACCACACCT-3′) (nt 2011264). Following PCR amplification of A. flavus genomic DNA with Ex Taq HS polymerase (TaKaRa), PCR products of the expected sizes of 745 bp for the 5′ nsdD amplification and 620 bp for the 3′ nsdD amplification were obtained. PCR products were subcloned into TOPO pCR2.1 (Invitrogen, Carlsbad, CA) and verified by DNA sequencing. The 745-bp PCR product was digested with KpnI and XhoI and subcloned into KpnI-XhoI-digested pBluescript II SK (Agilent Technologies, Santa Clara, CA). Subsequently, pBluescript II SK-5′-745-bp KpnI-XhoI plasmid DNA was XbaI/NotI digested and ligated to the XbaI-NotI-digested 620-bp 3′ nsdD PCR product. The resulting pBluescript II SK-nsdD 5′ and 3′ PCR product plasmid DNA was isolated and digested with XhoI-XbaI. Plasmid pSL82 harboring the A. parasiticus niaD gene was also digested with SalI-XbaI to release a fragment of about 6.7 kb containing the niaD gene and flanking regions. The 6.7-kb niaD gene was then ligated to XhoI-XbaI-digested pBluescript II SK-nsdD 5′ and 3′ PCR plasmid DNA to generate the final pNsdD-niaD knockout vector. The 5′ nsdD-niaD-3′ nsdD DNA region could be released from the pBluescript vector by NotI digestion for subsequent fungal transformation (see Fig. S1A in the supplemental material). Essentially the same protocol was used to prepare the nsdC knockout vector, NsdC-pyrG, except that the gene coding sequence was disrupted with the A. parasiticus pyrG gene (see Fig. S2A in the supplemental material). The nsdC complementation vector, pPTRI-NsdC, was constructed by amplifying an approximately 3.8-kb region of CA14 genomic DNA representing 2.0 kb of the nsdC coding region and 1.3 kb of upstream and 590 bp of downstream sequence using primers 5′ nsdC (ATTAAAGCTTTGCCTGGCGACCCTCAC) and 3′ nsdC (GAGTAAGCTTCACATTTTGCAACCGTCATACTCC). This PCR product was digested with HindIII and ligated into HindIII-digested pPTRI vector (TaKaRa) harboring the pyrithiamine resistance gene for selection of transformants. The nsdD complementation vector, pPTRI-NsdD, was constructed by amplifying an approximately 3.0-kb region of CA14 genomic DNA representing 1.6 kb of the nsdD coding region and 890 bp of upstream and 530 bp downstream sequence using primers 5′ nsdD (GTACCTGCAGACTGCCTGATCTTCCT) and 3′ nsdD (GTACCAGCTGGGAGTGTAAGGCAATCCAAG). This PCR product was digested with PstI-PvuII and ligated into PstI-SmaI-digested pPTRI vector.
Preparation, transformation, and regeneration of fungal protoplasts were carried out essentially as described by Cary et al. (11). Conidia were inoculated in potato dextrose broth supplemented with 1 mg/ml uracil and 1 mg/ml uridine (PDB-U), and transformants were regenerated on Czapek solution agar (CZ) (Difco) supplemented with 10 mM ammonium sulfate (CZ-AS) (pNsdC-pyrG transformation) or 1 mg/ml uracil and uridine (CZ-U) (pNsdD-niaD transformation). For complementation experiments, A. flavus ΔnsdC 17 and ΔnsdD 3 strains were grown in V8 broth supplemented with 10 mM ammonium sulfate (ΔnsdC 17) or 1 mg/ml uracil and uridine (ΔnsdD 3) for protoplasting and regenerated on CZ-AS agar [ΔnsdC 17(pPTRI-NsdC)] or CZ-U [ΔnsdD 3(pPTRI-NsdD)] and 0.1 μg/ml pyrithiamine. Putative Δnsd transformants were screened for nitrate (ΔnsdD) or uracil (ΔnsdC) prototrophy and single-spore isolated prior to further analyses. Putative Δnsd complementation transformants were screened by additional transfer on CZ-AS or CZ-U agar supplemented with 0.1 μg/ml pyrithimine and single-spore isolated prior to further analyses.
Nucleic acid isolation and analysis.
Fungal genomic DNA for PCR was prepared from mycelia following 48 h of incubation with shaking (200 rpm) at 30°C in YGT-U broth. Genomic DNA was extracted using a 0.1 M lithium–20 mM EDTA–0.5% SDS (LETS) buffer method (10). For real-time quantitative PCR (qPCR) of aflatoxin gene expression, 100-ml PDB-U cultures were inoculated with a suspension (105 spores/ml) of CA14 or Δnsd mutant conidia and incubated at 30°C with shaking (180 rpm) in the dark. Mycelia were harvested at 24, 48, and 72 h, immediately frozen in liquid nitrogen, and stored at −80°C for RNA extraction. For qPCR of developmental gene expression, 150-mm plates of YGT-U agar were overlaid with 7.5 ml of a suspension (106 spores/ml) of conidia in molten 0.7% water agar. The top agar layer containing the developing fungi was collected at 12, 24, and 36 h, immediately frozen in liquid nitrogen, and stored at −80°C for RNA extraction. RNA was isolated from about 100 mg of ground mycelia using the Aurum total RNA minikit (Bio-Rad, Hercules, CA) and treated with RNase-free DNase I (Bio-Rad). RNA quality and quantity were determined using the Experion automated electrophoresis station (Bio-Rad). RNA aliquots were preserved at −80°C. cDNA was synthesized from RNA using the iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR was performed using SYBR green I chemistry and the iCycler iQ5 multicolor real-time PCR detection system (Bio-Rad) (13). The sequences of the oligonucleotide primers used for qPCR amplification are listed in Table S1 in the supplemental material. When possible, all primers were designed to span an intron. Samples were run in triplicate, and gene expression levels at each time point were normalized (ΔΔCT analysis) to A. flavus 18S rRNA gene expression levels utilizing the gene expression analysis software package for the Bio-Rad iQ5 system. Reverse transcription-PCR (RT-PCR) of RNA isolated from complemented ΔnsdC and ΔnsdD mutants was used to confirm expression of nsdC and nsdD in these transformants. Template cDNA (∼100 ng) was prepared as described above for qPCR and amplified with Ex Taq HS polymerase using primers nsdC RT-PCR F (CGAGAAGCAACAACTAAATACAAT) and nsdC RT-PCR R (GTCAAGTTACCGGGCTGCGAGATG) and primers nsdD RT-PCR F (GTCACGACTCAAGATTCGCGAAACCACTC) and nsdD RT-PCR R (CTCGTATACGTAGTAAGGCATCCTGGTTC). Amplification conditions consisted of 94°C for 2 min followed by 35 cycles of 98°C for 15 s, 60°C for 30 s, and 72°C for 30 s and a final extension at 72°C for 7 min. The PCRs were separated on a 1% agarose gel and observed following ethidium bromide staining. Gel images were captured on a Fujifilm LAS-3000 luminescent image analyzer (Fujifilm Life Science, Stanford, CT).
Morphological studies.
Fungal growth was estimated as colony diameter measured after growth for 5 days following point inoculation of 1 μl of a spore suspension (103 spores/ml) onto YGT-U agar. Conidium production was determined after growth for 5 days on YGT-U agar overlaid with 3 ml of a suspension of conidia (106 spores/ml) in molten 0.7% water agar. Conidia were collected in duplicate from 7-mm cores taken from equivalent regions of the fungal mat on the agar surface. The cores were individually homogenized in 1 ml 0.01% Triton X-100 and appropriately diluted, and conidia were counted using a hemocytometer. Counts were done on three separate transformants and the mean and standard deviation determined from the combined data for the 6 samples. For determination of sclerotium production, 1 μl of a suspension of 103 spores/ml was center point inoculated onto YGT-U or PDA-U plates. Plates were incubated at 30°C for 14 days in the dark (a condition that promotes sclerotium formation). Following incubation, the plates were sprayed with 70% ethanol to collapse the mycelial mat and allow enumeration of sclerotia. The total number of sclerotia per 100-mm petri plate was determined. Sclerotia from triplicate plates were counted and the mean and standard deviation determined. Statistical differences in numbers of conidia and sclerotia were calculated using a two-sample t test, assuming equal variances. Pigment production was assayed for cultures on YGT-U agar supplemented with 100 μg/ml tricyclazole (a gift from Dow AgroSciences, Indianapolis, IN).
Microscopy.
Cultures for microscopy studies were center point inoculated with 1 μl of a suspension of conidia (103 spores/ml) and grown for 7 days at 30°C in the light on PDA-U. Conidiophore morphology was directly examined from colonies growing on PDA-U plates using an Olympus SZH10 stereomicroscope and Nikon DS-Qi1 camera. Images were captured using the NIS-Elements imaging software (Nikon). Measurements of conidiophore stipes and conidia from agar media or of conidiophore morphology from submerged cultures were obtained using an Olympus BH-2 microscope equipped with a Nikon Digital Sight DS-L1 camera. Samples were suspended in mounting medium (1.3 ml Aerosol OT and 24 ml 95% ethanol brought to a final volume of 100 ml with water).
Aflatoxin analysis.
Aflatoxin production was analyzed by extracting metabolites from the combined mycelia and broth of cultures grown in 100 ml PDB-U with shaking in the dark at 30°C for 72 h. Following incubation, 50 ml acetone was added to the cultures and then gently agitated at room temperature for a minimum of 1 h. AF was extracted and analyzed by thin-layer chromatography (TLC) as previously described (11). The extract residue was resuspended in acetone and spotted on a TLC plate (Si250F; J.T. Baker). The plate was then developed in toluene-ethyl acetate-formic acid (5:4:1, vol/vol/vol), air dried, and examined for the presence of AF under UV light. A second solvent system consisting of ethyl acetate, methanol, and water (40:1:1, vol/vol/vol) was also used to confirm the presence of AF in the samples. Liquid chromatography-mass spectrometry (LC-MS) was performed in both the positive- and negative-ion modes by atmospheric pressure chemical ionization (APCI). LC conditions were a 10-min gradient of 10% acetonitrile in 0.025% aqueous trifluoroacetic acid (TFA) to 90% acetonitrile in 0.025% aqueous TFA on a C18 column. LC detection was with a photodiode array detector.
Western blot analysis.
Mycelia of the CA14 and the ΔnsdC and ΔnsdD strains were harvested from 48-h PDB-U shake cultures, frozen in liquid nitrogen, and stored at −80°C. Frozen mycelia were ground to a powder in liquid nitrogen and resuspended in Tris-saline buffer (10 mM Tris [pH 8.0], 150 mM NaCl) supplemented with Halt protease inhibitor (Thermo Scientific, Rockford, IL) and phenylmethylsulfonyl fluoride (PMSF) (125 μM; Sigma). Supernatants were collected following centrifugation at 10,000 × g for 20 min and concentrated approximately 10-fold using an Amicon Ultra-15 centrifugal filter unit (Millipore Corp., Billerica, MA). Total protein samples (∼15 μg) were electrophoresed on NuPAGE 4 to 12% Bis-Tris gels (Invitrogen) and subsequently electroblotted onto Hybond-P polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Buckinghamshire, United Kingdom). Membranes were blocked with 3% ECL Advance blocking agent (GE Healthcare), and protein detection was carried out using anti-AflD (Nor-1), -AflM (Ver-1), or -AflP (OmtA) polyclonal antiserum (1:500 dilution; a gift from Ludmila Roze) (15) and peroxidase-labeled anti-rabbit secondary antibody (1:50,000 dilution; GE Healthcare) following the manufacturer's protocols (Amersham ECL Advance western blotting kit; GE Healthcare).
RESULTS
Generation and molecular analysis of A. flavus nsdC and nsdD knockout mutants.
Orthologs in A. flavus to A. nidulans nsdC and nsdD were identified by BLASTP searches of the Broad Comparative Aspergillus Database (http://www.broadinstitute.org/annotation/genome/aspergillus_group/MultiHome.html). The predicted cDNA and genomic sequences were also obtained from the database. The A. flavus nsdD gene ortholog (AFL2G_03635.2) is predicted to be 1,580 bp, with a deduced protein sequence of 453 amino acids. The A. flavus nsdC gene ortholog (AFL2G_06495.2) is 1,914 bp, with a predicted protein sequence of 638 amino acids. Clustal alignment of the A. nidulans and A. flavus NsdC and NsdD amino acid sequences demonstrated 72.2% and 71.3% identity, respectively. Deletion mutants of A. flavus CA14 were constructed (see Fig. S1A in the supplemental material) using niaD selection for nsdD knockout and pyrG selection for nsdC knockout (see Fig. S2A in the supplemental material). Three isolates were confirmed to be knockouts by PCR with primers P1 and P2, in which a CA14 nontransformed control generated a product of 1.1 kb whereas the ΔnsdD mutants had a product of 6.9 kb as the expected size for insertion of niaD (see Fig. S1B in the supplemental material). The three putative ΔnsdC mutants gave PCR products of the expected size of 3.6 kb for the pyrG-disrupted nsdC gene, while the nontransformed CA14 strain gave a product with the expected size of 2.8 kb (see Fig. S2B in the supplemental material). Since mutant phenotypes of each gene knockout were similar, only ΔnsdC 17 and ΔnsdD 3 were used for the experiments described below. The ΔnsdC 17 and ΔnsdD 3 strains were complemented with the nsdC and nsdD wild-type alleles using pyrithiamine resistance for selection after transformation with pPTRI-nsdC and pPTRI-nsdD, respectively. Integration of these vectors was confirmed by PCR analysis (see Fig. S3A in the supplemental material) and RT-PCR, indicating that both strains expressed the respective complemented genes for nsdC and nsdD (see Fig. S3B in the supplemental material). The genetically complemented strains gave PCR products of sizes similar to that for the CA14 isolate, and no PCR product was generated from the Δnsd knockout strains.
ΔnsdC and ΔnsdD mutants have altered colony morphology.
Colony phenotypes of ΔnsdC and ΔnsdD mutants were markedly different from those of the control strains when grown on YGT-U medium (Fig. 1). The ΔnsdC mutants displayed an ochre conidial pigmentation, compared to the dark green of the CA14 strain, while the ΔnsdD mutants were a slightly lighter green than CA14. The CA14 strain presented a light beige pigment on the underside of the colony, while the ΔnsdC and ΔnsdD mutants produced a black pigment that did not diffuse into the surrounding medium. The presence of this pigment was more pronounced earlier in growth of the ΔnsdC mutants, but upon extended incubation the black pigmentation in the ΔnsdD mutants almost equaled that in the ΔnsdC mutants. Pigmentation was best demonstrated during growth on YGT-U; however, it was also produced during growth on PDA-U and CZ-U (data not shown). Although the pigment appeared to have melanin-like characteristics based upon its insolubility in water, precipitation by 3 M HCl, UV spectrophotometry, and decolorization by hydrogen peroxide (data not shown), pigment production was not inhibited when ΔnsdC and ΔnsdD mutants were grown on YGT-U supplemented with 100 μg/ml tricyclazole, a dihydroxynaphthalene (DHN)-melanin inhibitor (see Fig. S4 in the supplemental material). Genetically complemented Δnsd strains did not produce the pigment. The ΔnsdC mutants showed radial growth that was comparable to that of the CA14 control strain after 4 days of growth on YGT-U agar, while the ΔnsdD mutants presented decreased radial growth (Fig. 1).
Fig 1.
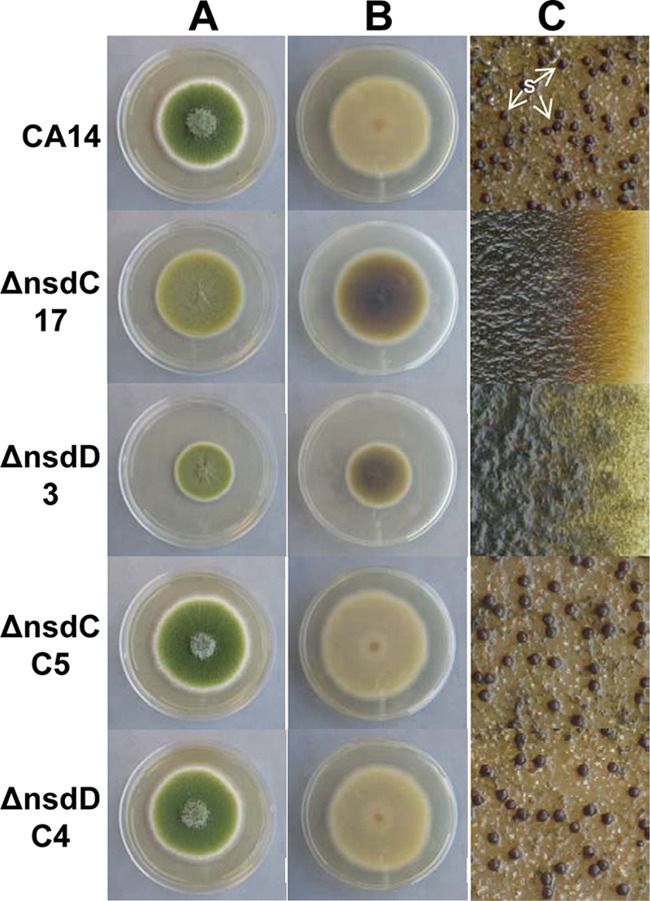
Colony growth, pigmentation, and sclerotium production in A. flavus CA14 ΔnsdC and ΔnsdD mutants and ΔnsdC and ΔnsdD complementation strains. CA14, ΔnsdC 17 and ΔnsdD 3 mutants, and genetically complemented ΔnsdC C5 and ΔnsdD C4 strains were grown on YGT-U medium for 5 days with fluorescent light illumination. (A) View from top of colony. Conidial pigmentation was altered in the Δnsd mutants. Decreased growth of the ΔnsdD 3 strain was observed. (B) View of underside of colony. Pigmentation was readily observed on the undersides of the ΔnsdC 17 and ΔnsdD 3 mutant colonies. (C) YGT-U plates demonstrating sclerotium production were grown for 14 days in the dark. The plates were sprayed with 70% ethanol to allow visualization of sclerotia. Sclerotia were absent in the ΔnsdC and ΔnsdD mutants and were produced in the wild-type CA14 and ΔnsdC and ΔnsdD complementation strains. S, sclerotia.
Altered conidiophore morphology in Δnsd mutants.
Microscopic examination of the ΔnsdC and ΔnsdD mutants revealed marked alterations in conidiophore morphology (Fig. 2A). The mutants exhibited a columnar arrangement of the conidial chains on the conidiophores, while the CA14 parental strain presented radiate conidial heads. Conidiophore stipes were significantly shorter for the mutants than the CA14 strain. The average stipe lengths for the ΔnsdC 17 and ΔnsdD 3 mutants were 80.0 ± 28.7 μm and 84.8 ± 29.3 μm, respectively, while the CA14 stipe length was 862.5 ± 176.9 μm (Fig. 2B). Both mutants produced degenerate conidiophores following 5 days of growth in submerged culture (Fig. 2A). While the CA14 strain produced few or no degenerate conidiophores and no observable conidia in submerged culture, the ΔnsdC 17 mutant produced numerous conidiophores which usually consisted of multiple, single-conidium-harboring phialides present on the vesicle. The ΔnsdD 3 strain also produced numerous conidiophores, yet these were usually observed as vesicles harboring a single phialide and conidium. The ΔnsdC C5 and ΔnsdD C4 complementation strains produced stipes of almost normal length (536.5 ± 129.0 and 504.5 ± 67.5 μm, respectively). Measurement of conidial diameter did not reveal any significant differences between the CA14, nsd mutant, and genetically complemented strains (data not shown).
Fig 2.
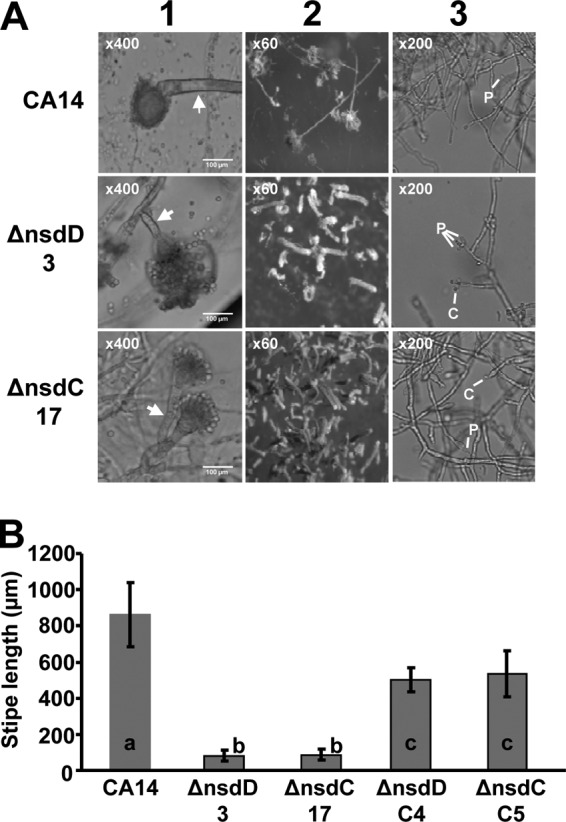
Light microscopy of Δnsd mutants reveals altered conidiophore morphology. (A) Column 1, examination of conidiophore stipe length. Fungal mycelia were harvested from PDA-U plates and placed in mounting medium solution on a microscope slide. Magnification, ×400. Arrows denote the conidiophore stipe. Note that the CA14 image does not demonstrate the total stipe (see column 2). Column 2, examination of conidial head structure. Images were captured from cultures growing on surfaces of PDA-U agar plates. Magnification, ×60. Column 3, examination of conidiophore production under submerged culture in PDA-U broth. Magnification, ×200. Phialides (P) and conidia (C) on the degenerate conidiophores are indicated. (B) Analysis of stipe length was done by microscopy on the wild-type CA14, the ΔnsdC 17 and ΔnsdD 3 mutants, and the ΔnsdC C5 and ΔnsdD C4 complementation strains. Values are the means and standard deviations of 20 stipe length measurements for each Δnsd mutant, each complementation isolate, and the wild-type CA14 isolate. Bars with different letters have values that are significantly different (P < 0.001).
Decreased conidium production and loss of sclerotia in ΔnsdC and ΔnsdD mutants.
Quantitative analysis of conidium production showed a statistically significant decrease in the number of conidia in both the ΔnsdC and ΔnsdD mutant colonies compared to that of CA14 following growth on YGT-U medium (Fig. 3). Complementation of the ΔnsdC and ΔnsdD mutants produced transformant isolates with conidium production comparable to that of the CA14 strain (Fig. 3). Initiation of conidium production at 30°C with illumination in the CA14 strain occurred at about 29 h, while initiation in the ΔnsdD 3 and ΔnsdC 17 mutants occurred at about 21 h and 14 h, respectively. qRT-PCR expression analysis of the A. flavus brlA ortholog (AFL2G_00999.2) revealed that its mRNA levels were about 100- and 200-fold higher in the ΔnsdD 3 and ΔnsdC 17 mutants than in CA14 at 12 and 24 h, respectively, on YGT-U (Fig. 4). Expression of brlA at 36 h in the ΔnsdC 17 mutant was about 2-fold higher than that in CA14 and the ΔnsdD 3 mutant. Expression of the A. flavus abaA ortholog (AFL2G_02163.2) was also higher at 12 and 24 h in the nsd mutants than in CA14, although expression in CA14 was higher than that in both mutants at 36 h.
Fig 3.
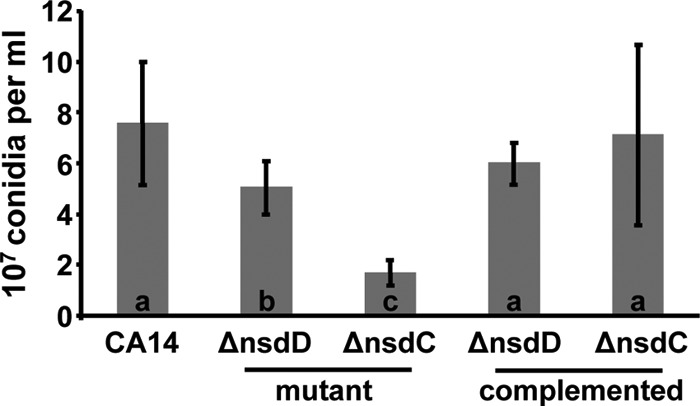
Deletion of nsdC and nsdD results in reduced conidiation. The wild-type CA14, three ΔnsdC (16, 17, and 21) and ΔnsdD (2, 3, and 7) mutants, and two ΔnsdC (C2 and C5) and ΔnsdD (C4 and C5) complementation isolates were grown on YGT-U agar for 5 days at 30°C with illumination. Values are the means and standard deviations from the two samplings each of conidia from the three separate transformants of the deletion mutants and two transformants of the complementation isolates. Bars with different letters have values that are significantly different (P < 0.003).
Fig 4.
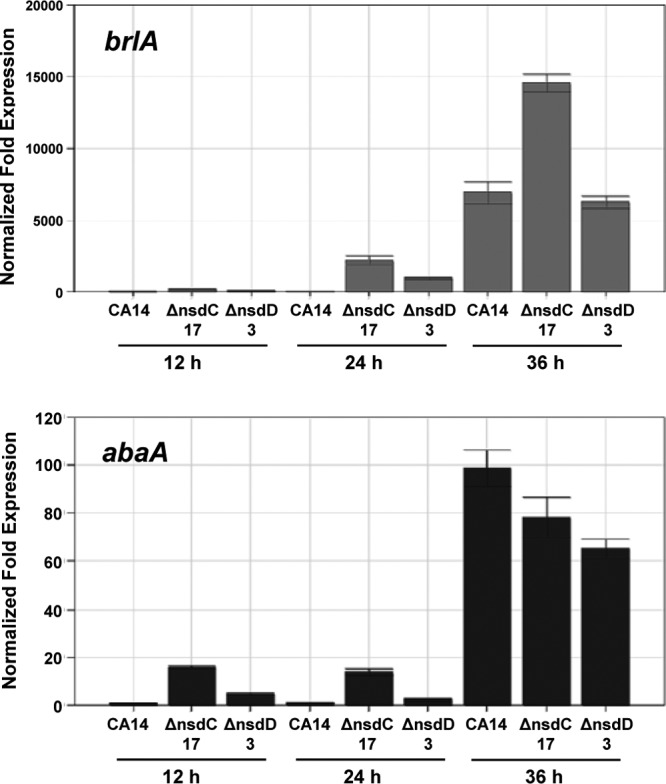
qPCR of the conidiation-specific transcription factors brlA and abaA. Gene expression levels at each time point were normalized (ΔΔCT analysis) to A. flavus 18S rRNA gene expression levels utilizing the gene expression analysis software package for the Bio-Rad iQ5.
In addition to the defects observed in conidiphore formation, ΔnsdC and ΔnsdD mutants did not produce sclerotia on PDA-U or YGT-U after 14 days at 30°C either in the light or in the dark (Fig. 1). The CA14 strain grown in the dark produced sclerotia on both PDA-U (634.7 ± 25.5 sclerotia/100-mm plate) and YGT-U (753.3 ± 116.0 sclerotia/100-mm plate) at 14 days. Genetically complemented strains ΔnsdC C2 and C5 produced 922.7 ± 73.5 and 1,264.0 ± 280.0 sclerotia/plate, respectively, on PDA-U, and ΔnsdD C4 and C5 produced 517.3 ± 22.6 and 506.0 ± 8.5 sclerotia/plate, respectively, on PDA-U. The genetically complemented strains also produced sclerotia on YGT-U (Fig. 1C).
Aflatoxin biosynthesis, gene transcription, and immunoblotting.
AFB1 or AFB2 was not detected by TLC analysis of extracts from 72-h PDB-U broth cultures of the ΔnsdC 17 mutant. Significantly lower levels of AFs were detected in the ΔnsdD 3 mutant than in extracts of CA14 (Fig. 5A). AF accumulation in the CA14, ΔnsdC 17, and ΔnsdD 3 strains observed at 120 h was similar to that observed at 72 h (data not shown). A small amount of a greenish-yellow metabolite was detected in the extract of the ΔnsdC 17 culture, which migrated close to the AFB1 band on TLC. However, TLC using a different solvent system (ethyl acetate-methanol-water at 40:1:1, vol/vol/vol) revealed that this metabolite migrated differently from that of the AFB1 standard (results not shown). The ability to synthesize AFB1 and AFB2 was restored in the genetically complemented ΔnsdC and ΔnsdD strains (Fig. 5A). The extracts from the mutant cultures were also analyzed by LC-MS (see Fig. S5 in the supplemental material). The AFB1 standard (m/z 313 [M + H]+ in the positive-ion spectrum) eluted at 3.63 to 3.90 min under the LC gradient conditions and was detected in an extract of A. flavus CA14 and in extracts of the ΔnsdD 3 mutant. The m/z 313 ion profile of the ΔnsdC strain showed multiple m/z 313 peaks but showed no peak at the expected elution time for AFB1 or with the signal strength of that of the ΔnsdD mutant or the parental strain.
Fig 5.

Aflatoxin production and expression of aflatoxin biosynthetic genes in Δnsd mutants and Δnsd complementation strains. (A) Extracts of the wild-type CA14, ΔnsdC 17 and ΔnsdD 3 mutants, and ΔnsdC (C2 and C5) and ΔnsdD (C4 and C5) genetically complemented strains grown for 3 days on PDB-U. Extracts (5 μl) were spotted onto 250-μm silica gel TLC plates, and metabolites were separated in toluene-ethyl acetate-formic acid (5:4:1, vol/vol/vol). Aflatoxin standards were also spotted on the plate. The plates were visualized under 310-nm UV light. (B) qPCR of aflatoxin gene transcripts from 24-, 48-, and 72-h cultures. Gene expression levels at each time point were normalized (ΔΔCT analysis) to A. flavus 18S rRNA gene expression levels utilizing the gene expression analysis software package for the Bio-Rad iQ5. Bars: white, CA14; gray, ΔnsdD 3 mutant; black, ΔnsdC 17 mutant. (C) Western blot analysis of AF enzymes (AflM, AflD, and AflP) present in total protein extracts of 48-h cultures of strains grown in PDB-U shake cultures.
Transcript levels of several aflatoxin biosynthetic genes were analyzed by qPCR of total RNA isolated from mycelia harvested at 24, 48, and 72 h (Fig. 5B). The genes aflD (nor-1), aflM (ver-1), aflP (omtA), and aflR were expressed in both the ΔnsdD 3 and ΔnsdC 17 mutants. For aflD, aflM, and aflP at 48 h, CA14 mRNA levels were higher than those of the ΔnsdC 17 mutant and ΔnsdC 17 levels were not significantly different from those of the ΔnsdD 3 mutant, though the trend indicated slightly higher levels of these transcripts at this time point in the ΔnsdC mutant. At 72 h, the ΔnsdD 3 strain showed the highest levels of expression of these genes, followed by CA14 and then the ΔnsdC 17 strain. At 24 h, little to no expression of these three genes was detected. The level of aflR mRNA was highest in the ΔnsdD 3 mutant at all time points, followed by that in the ΔnsdC 17 mutant, and it was lowest in CA14. Agarose gel electrophoresis of the PCR products showed that all transcripts were processed completely in CA14 as well as the mutant strains at all of the assay times (data not shown).
Western blot analysis of AflM, AflD, and AflP after growth for 48 h in PDB-U shake culture showed that the ΔnsdC 17 strain produced no detectable levels of these proteins, while the ΔnsdD 3 strain produced detectable levels of these proteins but significantly lower levels than CA14 (Fig. 5C). The levels of protein production correlated well with the results for aflatoxin production by these strains (Fig. 5A).
DISCUSSION
nsdD and nsdC are required for normal conidiophore development and sclerotium production in A. flavus.
In A. nidulans, nsdC and nsdD are predicted to encode C2H2-type DNA-binding proteins that act as positive regulators of sexual (cleistothecial) and negative regulators of asexual (conidial) development (23, 30). What function the orthologs of these genes have in the predominately asexual A. flavus has not been determined. In the absence of either nsdC or nsdD, stipe formation and ultimately conidiophore head morphology were profoundly altered (Fig. 2A), suggesting that these transcription factors activate genes involved in early developmental processes common to both sexual and asexual fungi. In both ΔnsdC and ΔnsdD mutants, the altered stipes caused the conidia to form in long columnar stacks on the phialides, in contrast to the normal radial arrangement on stipes in the parental strain. This change caused a reduction of the number of conidia produced by the mutants. Alterations to conidiophore morphology in A. nidulans nsdC or nsdD mutants have not been reported.
The lack of ability to produce sclerotia of the A. flavus ΔnsdC and ΔnsdD mutants observed in this study suggests that formation of these structures is controlled similarly to the formation of cleistothecia in A. nidulans (30) and A. fumigatus (40). The absence of sclerotia in the mutants could be caused by defects in hyphal fusion, as was observed previously in A. fumigatus ΔnsdD mutants (40). This observation suggested that NsdD may be necessary for transcriptional activation of genes required for production of proteins involved in hyphal anastomosis as well as cell wall integrity and maintenance. Defects in cell wall structure could also contribute to the observed defects in conidiophore development in A. flavus ΔnsdD as well as ΔnsdC mutants. Both the A. flavus ΔnsdC and ΔnsdD mutants accumulate a dark pigment during growth on agar media (Fig. 1) that appears to resemble the dark pigments produced by A. nidulans nsdC and nsdD mutants (22) and A. fumigatus nsdD mutants (40). The pigment in the A. flavus mutants appears to resemble melanin, but because its production was not affected by the melanin biosynthesis inhibitor tricyclazole, it does not appear to be the product of the DHN-melanin pathway (7). The observed pigment may be the product of the 3,4-dihydroxyphenylalanine (DOPA)-melanin biosynthetic pathway, which is not sensitive to tricyclazole. The pigment did not appear to be secreted into the medium, as it was not observed in the agar beyond the colony margin. The pigmentation observed may be similar to the brown pigment produced by aging colonies of A. oryzae, an A. flavus variant that is also incapable of sclerotium production (4, 31). We hypothesize that the brown pigment may be an oxidation product of a pigment specifically targeted to sclerotia and that the pigment is still produced in the nsdC and nsdD mutants. In the absence of sclerotia, it accumulates in the mycelia.
NsdD and NsdC repress expression of genes that activate asexual sporulation.
Our study revealed that NsdD and NsdC repress expression of genes that activate asexual sporulation. The A. flavus ΔnsdC and ΔnsdD mutants showed a precocious, abnormal increase in expression of brlA and abaA, genes encoding transcriptional activators of asexual sporulation (1). This increase likely caused the earlier conidiophore development than that observed for the parental strain. Premature expression of brlA was also noted in A. nidulans nsdC mutants (30), and earlier conidium formation was also observed in A. nidulans nsdC and nsdD mutants (23, 30). The increased and earlier expression of brlA and abaA in the A. flavus Δnsd mutants suggests that both NsdC and NsdD negatively regulate asexual reproduction by repressing brlA expression, which in turn delays expression of abaA. BrlA functions in the early stages of conidial development, while AbaA functions in the intermediate stages (3). Despite an apparent increase in brlA and abaA expression during growth of the A. flavus ΔnsdC and ΔnsdD mutants, conidiation decreased (Fig. 3). As stated above, we postulate that the observed decrease in conidium production in these mutants is a direct result of the aberrant architecture observed in the conidiophores in the Δnsd mutants. During growth in submerged culture, both ΔnsdC and ΔnsdD mutants produced numerous degenerate conidiophores usually harboring a single conidium attached to the phialide. This observation also is consistent with the loss in normal controls for conidiophore development in the mutants (Fig. 2A, lane 3) and with the observation in A. nidulans that NsdC is involved in repressing asexual sporulation under hypoxic conditions that exist in submerged cultures (30). However, unlike the A. flavus mutants, A. nidulans nsdD mutants did not produce conidia under submerged culture.
The transcription factors NsdD and NsdC affect expression of genes involved in aflatoxin production.
Fungal development and production of many secondary metabolites (such as AFs) are known to be coregulated in filamentous fungi (9, 38). Interestingly, in addition to their regulatory role governing morphological differentiation in A. flavus, the transcription factors NsdD and NsdC are required for normal production of the potent carcinogen aflatoxin. To our knowledge, this is the first report on the effect of NsdC and NsdD on secondary metabolism. Our results show that AF production does not occur in nsdC mutants even though the regulatory gene, aflR, is expressed at elevated levels compared to those in the parental strain. The markedly downregulated expression of tested AF biosynthetic genes and the inability to detect key biosynthetic proteins together with the absence of AF biosynthesis suggest that NsdC may be required for expression of a transcriptional cofactor necessary for activation of expression of AF cluster genes by AflR. The only fungal cofactors so far described that are known to affect AF production are VeA and LaeA. However, both LaeA and VeA have also been shown to be necessary for aflR expression, so a previously undescribed cofactor may be involved in the inability of ΔnsdC mutants to make AFs. Loss of nsdD also led to a marked decrease in AF production, but even though expression of aflR was also elevated in these mutants, expression of key biosynthesis genes and their encoded proteins occurred at levels consistent with the levels of AF produced.
Taken together, these results suggest that the loss of NsdC or NsdD results in developmental alterations that impact the ability of AflR to activate expression of the AF biosynthesis genes. It is possible that posttranslational modifications of AflR, which are necessary for AflR's full activity, could be altered in the nsdC and nsdD knockout mutants. A similar phenomenon was observed in both A. flavus and A. parasiticus strains following treatment with 5-azacytosine or following numerous serial mycelial transfers (16, 28, 44). Despite partial expression of aflR and AF biosynthetic genes, these strains no longer produced AF (or produced AF at significantly lower levels in the case of ΔnsdD mutants) and often demonstrated morphological changes such as reduced sporulation and sclerotium production. Future research will further contribute to the understanding of the observed reduction/loss of AF production in the A. flavus Δnsd mutants and possible genetic elements associated with this pattern.
In conclusion, our results provide evidence that NsdC and NsdD regulate expression of early genes required for the formation of the conidiophore. Both NsdC and NsdD are also required for sclerotium production and, importantly, for normal AF production. Orthologs of NsdD and NsdC are present in numerous filamentous fungi. Therefore, agents or environmental conditions that alter proper fungal development could prevent AF production. These considerations may be of importance for development of effective strategies for remediation of preharvest AF contamination in crops.
Supplementary Material
ACKNOWLEDGMENTS
Our thanks go to Maren Klich, Shannon Beltz, and Bruce Ingber for their assistance with conidiophore microscopy. We also acknowledge Ludmila Roze for providing the antisera used in immunoblotting.
Footnotes
Published ahead of print 13 July 2012
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1. Adams TH, Boylan MT, Timberlake WE. 1988. brlA is necessary and sufficient to direct conidiophore development in Aspergillus nidulans. Cell 54:353–362 [DOI] [PubMed] [Google Scholar]
- 2. Amaike S, Keller NP. 2009. Distinct roles for VeA and LaeA in development and pathogenesis of Aspergillus flavus. Eukaryot. Cell 8:1051–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrianopoulos A, Timberlake WE. 1994. The Aspergillus nidulans abaA gene encodes a transcriptional activator that acts as a genetic switch to control development. Mol. Cell. Biol. 14:2503–2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barbesgaard P, Heldt-Hansen HP, Diderichsen B. 1992. On the safety of Aspergillus oryzae: a review. Appl. Microbiol. Biotechnol. 36:569–572 [DOI] [PubMed] [Google Scholar]
- 5. Bhatnagar D, Cotty PJ, Cleveland TE. 2001. Genetic and biological control of aflatoxigenic fungi, p 208–240. In Wilson CL, Droby S. (ed), Microbial food contamination. CRC Press, Boca Raton, FL [Google Scholar]
- 6. Bok J-W, Keller NP. 2004. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 3:527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Butler MJ, Day AW. 1998. Fungal melanins: a review. Can. J. Microbiol. 44:1115–1136 [Google Scholar]
- 8. Calvo AM. 2008. The VeA regulatory system and its role in morphological and chemical development in fungi. Fungal Genet. Biol. 45:1053–1061 [DOI] [PubMed] [Google Scholar]
- 9. Calvo AM, Wilson RA, Bok JW, Keller NP. 2002. Relationship between secondary metabolism and fungal development. Microbiol. Mol. Biol. Rev. 66:447–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cary JW, Ehrlich KC, Beltz SB, Harris-Coward P, Klich MA. 2009. Characterization of the Aspergillus ochraceoroseus aflatoxin/sterigmatocystin biosynthetic gene cluster. Mycologia 101:352–362 [DOI] [PubMed] [Google Scholar]
- 11. Cary JW, Ehrlich KC, Bland JM, Montalbano BG. 2006. The aflatoxin biosynthesis cluster gene, aflX, encodes an oxidoreductase involved in conversion of versicolorin A to demethylsterigmatocystin. Appl. Environ. Microbiol. 72:1096–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cary JW, Linz JE, Bhatnagar D. 2000. Aflatoxins: biological significance and regulation of biosynthesis, p 317–361. In Cary JW, Linz JE, Bhatnagar D. (ed), Microbial foodborne diseases: mechanisms of pathogenesis and toxin synthesis. Technomic Publishing Co., Lancaster, PA [Google Scholar]
- 13. Cary JW, et al. 2007. Elucidation of veA-dependent genes associated with aflatoxin and sclerotial production in Aspergillus flavus by functional genomics. Appl. Microbiol. Biotechnol. 76:1107–1118 [DOI] [PubMed] [Google Scholar]
- 14. Cary JW, Szerszen L, Calvo AM. 2009. Regulation of Aspergillus flavus aflatoxin biosynthesis and development, p 183–203. In Appel M, Kendra DF, Trucksess MW. (ed), Mycotoxin prevention and control in agriculture. American Chemical Society, Washington, DC [Google Scholar]
- 15. Chanda A, et al. 2009. A key role for vesicles in fungal secondary metabolism. Proc. Natl. Acad. Sci. U. S. A. 106:19533–19538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang PK, et al. 2007. Genes differentially expressed by Aspergillus flavus strains after loss of aflatoxin production by serial transfers. Appl. Microbiol. Biotechnol. 77:917–925 [DOI] [PubMed] [Google Scholar]
- 17. Cotty P. 1988. Aflatoxin and sclerotial production by Aspergillus flavus: influence of pH. Phytopathology 78:1250–1253 [Google Scholar]
- 18. Duran R, Cary J, Calvo A. 2007. Production of cyclopiazonic acid, aflatrem, and aflatoxin by Aspergillus flavus is regulated by veA, a gene necessary for sclerotial formation. Appl. Microbiol. Biotechnol. 73:1158–1168 [DOI] [PubMed] [Google Scholar]
- 19. Garber RK, Cotty PJ. 1997. Formation of sclerotia and aflatoxins in developing cotton bolls infected by the S strain of Aspergillus flavus and potential for biocontrol with an atoxigenic strain. Phytopathology 87:940–945 [DOI] [PubMed] [Google Scholar]
- 20. Geiser DM, Timberlake WE, Arnold ML. 1996. Loss of meiosis in Aspergillus. Mol. Biol. Evol. 13:809–817 [DOI] [PubMed] [Google Scholar]
- 21. Han K-H, et al. 2003. Environmental factors affecting development of Aspergillus nidulans. J. Microbiol. 41:34–40 [Google Scholar]
- 22. Han KH, Cheong SS, Hoe HS, Han DM. 1998. Characterization of several NSD mutants of Aspergillus nidulans that never undergo sexual development. Kor. J. Genet. 20:257–264 [Google Scholar]
- 23. Han KH, et al. 2001. The nsdD gene encodes a putative GATA-type transcription factor necessary for sexual development of Aspergillus nidulans. Mol. Microbiol. 41:299–309 [DOI] [PubMed] [Google Scholar]
- 24. Han KH, et al. 2003. Regulation of nsdD expression in Aspergillus nidulans. J. Microbiol. 41:259–261 [Google Scholar]
- 25. Horn BW, Moore GG, Carbone I. 2009. Sexual reproduction in Aspergillus flavus. Mycologia 101:423–429 [DOI] [PubMed] [Google Scholar]
- 26. Horn BW, Ramirez-Prado JH, Carbone I. 2009. Sexual reproduction and recombination in the aflatoxin-producing fungus Aspergillus parasiticus. Fungal Genet. Biol. 46:169–175 [DOI] [PubMed] [Google Scholar]
- 27. Kafer E. 1977. The anthranilate synthetase enzyme complex and the trifunctional trpC gene of Aspergillus. Can. J. Genet. Cytol. 19:723–738 [DOI] [PubMed] [Google Scholar]
- 28. Kale SP, et al. 2007. Analysis of aflatoxin regulatory factors in serial transfer-induced non-aflatoxigenic Aspergillus parasiticus. Food Addit. Contam. 24:1061–1069 [DOI] [PubMed] [Google Scholar]
- 29. Kale SP, et al. 2008. Requirement of LaeA for secondary metabolism and sclerotial production in Aspergillus flavus. Fungal Genet. Biol. 45:1422–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim HR, Chae KS, Han KH, Han DM. 2009. The nsdC gene encoding a putative C2H2-type transcription factor is a key activator of sexual development in Aspergillus nidulans. Genetics 182:771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klich MA, Pitt JI. 1988. Differentiation of Aspergillus flavus from A. parasiticus and other closely related species. Trans. Br. Mycol. Soc. 91:99–108 [Google Scholar]
- 32. Kosalkova K, et al. 2009. The global regulator LaeA controls penicillin biosynthesis, pigmentation and sporulation, but not roquefortine C synthesis in Penicillium chrysogenum. Biochimie 91:214–225 [DOI] [PubMed] [Google Scholar]
- 33. Olarte RA, et al. 2012. Effect of sexual recombination on population diversity in aflatoxin production by Aspergillus flavus and evidence for cryptic heterokaryosis. Mol. Ecol. 21:1453–1476 [DOI] [PubMed] [Google Scholar]
- 34. Probst C, Schulthess F, Cotty PJ. 2009. Impact of Aspergillus section Flavi community structure on the development of lethal levels of aflatoxins in Kenyan maize (Zea mays). J. Appl. Microbiol. 108:600–610 [DOI] [PubMed] [Google Scholar]
- 35. Purschwitz J, et al. 2008. Functional and physical interaction of blue- and red-light sensors in Aspergillus nidulans. Curr. Biol. 18:255–259 [DOI] [PubMed] [Google Scholar]
- 36. Robens J, Cardwell K. 2003. The costs of mycotoxin management to the USA: management of aflatoxins in the United States. J. Toxicol. Toxin Rev. 22:139–152 [Google Scholar]
- 37. Sarikaya Bayram O, et al. 2010. LaeA control of velvet family regulatory proteins for light-dependent development and fungal cell-type specificity. PLoS Genet. 6:e1001226 doi:10.1371/journal.pgen.1001226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shwab EK, Keller NP. 2008. Regulation of secondary metabolite production in filamentous ascomycetes. Mycol. Res. 112:225–230 [DOI] [PubMed] [Google Scholar]
- 39. Strauss J, Reyes-Dominguez Y. 2011. Regulation of secondary metabolism by chromatin structure and epigenetic codes. Fungal Genet. Biol. 48:62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Szewczyk E, Krappmann S. 2010. Conserved regulators of mating are essential for Aspergillus fumigatus cleistothecium formation. Eukaryot. Cell 9:774–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Turner PC, Moore SE, Hall AJ, Prentice AM, Wild CP. 2003. Modification of immune function through exposure to dietary aflatoxin in Gambian children. Environ. Health Perspect. 111:217–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wicklow DT, Cole RJ. 1982. Tremoragenic indole metabolites and aflatoxins in sclerotia of Aspergillus flavus: an evolutionary perspective. Can. J. Bot. 60:525–528 [Google Scholar]
- 43. Wiemann P, et al. 2010. FfVel1 and FfLae1, components of a velvet-like complex in Fusarium fujikuroi, affect differentiation, secondary metabolism and virulence. Mol. Microbiol. 77:972–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wilkinson JR, Kale SP, Bhatnagar D, Yu J, Ehrlich KC. 2011. Expression profiling of non-aflatoxigenic Aspergillus parasiticus mutants obtained by 5-azacytosine treatment or serial mycelial transfer. Toxins 3:932–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu F. 2006. Mycotoxin reduction in Bt corn: potential economic, health, and regulatory impacts. Transgenic Res. 15:277–289 [DOI] [PubMed] [Google Scholar]
- 46. Yu JH, Keller N. 2005. Regulation of secondary metabolism in filamentous fungi. Annu. Rev. Phytopathol. 43:437–458 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.