

Abstract
During their progression from intranuclear capsids to mature trilaminar virions, herpesviruses incorporate an extensive array of viral as well as a smaller subset of cellular proteins. Our laboratory previously reported that rhesus monkey rhadinovirus (RRV), a close homolog of the human pathogen Kaposi's sarcoma-associated herpesvirus (KSHV), is comprised of at least 33 different virally encoded proteins. In the current study, we found that RRV infection activated the extracellular signal-regulated kinase (ERK) pathway and nascent virions preferentially incorporated the activated form of ERK2 (pERK2) into the tegument. This was evident even in the face of greatly diminished stores of intracellular ERK2, suggesting a clear bias toward the incorporation of pERK2 into the RRV particle. Similar to earlier findings with KSHV, activation of ERK was essential for the production of lytic viral proteins and virions. Knockdown of intracellular ERK, however, failed to inhibit virus production, likely due to maintenance of residual pools of intracellular pERK2. Paradoxically, selective knockdown of ERK1 enhanced virion production nearly 5-fold and viral titers more than 10-fold. These data are the first to implicate ERK1 as a negative regulator of lytic replication in a herpesvirus and the first to demonstrate the incorporation of an activated signaling molecule within a herpesvirus. Together, the results further our understanding of how herpesviruses interact with host cells during infection and demonstrate how this family of viruses can exploit cellular signal transduction pathways to modulate their own replication.
INTRODUCTION
The gammaherpesvirus, Kaposi's sarcoma-associated herpesvirus (KSHV), is the causative agent of three human malignancies, most notably Kaposi's sarcoma (KS), an AIDS-defining disease (10, 11, 21, 22, 44, 45, 58, 59). Since even the best source of KSHV virions tends to yield titers that are often less than 10E5/ml (1), primate homologs such as rhesus monkey rhadinovirus (RRV), which favor lytic rather than latent infection in culture, often serve as a model system for studying virus structure and protein composition (16, 50). Identifying the proteins that comprise the gammaherpesvirus particle allows a better understanding of the processes and pathways that are important in infection (2, 15–17, 19, 50, 52, 67, 80).
It is clear that herpesviruses are not solely made up of virally encoded proteins. Instead, the tegument layer of a herpesvirus is a collection of both viral and cellular components (5, 31, 40, 81). In our previous study, we identified 33 distinct virally encoded proteins (51), which closely paralleled and extended earlier findings with KSHV (5, 81). More recently, detailed MS analyses of the protein components of RRV have revealed that the virions also contain a small subset of cellular proteins. We were particularly intrigued to discover, using detailed mass spectrometry (MS) analyses of RRV particles, the consistent presence of extracellular signal-regulated kinase 2 (ERK2), one of the major isoforms of ERK and a prototypical member of the family of mitogen-activated protein kinases (MAPK) that comprise the Raf-MEK-ERK pathway (64).
The MAP kinases are a family of serine-threonine kinases that are central to cell signaling pathways, allowing cells to respond to a variety of extracellular stimuli, including viral infection. These pathways integrate signals from the extracellular environment into biological responses within the cell such as cell proliferation, differentiation, and cell cycle progression. A cascade of sequential phosphorylation events, including those involving the MAP kinase kinase kinases (MAPKKK; e.g., Raf), MAP kinase kinases (MAPKK; e.g., MEK), and MAP kinases (MAPK; e.g., ERK), leads to the activation of substrates such as transcription factors, promoting both cellular and viral gene expression (63, 64, 68, 78).
The two main isoforms of ERK, ERK1 and ERK2 (ERK1/2), share approximately 85% amino acid sequence identity, become activated by similar stimuli, and display identical kinetics of activation, similar substrate specificity, and subcellular localization (39, 63, 64, 68, 78); thus, early investigations had assumed that ERK1 and ERK2 were likely functionally equivalent. Though that conclusion was reasonable, more recent studies suggest that, in addition to their overlapping functions, each of these isoforms may also exhibit unique and distinct functions (32, 33, 38, 39, 41, 71). One of the first observations that pointed to a potential functional difference between these two isoforms was the difference in expression profiles within different tissues (55). Marchi et al. also described a unique domain in the N-terminal region of ERK1 (between residues 8 and 39) that accounts for differences in nucleocytoplasmic shuttling rates between ERK1 and ERK2 as well as changes in overall signaling output and function (41). Further, knockout mouse studies have demonstrated that ERK1 cannot compensate for ERK2 (77), again pointing toward unique functions of each isoform. Despite ERK1's inability to replace ERK2 function, both in vivo and in vitro studies have shown that the loss of ERK1 resulted in improved functions or a gain-of-function phenotype, suggestive of an inhibitory role for ERK1 in many cellular processes (39).
Many viruses exploit the MAPK pathways to promote their own survival by activating both the cellular and viral genes necessary for productive infection (3, 4, 8, 46, 57, 66), and herpesviruses are no exception (12, 18, 30, 61). Primary KSHV infection and reactivation trigger all three MAPK pathways (24, 29, 37, 47, 56, 60, 65, 69, 74, 75, 79), and the virus depends on these pathways for the initial establishment of infection and early gene expression (13, 24, 34, 35, 47, 48, 56, 69). Though previous studies revealed that ERK2 associates with lentiviral particles (9, 25, 26, 28, 53), this is the first report of which we are aware that has characterized the association of any component of the MEK/ERK pathway with the virions of a herpesvirus. In the present study, we describe the incorporation of ERK2 in preference to ERK1 in the tegument of RRV particles and, for the first time, define distinct roles for these two isoforms in a herpesvirus infection.
MATERIALS AND METHODS
Cell culture.
Telomerase-immortalized rhesus fibroblasts (RhF) were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), 110 mg/liter sodium pyruvate, and 500 ng/ml puromycin, as described previously (51).
Virus stocks.
For RRV stocks, when RhF were completely confluent in a T182 flask (approximately 2 × 107 cells), cells were infected with RRV strain H26-95 at a multiplicity of infection (MOI) of 0.05 for 1 h, followed by supplementation of an additional 100 ml of complete media/flask. Supernatants were harvested at 5 days postinfection (p.i.). Viral supernatants were cleared of cellular debris by low-speed centrifugation and subsequently passed through a 0.45-μm-pore-size filter. Virus was then formed into a pellet by high-speed centrifugation for 3 h at 12,855 × g in a Sorvall SL250T rotor, and the resulting pellet was resuspended overnight at 4°C in a final volume of 1.0 ml of TNE (20 mM Tris [pH 7.5], 100 mM NaCl, 1 mM EDTA).
As described previously (51), for highly purified viral stocks, viral particles were further purified over Sepharose CL-4B (Sigma-Aldrich) packed size exclusion chromatography columns (Econo-Column; Bio-Rad) and peak fractions were concentrated in a microcentrifuge at 4°C for 2 h at 35,000 × g. Viral particles were then treated with RNase-free DNase I (Stratagene) (20 U) and proteinase K (PK; Sigma-Aldrich) (150 ng/ml), followed by two sequential 60-μl 20% to 50% sucrose-TNE step gradients, each performed for 45 min at 60,000 × g in an SW-55 Ti rotor. Fractions were collected by bottom puncture, processed on 10% NuPAGE Bis-Tris gels (Invitrogen), and stained with Coomassie blue. Peak fractions were determined by the intensity of the major capsid protein (MCP) bands and pooled.
Reagents. (i) Abs.
Antibodies (Abs) detecting total ERK1 and ERK2 (ERK1/2) (1:750) and phospho-p38 (1:750) were purchased from Cell Signaling Technology (tested by the manufacturer to detect these proteins across species by Western blot analysis); phospho-ERK1/2 (pERK) (1:10,000) antibody was purchased from Sigma-Aldrich (the epitope recognized by the antibody is in the regulatory site of active MAP kinase and is completely conserved in monkey); and RanBP (1:7,500) antibody was purchased from BD Biosciences. Anti-RRV Major Capsid Protein (MCP) (1:200) was kindly provided by Scott Wong at Oregon Health and Science University. Anti-RRV ORF65 (SCIP) (1:2,500) was raised in mice in the Lymphocyte Culture Center at the University of Virginia. Anti-RRV ORF45 Ab (1:7,500) was raised in rabbits and purchased from Open Biosystems, Inc. Infrared Dye 800 anti-mouse, Infrared Dye 800 anti-rabbit, and Infrared Dye 680 anti-rabbit (all 1:10,000) were purchased from LiCor Biosciences and Rockland Immunochemicals.
(ii) Drugs.
The MEK inhibitor U0126 (1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio] butadiene) was purchased from Cell Signaling Technology. 12-O-Tetradecanoylphorbol 13-acetate (TPA) was purchased from Sigma-Aldrich.
(iii) siRNAs.
ON-TARGETplus SMARTpool siERK1, siERK2, and siGENOME nontargeting small interfering RNA (siRNA) 5 (siRNA negative control) were purchased from Thermo-Scientific.
Protein electrophoresis and immunoblot analyses.
Cells were lysed with whole-cell lysis buffer (50 mM Tris [pH 7.3], 150 mM NaCl, 1% Nonidet P-40, 5 mM EDTA, 10% glycerol) supplemented with 1 mM sodium orthovanadate (Na3VO4), 40 mM β-glycerophosphate, 30 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, and 1× protease inhibitor cocktail (Roche Applied Science) just prior to use. Cell lysates and virus samples were reduced in sample buffer (lithium dodecyl sulfate [LDS]) with reducing agent containing 0.5 M dithiothreitol (DTT) (NuPage; Invitrogen) and proteins separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 10% Bis-Tris gels (Invitrogen).
For immunoblot analyses, proteins separated by SDS-PAGE were transferred to nitrocellulose membranes for 60 min at 250 mA at 4°C. The membranes were blocked in 5% nonfat milk–TBS (20 mM Tris base, 150 mM NaCl, 3 mM Tris-HCl) for 1 h at room temperature and then incubated with primary antibodies for 2 h at room temperature or overnight at 4°C. After three washes with TBS-Tween (0.05%) at room temperature, membranes were incubated with secondary antibodies (45 min, room temperature). For semiquantitative enhanced chemiluminescence (ECL) immunoblotting (see Fig. 2), membranes bound to primary Abs were incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) (1:5,000 to 1:10,000) and Western Lightning chemiluminescent reagent (Perkin-Elmer) was used according to the manufacturer's protocol. For all immunoblotting other than that shown here (see Fig. 2), quantitative immunoblotting was employed and entailed incubating membranes with Infrared Dye 800-conjugated anti-mouse or anti-rabbit and Infrared Dye 680-conjugated anti-mouse diluted 1:10,000 in TBS-Tween (0.05%). Images were scanned and analyzed using an Odyssey infrared imaging system and 3.0 software (LiCor Biosciences).
Fig 2.
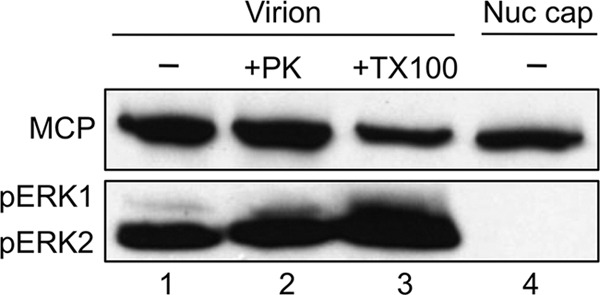
pERK is specifically incorporated into the tegument of RRV virions. Gradient-purified RRV was either left untreated (lane 1) or treated with proteinase K (150 ng/ml; lane 2) or Triton X-100 (2%; lane 3) and analyzed by immunoblotting alongside capsids purified from isolated nuclei (Nuc cap; lane 4), probing for the presence or absence of pERK (bottom panel) and MCP (top panel), the latter to approximate the relative particle number in each sample.
Determining relative ratios of viral proteins per particle.
Particles were harvested under siERK conditions and viral proteins separated by SDS-PAGE. Values obtained from the Odyssey imaging system for ORF45 and SCIP were divided by MCP values, which are present at a known stoichiometry (955 copies per particle), to determine the amounts of ORF45 and SCIP per particle. The normalized values for ORF45 and SCIP determined under each set of conditions were then compared to those determined for the control.
Linearity of the Odyssey imaging system.
To determine the linear range for the secondary antibodies used with Odyssey infrared imaging system, infected whole-cell lysates were diluted in a 2-fold series and the proteins separated by SDS-PAGE and then, once transferred to nitrocellulose membranes, incubated with primary antibodies and, finally, stained with Infrared Dye 800-conjugated anti-mouse or anti-rabbit monoclonal Ab (MAb) or Infrared Dye 680-conjugated anti-mouse MAb. Each band was quantified using the Odyssey infrared imaging system and software as described above. Odyssey values remained linear as a function of protein concentration within at least a 100-fold dilution series (r2 = 0.99). For experiments, exposure times-intensities on the scanner were adjusted to ensure that only values that fell within this range were employed in calculations to determine the relative level of each protein.
Determining correction factor for differential Ab sensitivities to pERK1 versus pERK2.
The antibody used to detect pERK recognizes pERK2 with greater sensitivity than pERK1 (per manufacturer's data sheet; Sigma-Aldrich). To determine this difference quantitatively, we generated parallel dilution series for pERK1 and pERK2, using known initial concentrations of purified reagent grade sources of the two proteins (Millipore). Standard curves determined with the Odyssey imaging system revealed that the values for pERK2 were 3 times greater than pERK1 values at the same concentrations. In our calculations to determine the relative amounts of pERK1 and pERK2 in our samples, we multiplied pERK1 values by 3 to compensate for this difference.
siRNA tranfection followed by RRV infection.
ERK1 and ERK2 siRNA SMARTpools (or siRNA control) were transfected into RhF using Lipofectamine RNAiMAX (Invitrogen) at concentrations between 10 and 25 nM per the manufacturer's protocol. At 24 h posttransfection, cells were infected at an MOI of 5 (unless noted otherwise) for 1 h at 37°C. Residual virus was removed by two sequential phosphate-buffered saline (PBS) washes at room temperature, followed by the addition of fresh media. Cells were harvested 48 h p.i. for immunoblot analysis.
Virus collection following siERK treatments.
Culture supernatants were cleared by low-speed centrifugation, viral pellets were collected by sedimentation through a 35% sucrose cushion for 45 min at 60,000 × g in an SW-55 Ti or SW-41 Ti rotor, and the resulting pellets were resuspended in 60 to 75 μl of PBS overnight at 4°C.
Drug treatments.
RhF were treated with 50 μM U0126 in dimethyl sulfoxide (DMSO) 4 h prior to infection, and drug remained in the media during the subsequent 1 h of RRV infection at 37°C. Virus was then removed and washed off with two PBS washes and replaced with fresh media containing the drug. At 24 h p.i., the media were refreshed with new U0126. Cells were collected at 48 h p.i.
Phase-contrast microscopy.
Images of RhF were obtained with a Nikon Eclipse TE-2000-E microscope and an ORCA-ER digital charge-coupled-device (CCD) camera (Hamamatsu) and saved as tiff files using Openlab 5 software (Improvision; Perkin Elmer).
Plaque assays.
As described previously (17), 48 h after RhF were plated in 12-well plates, precleared supernatant from each siRNA condition was serially diluted (5-fold) and applied to cells for 1 h at 37°C. Each dilution was performed in duplicate. Overlay media containing methylcellulose (0.6%) were added to plates containing the viral inoculum. Plates were incubated for 5 days at 37°C. Overlay media were then removed, and cells were stained with crystal violet for 10 min at room temperature. Plaques were counted under ×10 magnification using an inverted microscope (Nikon Eclipse TE-2000-E). Absolute titers were determined based on numbers of plaques and dilutions used. Relative titers were calculated by dividing the absolute titer under each condition by the absolute titer of the control. Likewise, relative virion infectivity values were calculated by dividing relative titers by relative MCP levels (determined by quantitative immunoblotting; see above) and setting the data from control plates (nontargeted siRNA) to an arbitrary value of 1.
Qualitative determination of relative protein levels in the cell and viral particle.
For cell lysates, ERK and viral proteins were first normalized to Ran and then compared to the control to determine relative expression levels. For viral particles, ERK and viral proteins were normalized to MCP.
For quantitative comparisons of ERK1 and ERK2 as well as their ratios within virions and cells under the indicated experimental conditions, we set ERK2 data in the control samples to an arbitrary value of 1 and expressed all other ERK1 and ERK2 values relative to this control value. Ab to total ERK detects ERK1 and ERK2 with the same sensitivity, so no further compensation was necessary. Similarly, we set pERK2 data in the control samples to a value o f1 and expressed all other pERK1 and pERK2 values relative to this control value while also adjusting the pERK1 values to correct for pERK Ab sensitivity bias (see above). For MCP, ORF45, and SCIP, the level of expression of each of these viral proteins in the control was set to 1 and all values in the experimental samples were compared to those of their respective controls.
Quantitative PCR (qPCR). (i) Viral supernatants.
Precleared supernatants were collected under each condition and particles formed into pellets through a 35% sucrose cushion as described above. The viral pellet was resuspended in PBS overnight at 4°C. Viral particles were dissociated by bath sonication, and samples were treated with 5 U DNase I (Stratagene) for 30 min at 37°C to leave only encapsidated DNA. DNase I was inactivated with 50 mM EDTA and an additional 10 min of incubation at 65°C. Samples were then treated with 0.11 mg/ml PK (Sigma-Aldrich) at 55°C overnight. DNA was extracted with phenol-chloroform and precipitated with ethanol along with 1 μg of glycogen carrier to aid in recovery (Glycoblue; Ambion). RRV genomic copy numbers were assayed in triplicate (siERK experiments) or quadruplicate by real-time PCR (SYBR green PCR Master Mix; Applied Biosystems) using primers specific to the ORF45 coding sequence (for ORF45F, 5′-TGATTCGTCCCATGTCTCAA-3′; for ORF45R, 5′-CCTGTTGTTGCTGGATCAAA-3′) and amplified and detected with an ABI Prism 7900 HT detection system instrument at the University of Virginia Biomolecular Research Center. Quantification was based on serial dilution of a plasmid bearing the ORF45 coding sequence, pCMV-Tag2A-R45.
(ii) RRV-infected nuclei.
As previously described (42), we used a modified protocol to isolate nuclei by sucrose gradient centrifugation. Nuclei were treated with PK and DNA extracted as described above. Viral genome equivalents were quantified in quadruplicate by real-time PCR using the ORF45 primers mentioned above. The standard curve was based on serial dilutions of an ORF45 plasmid, pCMV-Tag2A-R45. Primers for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH), GAPDH-F (5′-GAAGATGGTGATGGGATTTCCA-3′) and GAPDH-R (5′-GATTCCACCCATGGCAAATT-3′), were used to normalize the samples.
RT-qPCR.
Total RNA was extracted from approximately 1 × 106 infected RhF using Tri-Reagent (Ambion) per the manufacturer's protocol. Residual DNA was removed from samples by treatment with TURBO-DNase (Ambion), and 0.5 to 1.0 μg total RNA per condition was converted to first-strand cDNA using a 1:1 mixture of oligo(dT) primers and random hexamers and a RETROscript reverse transcription (RT) kit (Ambion), according to the manufacturer's protocol. Diluted cDNA equivalent to 5 to 20 ng of total input RNA, or equivalent no-RT controls, was assayed in quadruplicate by quantitative real-time PCR (SYBR green PCR Master Mix). Specific viral transcripts for ORF25, ORF37, and ORF50 were detected using previously designed primer sets (14). Results were normalized to GAPDH mRNA, using the primers described above, on sister aliquots of the cDNA.
Statistical analysis.
Quantitative data are presented as means and standard errors of the means (SEM). Statistically significant differences between conditions were determined using an unpaired Student's t test. All calculations were performed using GraphPad Prism online software. To denote statistical significance, we used the following P value conventions: P ≥ 0.05, not statistically significant (ns); *, P < 0.05; **, P < 0.01; and ***, P < 0.001. In calculating the variance in the ratios of ERK2 to ERK1 species within the virion and cell, we propagated the error of the quotient by the formula , where SD represents the standard deviation and A and B represent the respective values of the two ERK species being compared.
Mass spectrometry.
Highly purified virions were diluted in SDS sample-loading buffer and boiled for several minutes. The sample was then loaded onto a one-dimensional (1D) SDS-PAGE gel (25% in each of three lanes) and run for only 1 cm to purify the viral proteins. Two lanes were digested with trypsin, and one lane was digested with chymotrypsin. C18 and Ti-enriched runs were done for both enzymes.
The gel piece was transferred to a siliconized tube and washed and destained in 200 μl of 50% methanol overnight. The gel pieces were dehydrated in acetonitrile, rehydrated in 30 μl of 10 mM dithiothreitol in 0.1 M ammonium bicarbonate, and reduced at room temperature for 0.5 h. The DTT solution was removed and the sample alkylated in 30 μl of 50 mM iodoacetamide in 0.1 M ammonium bicarbonate at room temperature for 0.5 h. The reagent was removed and the gel pieces dehydrated in 100 μl acetonitrile. The acetonitrile was removed and the gel pieces rehydrated in 100 μl of 0.1 M ammonium bicarbonate. The pieces were dehydrated in 100 μl of acetonitrile, the acetonitrile removed, and the pieces completely dried by vacuum centrifugation. The gel pieces were rehydrated in 20 ng/μl enzyme in 50 mM ammonium bicarbonate on ice for 10 min. Any excess enzyme solution was removed and 20 μl of 50 mM ammonium bicarbonate added. The sample was digested overnight at 37°C, and the peptides formed were extracted from the polyacrylamide in two 30-μl aliquots of 50% acetonitrile–5% formic acid. These extracts were combined and evaporated to 15 μl for MS analysis.
The liquid chromatography-mass spectroscopy (LC-MS) system consisted of a Finnigan LTQ-FT mass spectrometer system with a Protana nanospray ion source interfaced to a self-packed Phenomenex Jupiter 10-um-pore-size C18 reversed-phase capillary column (8 cm by 75 μm inner diameter). Volumes of the extract (5 to 10 μl) were injected and the peptides eluted from the column by an acetonitrile–0.1 M acetic acid gradient at a flow rate of 0.4 μl/min. The nanospray ion source was operated at 2.8 kV. The digest was analyzed using the double-play capability of the instrument, acquiring full-scan mass spectra to determine peptide molecular weights and product ion spectra to determine amino acid sequences in sequential scans. This mode of analysis produces approximately 6,000 collisionally activated dissociation (CAD) spectra of ions ranging in abundance over several orders of magnitude. Not all CAD spectra are derived from peptides.
Ti enrichment was performed using the following procedure. The Ti column (3 cm by 100 μm) was washed with a solution of 0.5% NH4OH–40% ACN (pH 10.5) followed by equilibration with 2% formic acid–20% ACN. The sample was loaded in the equilibration buffer and then washed in equilibration buffer with 80% ACN. The final wash was performed with 2% formic acid. The sample was then eluted directly with 200 mM ammonium bicarbonate (pH 9.0) into a C18 precolumn, and this column was washed with 0.1 M acetic acid.
The data were analyzed by database searching using the Sequest search algorithm against RRV and the human International Protein Index (IPI). Any potential phosphopeptides were manually validated.
RESULTS
Selective incorporation of pERK2 in RRV virions.
In addition to detecting over 30 virally encoded proteins comprising RRV particles (51), analysis of our more recent tandem mass spectrometry (MS/MS) data using highly purified particles also revealed the presence of a number of cellular proteins (see Table S1 in the supplemental material), including a mitogen-activated protein kinase (MAPK), ERK2 (Fig. 1A). On each of two separate viral preparations, MS/MS analyses revealed 9 and 12 distinct tryptic peptides, respectively (for a total of 42.5% coverage), mapping to ERK2 (Fig. 1A, shaded in gray). Although previous proteomic analyses of other herpesviruses have not reported the presence of such signaling molecules within mature gammaherpesvirus virions (5, 81), we reasoned that RRV-associated ERK2 might simply arise from elevated intracellular concentrations of this signaling molecule during RRV assembly. Of note, however, this approach failed to detect evidence of ERK1 within the virions.
Fig 1.

Preferential incorporation of ERK2 into RRV particles. (A) MS and MS/MS analyses of sucrose gradient-purified RRV particles identified multiple tryptic peptides (shaded) that mapped to ERK2. MS/MS analysis of these samples following titanium enrichment identified a single ERK2 phosphopeptide (boxed sequence, with the phosphorylated residues [threonine 185 and tyrosine 187] in white with black outlining). These sites are the known MEK-specific target sites. (B) Supernatants and cell pellets from RRV-infected RhF were harvested at 72 h p.i., virions were formed into pellets and then gradient purified from the supernatant (lane 1), and whole-cell lysates from the RRV-infected RhF (Inf; lane 2) and mock-infected RhF (Mock; lane 3) were immunoblotted for viral structural proteins MCP, ORF45, and SCIP, as indicated, as well as for total ERK (ERK1/2). The cellular protein Ran served as a loading control for the cellular extracts (lanes 2 and 3). (C) The blots presented in panel B were stripped and reprobed for ERK2 to confirm its position on the immunoblot. (D) The blots presented in panel B were stripped and reprobed with an antibody that specifically recognizes diphosphorylated/activated (pT185/pY187) ERK1/2 to confirm the MS data. (E) The blot presented in panel B was also stripped and reprobed for another activated (nuclear) MAPK, phospho-p38 (pp38).
To confirm the MS data, we separated the virion-associated proteins of gradient-purified RRV by SDS-PAGE and performed immunoblot analyses, using a quantitative, nonenzymatic infrared detection system (see Materials and Methods) to probe for three known virally encoded structural proteins, the major capsid protein (MCP/ORF25), a tegument protein encoded by ORF45, and the small capsomer-interacting protein (SCIP/ORF65), as well as for ERK1 and ERK2 (ERK1/2), using an antibody that recognizes both isoforms equally (Fig. 1B, lane 1). The results of this representative experiment (performed using >5 separate viral preparations) indicated that all three structural proteins were evident in the particle, along with a relative abundance of the faster migrating of the two ERK species. The immunoblot results demonstrated that the virions contained only a trace amount of the slower-migrating species. Due to the migration of these two ERK bands, we assumed that the top band represented ERK1 (p44) and the dominant bottom band represented ERK2 (p42). To confirm their identities, we stripped the blot and reprobed with a monospecific antibody to ERK2 (Fig. 1C). The bands of ERK2 were superimposable on the lower bands of each ERK pair in Fig. 1B. The abundance of ERK2 compared to ERK1 within the virions provided a possible explanation for our consistent detection of only the former isoform in multiple MS analyses of the virions.
To determine if the ERK2 bias within virions resulted from preferential incorporation of one isoform over the other or, instead, simply reflected their relative abundances within the cell, we also analyzed, in parallel, the infected and uninfected cell lysates (Fig. 1B, lanes 2 and 3, respectively), probing the blots for the two ERK isoforms along with MCP, ORF45, and SCIP. ERK1, ERK2, and Ran (the cellular loading control) were all present in both the infected and uninfected cell lysates, while the viral proteins were evident only in infected cells. We noted that uninfected cells had approximately equal levels of ERK2 and ERK1 (Fig. 1B, lane 3), while infected cells had slightly more ERK2 than ERK1 (Fig. 1B, lane 2), but, as we noted above, purified virions showed clear evidence of ERK2, with only trace amounts of ERK1 (Fig. 1B, lane 1). This suggested that the virion content of the two ERK isoforms was not simply a sampling of the intracellular environment of the producer cell.
We also noted an upward mobility shift in the ERK2 band present in the infected compared to uninfected cells that was also evident and even further shifted in the virions (Fig. 1B and C). Since a variety of viruses, including KSHV, induce ERK phosphorylation upon infection (3, 4, 8, 62, 66), we hypothesized that this mobility shift in the ERK2 protein band might similarly be due to its phosphorylation status. To begin to test this idea, we stripped the ERK blot and reprobed with an antibody that specifically recognizes dually phosphorylated (fully activated) ERK (pERK in Fig. 1D). In the absence of activating stimuli, uninfected cells contained only minimal levels of pERK, but infected cells, and purified virions released from these cells, showed markedly increased levels of pERK2 and a relative paucity of pERK1 (compare lanes 1 to 3 in Fig. 1D). Of note, persistent intracellular activation of ERK required infectious virions. In contrast, exposure to UV-inactivated RRV led to a rapid but transient rise in the level of intracellular pERK2 that returned to baseline by 2 to 4 h (E. N. Woodson and D. H. Kedes, unpublished data). After normalizing for gel loading differences, we found that the levels of pERK1 in infected cells were only minimally higher than in uninfected controls (compare upper pERK bands and their respective Ran bands in lanes 2 and 3 of Fig. 1D to panel B data). Since the pERK2 bands were superimposable on the shifted ERK2 bands (lanes 1 and 2 in Fig. 1B and C), we concluded that the lower ERK bands in virions and infected RhF lysates contained pERK2 with or without additional posttranslational modifications.
To determine if ERK2 was phosphorylated at any other amino acids in addition to those recognized by the pERK antibody (pT185 and pY187), we used MS/MS to analyze highly purified virions after first enriching for phosphopeptides (see Materials and Methods). The majority of the phosphopeptides mapped to virally encoded structural proteins, but, notably, the only sites mapping to ERK2 were the MEK phosphorylation sites on ERK2 (T185 and Y187) (63, 64, 68, 78) (Fig. 1D and highlighted residues within the annotated sequence in panel 1A) whereas no phosphopeptide(s) mapped to ERK1. In contrast, even though phosphatase treatment eliminated the ability of the pERK-specific antibody to recognize the ERK2 species in the immunoblots, reprobing with ERK1/2 antibody showed that this treatment failed to reverse the shifts (not shown), suggesting that ERK2 species within the infected RhF and virions also underwent one or more additional posttranslational modifications that we have, to date, been unable to identify. Nevertheless, together, these data indicated that RRV infection led to activation of ERK2 and the preferential incorporation of this isoform during viral assembly. Finally, to further address whether virion incorporation of pERK2 was directed rather than reflecting a random incorporation of any one of the many activated signaling molecules within infected cells, we probed for another phosphoprotein, pp38. This activated protein also localizes to the nucleus in response to a variety of stimuli (6, 63) and became phosphorylated following RRV infection (Fig. 1E, lane 2). However, in contrast to our findings with pERK2, we were unable to detect pp38 within the virion (Fig. 1E, lane 1). Of note, we were similarly unable to detect Jun N-terminal protein kinase (JNK) within the virion (not shown). Taken together, these data argue for the specificity of pERK2 packaging within mature virions.
pERK2 localizes to the tegument of RRV.
Since cellular proteins within herpesviruses tend to localize to the tegument layer of the particle (5, 31), we hypothesized that pERK2 would reside within this same layer in RRV. To determine the specific location of pERK2, we subjected virions to different combinations of proteolytic as well as detergent treatments, followed by gradient purification of the resulting particles and analysis by immunoblotting. This approach served to localize pERK2 within the virion while also ensuring that it was not simply adhering nonspecifically to the virion surface. Since each viral capsid contains 955 molecules of MCP (20, 49, 51), we probed for and then quantified MCP in each sample to normalize for particle number in parallel to determining the relative abundance of pERK2 under each condition (Fig. 2). Again, untreated RRV particles predominantly contained pERK2 (Fig. 2, lane 1), and this signal persisted even after treatment with proteinase K (PK) (Fig. 2, lane 2), which removes proteins that adhere nonspecifically to the virion surface (51). As a control, we treated lysates from RRV-infected cells (48 h p.i.; MOI of 10) with increasing concentrations of PK under identical treatment conditions, and intracellular pERK was degraded (not shown). This suggested that pERK was protected from PK by the viral envelope. Next, we used Triton X-100 (Fig. 2, lane 3), a nonionic detergent, to solubilize the viral envelope and found that pERK2 remained associated with the purified particle. Even at higher-temperature incubations with Triton X-100 (1 h at 37°C), over 50% of the pERK2 signal remained with the purified particle (not shown). Together, these findings suggested that pERK2 was present within the tegument or, potentially, within the capsid. To distinguish between these two remaining possibilities, we derived untegumented capsids directly from the nuclei of infected cells. Since tegumentation occurs after egress from the nucleus, we reasoned that if pERK2 were in the tegument, the nuclear-derived particles would lack pERK2. We found this to be the case (Fig. 2, lane 4). Another known tegument protein encoded by ORF52 was also absent in the nuclear capsids, as we had anticipated from our earlier work (reference 51 and data not shown).
ERK activation is important in lytic viral gene expression.
Though this paper is the first to report the incorporation of ERK in a herpesvirus particle, previous studies have shown that activation of the MEK/ERK pathway is critical for the production of other herpesviruses (56, 62, 75). Blocking ERK activation with either pharmacological or molecular inhibition of MEK (e.g., siRNA or dominant-negative constructs) (24, 56) completely abrogated virus production. Since our data demonstrated that RRV infection activated ERK (Fig. 1D), we hypothesized that activation of this pathway was also essential for RRV production. To test this, we used the MEK inhibitor U0126 (23) to block ERK activation prior to and during RRV infection and then, with an immunoblot of cell extracts, measured the expression of viral proteins MCP, ORF45, and SCIP as well as cellular proteins ERK1/2, pERK1/2, and Ran. To ensure that U0126 retained its MEK inhibition for the duration of the experiment, we transiently stimulated RhF with the phorbol ester TPA 48 h after treating the cells with either the drug vehicle (DMSO) or U0126 (Fig. 3A, lanes 1 and 2). ERK activation (pERK1/2) was evident in cells treated with TPA in the absence of drug but greatly diminished in cells in the presence of drug, demonstrating its efficacy throughout the duration of the experiments. Even when overloaded on the immunoblot, lysates from uninfected cells showed only trace amounts of ERK activation (Fig. 3A, lane 3). In lysates from infected cells pretreated with vehicle, all three viral proteins were present and ERK was expressed and activated (pERK) (Fig. 3A, lane 4). However, in lysates from RRV-infected cells in the presence of U0126, little to no ERK activation was evident and these cells showed only a trace amount of MCP and no evidence of the other viral proteins on the immunoblot (Fig. 3A, lane 5). U0126 had no significant effect on cell viability even after 48 h of treatment (98% viability with DMSO alone compared to ∼93% viability with the drug). Moreover, U0124, a structurally related but inactive analog of U0126, did not inhibit ERK activation and did not affect expression of the viral lytic proteins and viral titers compared to the results seen with infected cells treated with DMSO alone (data not shown).
Fig 3.

The MEK inhibitor U0126 blocks the activation of ERK in RRV infection, inhibiting viral protein expression and virus production. (A) RhF were stimulated with TPA after 48 h of culture in the presence of drug vehicle DMSO (lane 1) or the MEK inhibitor U0126 (lane 2). RhF were cultured without exposure to RRV (lane 3; overloaded) or with exposure to RRV at an MOI of 5 in the presence of vehicle (lane 4) or U0126 (lane 5). For drug-treated samples, fresh inhibitor was added 24 h p.i. All cells were collected at 48 h p.i. for immunoblot analysis. Blots were probed with the following antibodies as indicated in the figure: anti-MCP, anti-ORF45, anti-pERK, anti-total ERK, anti-SCIP, and, to control for sample loading, anti-Ran. (B) Representative phase-contrast images of RhF that were uninfected (panel i) or infected with RRV (MOI of 5) and either pretreated with vehicle (panel ii) or U0126 (panel iii). (Original magnification for all panels, ×10). (C) Viral titers from the media collected under each condition as described for panel B. The columns represent the means ± SEM of the results of three independent experiments. UI, uninfected. (D) Values for viral genome equivalents in the media collected under each condition indicated were determined by qPCR. Data are from a representative experiment, with bars representing the SEM of the results from qPCR experiments performed in quadruplicate. ***, P < 0.001 (Student's t test).
To further evaluate the effects of blocking ERK activation on RRV production, we also monitored the cells by phase microscopy throughout infection. By 48 h, the uninfected, untreated monolayers remained intact, displaying swirls of elongated cells, characteristic of quiescent fibroblasts in culture (Fig. 3B, panel i). In contrast, infected cells treated with vehicle showed typical signs of lytic infection, with the majority of the monolayer disrupted with rounded cells (Fig. 3B, panel ii). In infected cells treated with drug, however (Fig. 3B, panel iii), the monolayer was mainly left intact, much like uninfected cells, with only a few foci of rounded cells, suggesting that the drug was blocking nearly all virus production. To quantify more precisely the effect this block of ERK activation had on viral production, we determined the titer of the virus released under each condition (see Materials and Methods) (Fig. 3C). In three separate experiments, absolute titers differed but, invariably, those of the U0126-treated RRV-infected cells were more than 2 logs lower than those from infected cells treated with vehicle alone (set to a relative value of 100). We also used qPCR to determine viral genome copy numbers in the supernatants of infected cells treated with or without U0126. As we expected based on the titer data (Fig. 3C), the viral genome copy number was significantly lower (∼30-fold; P = 0.0001) in cells treated with U0126 compared to those treated with the DMSO control (Fig. 3D). Collectively, these data pointed to a critical role for ERK activation in the viral life cycle of RRV, though they did not distinguish at which stage this activation was most important.
Sharma-Walia et al. have shown that ERK is activated at various time points after KSHV infection, including glycoprotein engagement of cellular receptors and entry; however, this activation was most necessary for the establishment of infection and early viral gene transcription (69). To determine if this is also a requirement for RRV, we monitored the effects of U0126 on viral entry by measuring intranuclear viral DNA postinfection in the presence of the drug. Using qPCR, we found no significant difference in the numbers of intranuclear viral genomes 4 h postinfection, whether in the presence or absence of U0126 (Fig. 4A; P = 0.07). In contrast, U0126 dramatically decreased the level of the immediate early gene ORF50 (Fig. 4B). Not surprisingly, we also found that early (ORF37) and late (ORF25) lytic mRNA levels were also greatly decreased in U0126-treated cells (Fig. 4B). As an additional control, we treated infected cells simultaneously with U0126 and a viral DNA polymerase inhibitor, phosphonoacetic acid (PAA), which blocks viral DNA replication, thereby preventing viral genome amplification. The decrease in levels of all lytic transcripts was even greater in the presence of PAA, though these differences did not reach statistical significance (Fig. 4B). Together, these data indicated that RRV, like KSHV, depends on the activation of ERK activation for immediate early gene expression.
Fig 4.

Expression of lytic RRV genes but not episome delivery to the nucleus is dependent on ERK activation. (A) RhF were infected in the absence or presence of U0126. At 4 h p.i, nuclei were isolated and DNA was extracted and analyzed by qPCR using RRV ORF45 primers. The RRV genome copy number was normalized using the human GAPDH copy number (see Materials and Methods). (B) RhF were infected in the absence or presence of U0126 with or without phosphonoacetic acid (PAA). At 48 h p.i., total RNA was extracted from cells with Tri-Reagent. RNA was treated with DNase and converted to first-strand cDNA. Diluted cDNA (5 to 20 ng) was assayed by real-time quantitative PCR (SYBR green) using primers for RRV ORF50 (left columns), ORF37 (middle columns), and ORF25 (right columns). Relative numbers of viral transcripts were normalized to GAPDH mRNA.
Altering the absolute or relative levels of intracellular ERK isoforms produces minimal changes in intracellular viral protein production.
Although initial studies often assumed that ERK1 and ERK2 were interchangeable and served redundant functions (68, 78), more recent data suggest that they may have distinct roles (39, 71). Since we noted a marked and reproducible bias toward ERK2 activation in RRV infection (Fig. 1D, lane 2, and Fig. 3A, lane 4), we chose to test whether ERK2 plays a critical and nonredundant role in RRV infection and virion production. We used a nontargeting siRNA as a control (siCNL) to establish the levels of ERK1 and ERK2 during RRV infection, pretreating and leaving the siRNA in the media during the 48-h infection, and then collected the whole-cell lysate (Fig. 5A, lane 1). In nearly all experiments, the steady-state levels of ERK2 were slightly higher than those of ERK1 in infected cells treated with or without siRNA. To establish relative ERK1 and ERK2 levels among repeated experiments with RRV-infected RhF, we chose to set the ERK2 level from siCNL-treated cells to 1.0 and compared all other ERK1 and ERK2 values to it. (Note that the anti-ERK antibody equally detects both isoforms.) Pretreatment of the cells with siRNA directed to ERK1 (siERK1; Fig. 5A, lane 2) led to a marked (∼84%) knockdown in ERK1 without a significant change in ERK2. siRNA knockdown of ERK2 (siERK2; Fig. 5A, lane 3) resulted in a similar (∼86%) decrease in ERK2 and a slight (∼19%) but not statistically significant increase in ERK1. Finally, siRNA targeting of both ERK1 and ERK2 resulted in ∼87% and ∼83% knockdown of ERK1 and ERK2, respectively (Fig. 5A, lane 4). Figure 5B shows the graphical representation of these data from four independent experiments. Since the loss of one isoform did not result in a compensatory increase in the expression of the other, the results were consistent with the possibility that ERK1 and ERK2 have functionally distinct roles.
Fig 5.

Differential suppression of ERK1 and ERK2 isoforms only minimally affects intracellular viral protein production. (A) RhF were transfected with nontargeting siRNA (siCNL), siERK1, siERK2, or siERK1 plus siERK2, as indicated. At 24 h posttransfection, cells were infected with RRV. Cells were harvested at 48 h p.i., and immunoblots of cell lysates were probed for total ERK (upper panel) as well as Ran to normalize for loading differences (lower panel). The quantitative immunoblot is from a representative experiment. (B) Graphical representation of the expression of each ERK isoform relative to its level in cells treated with siCNL. The ERK2 value was set at 1.0. Data represent the means ± SEM of the results of four separate experiments. P values were determined using Student's t test, comparing the level under the experimental condition to the control level for the corresponding ERK isoform. The values in columns lacking asterisks were not statistically distinguishable from corresponding siCNL values. With siERK1, the P value for ERK1 was 0.0013; with siERK2, the P value for ERK2 was 0.0001; and with siERK1 plus siERK2, the P values for ERK1 and ERK2 were 0.001 and 0.0001, respectively. (C) Cell lysates from the same four siRNA conditions were also probed with antibodies to viral proteins MCP, ORF45, and SCIP as well as to Ran, as indicated. (D) Changes in the expression of the viral proteins presented in panel C compared to that of CNL are represented graphically. Data represent the means ± SEM of the results of the same four separate experiments described for panel B. With siERK1 plus siERK2, the P value for ORF45 was 0.0064. The other levels were not statistically different from control values.
In light of previous studies performed with KSHV that showed that several ERK substrates bind to viral promoters to modulate viral protein expression (56, 72, 75), we predicted that knockdown of one or both of the ERK isoforms would downregulate the intracellular levels of structural viral proteins. However, we found that targeting of ERK1, ERK2, or both by siRNA knockdown failed to profoundly downregulate the expression of these structural viral proteins compared to the siRNA control-treated cell results (Fig. 5C, lanes 2 to 4 versus lane 1), with the exception of a modest decrease in levels of ORF45 in the dual ERK1 and ERK2 knockdown (Fig. 5C, lane 4). Figure 5D depicts quantitatively the combined data from four experiments, all normalized to Ran. Thus, although we found ERK activation to be essential for lytic viral gene and, thus, protein expression (Fig. 3 and 4), the siRNA ERK knockdown results decribed above demonstrated that relatively large changes in total ERK levels had only minor effects on the steady-state levels of viral structural proteins within infected cells, at least at 48 h postinfection.
Residual pools of pERK2 remain following knockdown of ERK1 and ERK2 during RRV infection.
Initially, the MEK inhibitor and siERK results seemed discordant. We were surprised to find persistent production of the viral structural proteins in siERK-treated cells, since blockade of ERK activation essentially abrogated viral protein and virion production (Fig. 3). Though each siERK condition led to marked knockdowns in intracellular ERK during RRV infection, we hypothesized that this might have proportionally lower effects on intracellular pools of pERK. If so, these residual pools of pERK might be sufficient to promote lytic gene expression. To test this idea, we probed immunoblots of infected siERK-treated RhF lysates for pERK (Fig. 6). In controls, we consistently detected more pERK2 than pERK1 even after correcting (see Materials and Methods) for the approximately 3:1-greater sensitivity of the pERK antibody to pERK2 compared to pERK1 (Fig. 6B). With ERK1 knockdown efficiencies of close to 85% (Fig. 5B, lane 2), we found that the average amount of remaining pERK1 was only ∼11% of control levels, while levels of pERK2 were actually slightly (1.22-fold) higher than control levels, but this increase did not reach statistical significance (Fig. 6A and B, lane 2). In contrast, despite >85% knockdown in ERK2 (Fig. 5B, lane 3), residual pools of pERK2 remained at ∼35% of the control levels (Fig. 6B, lane 3). Furthermore, the level of pERK1 rose to nearly double that in the control cells but this increase was shy of statistical significance. Finally, with dual knockdown of ERK1 and ERK2, at 87% and >80% efficiency, respectively (Fig. 5B, lane 4), the residual levels of the corresponding activated isoforms were 30% (for pERK1) and 48% (for pERK2) of their control values (Fig. 6A and B, lane 4). These data show that pERK2 pools were maintained under all siERK conditions and were likely sufficient to drive lytic gene expression and viral production (Fig. 3 to 5).
Fig 6.

Residual pools of pERK2 remain following the siRNA knockdown of ERK isoforms. (A) Representative blot of cell lysates from siERK experiment (Fig. 4) probed with anti-pERK antibody. (B) Relative changes in the levels of pERK1 and pERK2 shown graphically, with columns representing the means ± SEM of the results of four independent experiments. All values were normalized to a value of 1 for pERK2 for cells treated with siCNL, and relative levels of pERK1 were corrected quantitatively for a 3-fold-greater sensitivity for the pERK antibody to detect pERK2 compared to pERK1 (see Materials and Methods). With siERK1, the P value for pERK1 was 0.0013; with siERK2, the P value for pERK2 was 0.0001; and with siERK1 plus siERK2, the P values for pERK1 and pERK2 were 0.0041 and 0.0001, respectively. Columns lacking asterisks did not reach a statistically significance difference compared to corresponding control values.
ERK1 acts as a negative regulator of viral production.
Although knockdown of the expression of ERK1, ERK2, or both during RRV infection led to only minor changes in the intracellular levels of viral structural proteins, this approach did not measure virion formation, release, or infectivity. We reasoned that rapid egress and release of assembled virions would minimize potential differences in intracellular viral protein levels affected by changes in one or more of the ERK isoforms. Due to the preferential activation and incorporation of ERK2 in RRV, we initially hypothesized that decreased levels of ERK2 might result in a decrease in overall viral production. To this end, we harvested viral particles from the media of the siRNA experiments shown in Fig. 5. We loaded particles derived from equivalent volumes of media and then separated the proteins in each sample by SDS-PAGE and probed the immunoblots for structural proteins, MCP, ORF45, and SCIP. We set the control level of each protein to 1.0 (Fig. 7A and B, lane 1) and quantified the changes in protein levels from all other conditions relative to these controls. Remarkably, cells treated with siERK1 gave rise to markedly higher levels of viral particles, as evidenced by parallel elevations in the levels of all three structural viral proteins in the samples we collected from the media following centrifugation through a sucrose cushion (Fig. 7A, lane 2). In contrast, siERK2 knockdown led to few to no discernible differences in particle accumulation in the media (Fig. 7A, lane 3). Cells treated with the dual-ERK knockdown displayed a modest increase in the level of each viral protein (Fig. 7A, lane 4). Figure 7B displays a graphical representation of these data from three independent experiments. Although the overall pattern of particle release remained consistent among the different siRNA conditions, the levels of released particles showed appreciable interexperimental variability. Since the number of MCPs per particle is invariant (955 copies) and since the MCP comprises the structurally resilient capsid, we measured this protein in a total of 7 experiments. We found that levels of particle-associated MCP in the media from cells treated with siERK1 and with siERK1 plus siERK2 were 6- and 1.5-fold greater than in the control media, respectively (Fig. 7B). We also noted that under each of the siRNA conditions, the relative stoichiometries of ORF45 and SCIP per particle (normalized to MCP) did not change appreciably (Fig. 7B), suggesting that the overall structural and compositional integrity of the virions was conserved.
Fig 7.

Knockdown of cellular ERK1 markedly increases levels of virions released into the media. Equal volumes of media from the siERK experiments (Fig. 4) were harvested and cleared of cells and debris. Viral particles were formed into pellets through a 35% sucrose cushion and then analyzed by immunoblotting with antibodies to MCP, ORF45, and SCIP. (The MCP value served as a surrogate marker for the particle number; see text.) (A) Representative quantitative immunoblot of the viral particles released from the cells treated with the indicated siRNAs. (B) Changes in the levels of all three proteins described for panel A shown graphically, with columns representing the means ± SEM of the results of 3 (or 7 for MCP) separate experiments. P values were calculated by comparing the experimental levels to corresponding control (set at 1.0) protein levels: P values for MCP were 0.0273 and 0.017 for siERK1 and siERK1-plus-siERK2 conditions, respectively. Values in columns lacking asterisks did not reach statistical significance compared to control values.
Thus, in contrast to our initial hypothesis that ERK2 levels would play the dominant role in viral production and release, it appeared that ERK1 was perhaps the most critical modulator of these processes and suggested its role as a potent negative regulator. Nevertheless, lowering intracellular levels of ERK2 as well as ERK1 greatly dampened the overproduction of particles evident with ERK1 knockdown alone, suggesting that a minimal level of ERK2 is necessary for maximal virion production.
Intravirion ERK content reflects intracellular ERK expression.
Although the levels of ERK were suppressed within siERK-treated infected cells (Fig. 5A and B), the overall size and shape of the particles released under each condition appeared grossly similar to those in control samples (e.g., sucrose gradient-fractionated particles demonstrated structural protein peaks in parallel fractions; data not shown). In addition, the approximate stoichiometries of the tegument protein ORF45 and the capsid protein SCIP within the particles were similar regardless of the specific siERK condition (Fig. 7). However, it was unclear whether the intracellular ERK manipulations would lead to qualitative or quantitative disturbances in the incorporation of ERK in released virions. We reasoned that if any one of the ERK isoforms (or its corresponding activated form) were an essential structural component of the virus, the virions would incorporate similar numbers of ERK molecules. To approach this issue, we first probed virions produced under each of the siRNA conditions with antibody to total ERK and normalized each value to MCP in each sample, as we have described above. Virions released from cells treated with control siRNA had approximately 4-fold more ERK2 than ERK1 (Fig. 8A and B, lane 1). Those from siERK1-treated cells contained almost exclusively ERK2, with only trace amounts of ERK1, and the amount of ERK2 was nearly double the amount in controls (Fig. 8A and B, lane 2). Particles from siERK2-treated cells contained mostly ERK1 and only ∼5% of the amount of ERK2 present in controls (Fig. 8A and B, lane 3), while those from dual-siERK-treated cells contained slightly more ERK2 than ERK1 and only ∼10% of the ERK2 present in the controls (Fig. 8A and B, lane 4). Although the levels of ERK1 also differed in virions collected under each siERK condition, their differences from controls did not reach statistical significance (Fig. 8B).
Fig 8.

Intravirion ERK content reflects intracellular expression of ERK isoforms. (A) Representative blot of virions isolated from the media of RRV-infected cells treated with siRNA (Fig. 4) and probed with anti-total ERK antibody. (B) Graphical representation of the relative levels of ERK1 and ERK2 in the viral particle, normalized to MCP for each siRNA condition (not shown). Relative values were determined using quantitative immunoblot analysis. With siERK1, the P value for ERK2 was 0.0262; with siERK2, the P value for ERK2 was 0.0001; and with siERK1 plus siERK2, the P value for ERK2 was 0.0001. (C and D) Parallel analyses performed as described for panels A and B but for virion-associated levels of pERK1 and pERK2, respectively. The data represent the means ± SEM of the results of four independent experiments. With siERK1, the P value for pERK1 was 0.0227; with siERK2, the P value for pERK2 was 0.0001; and with siERK1 plus siERK2, the P values for ERK1 and pERK2 were 0.0247 and 0.0001, respectively. Columns without asterisks indicate the lack of statistically significance difference compared to their respective controls. Values for ERK2 (B) and pERK2 (D) were set to 1.0.
Though these data demonstrated that the intravirion total ERK content approximated the relative levels of the intracellular ERK isoform(s) following siRNA manipulations (compare Fig. 5A and 8A), it was still possible that proper assembly, structural integrity, or even infectivity might, instead, depend on the directed incorporation of pERK1 or, more likely, in light of our virion composition data, pERK2. If such a condition were critical for any of these or other unknown functions, we predicted that released infectious virions would likely preserve approximately equivalent amounts of pERK2 even in the face of siRNA perturbations in intracellular ERK levels. To address this possibility, we probed immunoblots of the virions from each siRNA condition for pERK (Fig. 8C and D). Control virions contained mainly pERK2, with minor amounts of pERK1 (Fig. 8C and D, lane 1). Virions from siERK1-treated cells contained only trace amounts of pERK1 but amounts of pERK2 approximately equivalent to those seen with controls (Fig. 8C and D, lane 2). The complementary knockdown with siERK2 led to released virions with 85% less pERK2 than in controls (Fig. 8C and D, lanes 3 and 1). In contrast, the amount of pERK1 in these virions was essentially unchanged (Fig. 8C and D, lanes 3 and 1), even though intracellular pERK1 levels more than doubled compared to the levels in control cells (Fig. 6B, lanes 3 and 1). Finally, we noted that these virions contained approximately equal amounts of the two pERK isoforms (Fig. 8C and D, lane 3) even though intracellular levels of pERK2 were nearly 2.5-fold lower than of pERK1 (Fig. 6B, lanes 1 and 3). Virions released from dually siERK-treated cells had levels of pERK1 and pERK2 that were lower than in control virions by ∼82% and ∼86%, respectively, but pERK2, again, remained the dominant species (Fig. 8C and D, lane 4).
Alterations in intravirion ERK content maintain or enhance virion infectivity.
We next tested whether the perturbations in the intracellular ERK content that led to differing amounts of particle production (Fig. 7A and B) might also affect viral infectivity. Since the various ERK knockdowns also affected intravirion ERK content (Fig. 8), we also tested whether the relative infectivities of virions in culture might change as a consequence of either quantitative or qualitative differences in their ERK content. We first determined the viral titer of the media from the infected RhF following knockdown of ERK1, ERK2, or both and compared these conditions to the control (Fig. 9A). To ensure that siRNA treatment did not negatively affect the efficiency of viral infection, we infected cells with or without control siRNA. We found no statistically significant difference in the number of infectious particles released from cells treated with or without control siRNA (2.8 ± 1.2 × 107 compared to 2.9 ± 1.2 × 107 PFU/ml, respectively). In contrast, compared to siRNA-treated controls, the titer in the media from the ERK1 knockdown condition was significantly greater, with an increase of 12.9-fold ± 5.0-fold relative to controls (Fig. 9A, columns 1 and 2), suggesting, at a minimum, that the large number of particles released following ERK1 knockdown (Fig. 7) retained their infectivity. (Note that, although the absolute titers determined between repetitions of these experiments showed biological variability [e.g., 1.92 × 10E6 to 1.44 × 10E7 PFU/ml for siCNL], the patterns of effects of the different siRNA conditions within each experiment were remarkably consistent.) Though the titers from the ERK2 and dual-knockdown conditions were slightly higher than the controls, these differences did not reach statistical significance (Fig. 9A, columns 3 and 4).
Fig 9.

Alterations in intravirion ERK content maintain or enhance virion infectivity. (A) Viral titers in the media of RRV-infected RhF treated with the indicated siRNA were determined in duplicate. For each experiment, relative titers were calculated by dividing the titer determined under each condition by the titer in the corresponding siCNL. With siERK1, the P value for the difference in titer was 0.01. (B) Relative virion infectivity (see Materials and Methods) was calculated by dividing the titers determined as described for panel A by the respective relative values of MCP (Fig. 5) and then comparing these values to those from siCNL. Each column represents the mean ± SEM of the results of 3 independent experiments. With siERK1 and siERK2, the P values for the titers compared to those of controls were were 0.03 and 0.04, respectively.
In separate experiments, we also compared titers to viral genome equivalents in the media from each siRNA condition. These experiments, again, showed that the ERK1 knockdown condition gave the highest titer (though it was only 4.5-fold higher in this series) compared to the control siRNA condition. However, surprisingly, the qPCR measurements of genome equivalents were not significantly different for any of the conditions (siCNL, 1.0 × 107; siERK1, 8.4 × 106; siERK2, 1.3 × 107; and siERK1/2, 8.3 × 106 genome equivalents/ml). Since differences in the encapsidated DNA content did not mirror the increased titer in the media from the ERK1 knockdown condition (at least within the precision of qPCR), these data suggested that ERK1 knockdown may lead to the production of particles with higher infectivity.
As we discussed above, the intensity of the MCP signal on a quantitative immunoblot of virions correlates well with particle number. We exploited this linear relationship to calculate the relative amounts of MCP in each sample, allowing us to normalize the titers to the relative numbers of particles. The titers (Fig. 9A) and corresponding MCP immunoblot signals (Fig. 7B) represented parallel analyses of sister aliquots in each experiment. For each experiment, we divided the absolute titer under each condition by its corresponding MCP value and compared each to the control, which we set to 1.0 (Fig. 9B, column 1). Particles from the siERK1 condition (Fig. 9B, column 2) were slightly more than 3 times more infectious than control particles. In addition, virions produced from the siERK2 condition (Fig. 9B, column 3) were also more infectious (at almost 2 times greater than the control level). The infectivity of virions from the dual-siERK condition, however, was not significantly different from those from control samples (Fig. 9A and B, columns 4). We noted that the viral species that were most infectious (those from siERK1) contained approximately twice as much ERK2 as and significantly less pERK1 than the control, whereas the only distinguishing component of virions from siERK2 was that these virions maintained the levels of pERK1 evident in control virions. Thus, it seems that the amount and level of activation of the ERK isoforms were not sufficient to predict these modest differences in relative infectivity of the virions. In the end, the data argue that RRV can tolerate dramatic reductions in both total and activated ERK isoforms as well as variability in the relative abundances of these species without a significant loss in infectivity.
Biased incorporation of ERK2 and pERK2 into RRV particles.
We noted that the relative amounts of ERK1 and ERK2 as well as pERK1 and pERK2 within virions generally paralleled their relative abundances in the infected RhF under each siERK condition (compare Fig. 8B with 5B and 8D with 6B, respectively). However, it was also evident that virions contained a disproportionate amount of ERK2 and pERK2, suggesting a bias toward incorporation of these species in preference to ERK1 and pERK1, respectively, during virion assembly. To more precisely evaluate this potential selectivity, we determined the ratio of ERK2 to ERK1 as well as pERK2 to pERK1 in cells and virions under each siERK condition.
In RRV-infected cells treated with nontargeting siRNA (siCNL), we found that the ratios of ERK2 to ERK1 and pERK2 to pERK1 were 1.81 ± 0.26 and 2.61 ± 0.38, respectively; however, the corresponding ratios were consistently and markedly higher in virions (7.92 ± 2.38 and 7.71 ± 1.0, respectively) (Fig. 10, upper and lower panels, first column sets). This bias favoring the packaging of ERK2 (and pERK2) in newly formed virions remained evident following marked reductions in intracellular ERK1 levels (Fig. 5A and B, lane 2). In this setting, the ratio of ERK2 to ERK1 and pERK2 to pERK1 in cells rose to 10.2 ± 1.2 and 26.8 ± 5.0, respectively. Nevertheless, the virions released from these cells showed ratios that were, again, even higher (29.3 ± 5.1 and 64.35 ± 7.8, respectively) (Fig. 10, upper and lower panels, second column sets).
Fig 10.
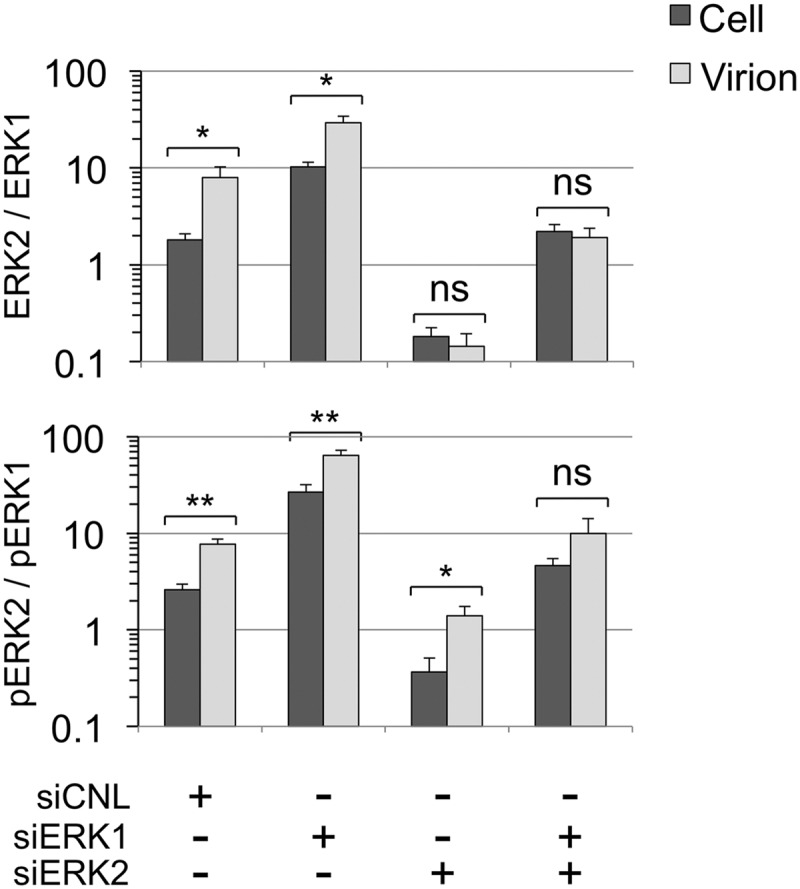
Biased incorporation of ERK2 and pERK2 in RRV virion assembly. Using data from Fig. 4, 6, and 7, involving 4 independent experiments, we determined ratios of ERK2 to ERK1 (upper panel) and pERK2 to pERK1 (lower panel) in cells and virions under each of the four knockdown conditions. The results are shown graphically on a log10 scale, since the ratios differed by nearly 2 orders of magnitude. The brackets and asterisks above each pair (cell and virion) indicate the level of statistical significance of the results of comparisons of virion and cellular ratios for each indicated knockdown condition. ns, not significant. THe P values for ERK2:ERK1 were 0.0434, 0.216, 0.552 (ns), and 0.6299 (ns) for siCNL, siERK1, siERK2, and siERK1 plus siERK2, respectively. The P values for pERK2:pERK1 were 0.003, 0.0079, 0.031, and 0.1953 (ns) for siCNL, siERK1, siERK2, and siERK1 plus siERK2, respectively.
The total ERK2 bias, however, was no longer evident following the ERK2 or dual knockdown in the cell (Fig. 10, upper panel, third and forth column sets). The ratios of ERK2 to ERK1 in the virions and cells were statistically indistinguishable in the ERK2 knockdown (0.14 ± 0.05 and 0.18 ± 0.04, respectively; Fig. 10, upper panel, third column set). In the double knockdown, the ratios were somewhat higher in both the virions and cells but, again, the values determined for virions and cells were statistically indistinguishable (1.91 ± 0.47 and 2.22 ± 0.39, respectively; Fig. 10, upper panel, fourth column set). In contrast, the virions continued to display a strong incorporation bias for pERK2 in preference to pERK1 even in the setting of low relative abundance of intracellular pERK2 (siERK2). The ratio of pERK2 to pERK1 was 1.40 ± 0.34 in virions but only 0.36 ± 0.14 in cells (Fig. 10, lower panel, third column set). This trend continued for ratios of pERK2 to pERK1 in the virions from the double knockdown, but the difference did not reach statistical significance (9.99 ± 4.12 and 4.61 ± 0.88 for virions and cell, respectively). Taken together, these data suggested that, compared to the intracellular milieu from which these virions derived, released virions appeared to preferentially incorporate total but, particularly, activated ERK2 (pERK2) over the corresponding ERK1 species by a margin of 3 or 4 to 1.
DISCUSSION
Increasingly detailed proteomic analyses of highly purified herpesviruses have revealed that both viral and cellular components contribute to virion structure (5, 31, 51, 81). This raises the intriguing notion that herpesviruses may have evolved to incorporate cellular proteins to play functional as well as structural roles during infection. In the current study, we initially focused our efforts on identifying the cellular components of highly purified RRV, using tandem mass spectrometry followed by quantitative immunoblot analyses. We found a subset of cellular proteins, some of which, such as actin, moesin, and heat shock proteins, are also present in KSHV (5, 81) (see Table S1 in the supplemental material). However, the present report is the first to identify the MAPK ERK2 within a herpesvirus particle. Furthermore, we found that at least some, if not the majority, of the intravirion ERK2 is in its phosphorylated/activated (pERK2) form. Using a biochemical approach, we were additionally able to localize pERK2 to the tegument layer. We also noted that, compared to cellular pERK2, intravirion pERK2 displayed slower electrophoretic mobility, suggesting the possibility of an additional posttranslational modification. Although we found that additional phosphorylation was not the culprit, the exact cause of this mobility shift remains unclear.
Our data also demonstrated that de novo RRV infection activates ERK (and particularly ERK2) and that activation of this pathway is necessary for RRV lytic protein and viral production. Pharmacologic inhibition of MEK blocked ERK activation, abrogating virus production (Fig. 3). Many viruses, including KSHV, exploit this pathway to promote virus production (3, 4, 8, 24, 29, 37, 46, 56, 62, 65, 69, 74). Therefore, we were not surprised to detect activated ERK in RRV-infected cells. High levels of pERK2 at times late in lytic infection could, in theory, be sufficient to drive its incorporation passively into virions, but our data suggest that incorporation is more specific, as we discuss below.
We found that RRV infection led to preferential activation of ERK2 and that this activation was necessary for the expression of ORF50, the immediate early gene which encodes the replication and transcription activator (RTA) that is both necessary and sufficient to initiate lytic replication in gammaherpesviruses (14, 16, 70). Since lytic replication in herpesviruses follows an ordered cascade of gene expression (14), we expected the decrease in both early and late gene transcription (Fig. 4) as well in the levels of lytic proteins and, ultimately, virion production (Fig. 3). Nevertheless, siRNA knockdown of the ERK isoforms indicated that as little as 10% of the amounts in control cells was sufficient to maintain virion production at levels indistinguishable from those in controls (Fig. 7). We submit that this seemingly contradictory finding likely reflected the attenuated effect of the knockdown approach on the activated form of ERK2. Levels of pERK2 in the cell remained at 35% to 50% of control values even with up to 90% knockdown of total ERK2 (Fig. 6). Thus, it seems that, even in the face of a relative scarcity of total cellular ERK2, RRV infection drives MEK to phosphorylate and, thus, activate a greater proportion of the residual ERK2, contributing to the maintenance of the pERK2 levels necessary for lytic gene expression.
In marked contrast to the modest effects we observed with suppression of ERK2 levels, siRNA targeting of ERK1 effectively reduced residual levels of both total and phosphorylated ERK1 to ≤5% of control levels and resulted in marked overproduction of infectious virions. These data strongly implicated ERK1 as a negative regulator of RRV production. Decreasing levels of both ERK isoforms led to an intermediate increase in virus production, suggesting that maximal (over)production of RRV requires full levels of pERK2 and that pERK2 at 40% to 50% of the control levels is limiting in the absence of the normally inhibitory effects of ERK1 (Fig. 7).
Selective incorporation of ERK2 into RRV.
Since the number of different types of cellular proteins in the purified RRV virions is highly restricted and excludes most abundant cellular proteins, this suggests a priori that the incorporation of pERK2 is likely selective. Buttressing this conclusion, we found that even though de novo RRV infection activates other signaling proteins (e.g., MAPK [pp38]) with localization patterns similar to those of pERK, these proteins are consistently absent from the virions (Fig. 1D and E). Nevertheless, the consistent presence of pERK2 in the RRV tegument might, in theory, simply result from the high intracellular concentration of this signaling molecule during the later stages of lytic infection. Arguing against this idea of passive incorporation, however, is our observation that the virions contain approximately 3-fold more pERK2 than pERK1 even after our suppression of pERK2 within the cell to levels lower than those of pERK1 (Fig. 10).
Of note, we also detected an ERK2 incorporation bias using the less-specific antibody that detects total (not just activated) ERK. However, the bias was evident only in virions from the control and ERK1 knockdown conditions. This suggests that the mechanism of incorporation likely targets pERK2 and not total ERK2. We speculate that, under the ERK2 and ERK1/ERK2 knockdown conditions, there might have been relatively greater amounts of the inactive (lacking phosphorylation at T202/Y204 [ERK1] and T185/Y187 [ERK2]) forms compared to the activated ERK isoforms within the virions. Thus, if the two inactive isoforms were present within the virions in amounts that were approximately equal but still significantly greater than those of the activated forms, the antibody to total ERK would be unable to reveal any differences in pERK1 and pERK2 content, thereby masking the pERK2 incorporation bias.
Consistent with our observations of selective incorporation of pERK2 specifically and cellular proteins in general, both the Ganem and Yuan laboratories have demonstrated that KSHV incorporates only a small subset rather than a more random sampling of the viral and cellular proteins present in the producer (PEL) cell lines (5, 81). The Ganem group, for example, showed that KSHV incorporates a slower-migrating form of RTA (ORF50) even though it is less abundant than the faster-migrating isoform during lytic induction (5). Likewise, Winkler and Stamminger demonstrated that human cytomegalovirus (HCMV) particles incorporate only the phosphorylated form of UL69 (73). These studies, together with our findings, suggest that herpesviruses selectively package at least a subset of cellular proteins.
The mechanism mediating the pERK2 incorporation bias remains unclear. One possibility, however, is that it reflects an increased affinity of another, structurally essential protein for ERK2/pERK2 in preference to ERK1/pERK1. One such candidate for RRV is the protein encoded by ORF45, since recent work with KSHV bacterial artificial chromosomes (BACs) and single-gene transfections into heterologous cell lines has indicated that the KSHV ORF45 protein interacts with both pRSK, a downstream target of pERK, and pERK, forming a triplex that protects the two phosphoproteins from host phosphatases, such as negative regulators of the ERK pathway PP2A and MKP-1 and -3 (7, 78). In this model system, the investigators observed sustained intracellular levels of pERK during both primary infection and reactivation. Of note, they also showed that preferential activation of ERK2 and preferential binding of RSK, the bridging protein, to ERK2 occurred only in the presence of KSHV ORF45 (36, 37). Since our earlier work showed that RRV ORF45 is also a tegument protein (51) and that severe knockdown of ERK2 led to a minor drop in the level of intracellular ORF45 (at least as evident in the dual-ERK1/ERK2 knockdown), it is plausible that the homologous ORF45-RSK-ERK2 triplex may also form in de novo RRV infection and serve as the means of governing pERK2 incorporated into the particle. Although we have also detected an RRV ORF45-pERK2 interaction, our current studies are aimed at verifying direct interaction between RRV ORF45 and RSK in infected cells or the particle (Woodson and Kedes, unpublished).
Intravirion pERK2: preparing the cell for infection or modulating viral infectivity?
Several studies have shown that both cellularly derived and virally encoded kinases target proteins within the virion and the cell (9, 25–28, 53, 76). Thus, it is possible that intravirion pERK2 could target adjacent viral or cellular proteins. Using an antibody that detects MAPK substrate consensus sequences (phosphoserine in a PXS*P or an S*PXR/K motif as well as a PXS*PXR/K motif), we recently found that there are several potential ERK targets within the virion (Woodson and Kedes, unpublished), but whether or not the phosphorylation occurs prior to or after virion assembly and the functional outcome of these phosphorylations remain unclear. Since pERK2 resides within the tegument, membrane fusion-mediated viral entry likely results in immediate release of pERK2 into the cell, where it could begin to modulate the host cell environment even before the capsid delivers the viral genome to the nucleus (5, 31, 81). It is reasonable to speculate, for example, that intravirion pERK2 might target cellular transcription factors necessary to promote early viral gene expression (35, 47, 56, 69). Such a model would, again, imply that the incorporation of pERK2 into virions represents not a passive event but an evolutionarily selected process. If true, it would predict that lowering the levels of intravirion pERK2 would disrupt the early stages of infection and possibly the efficiency of infection, including the levels of viral production.
ERK2 does modulate infectivity in other viruses. Work with HIV-1, for example, has revealed that ERK2 phosphorylates p6gag, a viral protein that functions in virion release. Mutation of the phosphosite in p6gag leads to the accumulation of immature virions unable to separate from the host cell membrane (26). Further, a more recent paper by Gupta et al. also showed that ERK2 is present in HIV-1 and simian immunodeficiency virus (SIV) particles and plays a role in phosphorylating viral proteins such as Vpr and Vpx that may be important for the replication of these viruses in nonreplicating primary cells (25).
To determine whether this is also true for RRV, we first showed that the protein composition of the virions released from infected cells following knockdown of ERK1, ERK2, or both remained (at least within the precision of the quantitative immunoblot assays) fundamentally unchanged (Fig. 7). This suggests that virions having various amounts and ratios of the ERK species were otherwise structurally similar to control virions. Nevertheless, we predicted that, if early release of pERK2 from the entering virion were important in effectively jump-starting the lytic cascade of viral gene expression, then the virions with low absolute amounts of pERK2 might lead to less-robust infections, i.e., lower infectivity per particle. Instead, our data indicated that virions with these experimentally induced perturbations in their ERK content were as infectious as or even somewhat more infectious per particle than control virions (Fig. 8 and 9). Virions containing amounts of pERK2 equivalent to those seen with control virions but with lower levels of ERK1 were the most infectious on a per-particle basis. However, titers of virions harvested under siERK1-plus-siERK2 conditions, containing significantly less pERK2 (and pERK1), were statistically indistinguishable from those of control virions. Interestingly, we also determined viral genome copy numbers in the media and found that there were similar amounts of genome equivalents under each condition. The increase in titer from the siERK1 condition, despite the approximately equal amounts of encapsidated genomes seen under all conditions, suggested that there might be more defective particles released under the siCNL, siERK2, and siERK1/2 conditions than under the siERK1 condition. In addition, we found that the per-particle infectivity from the ERK1 knockdown was also greater. However, since the particles produced under the siERK2 condition were also more infectious on a per-particle basis, we cannot attribute virion infectivity to either the amounts of or the ratio between ERK1 and ERK2 or their activated forms, suggesting that viral infectivity may be only minimally dependent on the nature of intravirion ERK content. Nevertheless, ERK1 knockdown may, instead, lead to subtler changes in the protein content or structure of the virions that contribute to infectivity. Finally, it remains possible that a potential role for intravirion pERK2, for example, might be evident only in a different cell type or in an in vivo setting.
Distinct functions for ERK1 and ERK2 in RRV-infected cells.
Increasing numbers of studies have shown that ERK1 and ERK2 have distinct functions in a wide variety of cell types (32, 33, 38, 39, 41, 43, 54, 55, 77). The clear bias toward the incorporation of ERK2 in preference to ERK1 in RRV that we detected, together with the potential that these two isoforms could have nonredundant functions, led us to test the interchangeability of these two kinases in RRV infection and production. We were able to dramatically knock down intracellular levels of both ERK isoforms, with minimal to no changes in the steady-state levels of structural viral proteins within the infected cells. Nevertheless, ERK1 knockdown by itself led to marked virion overproduction and release into the media, identifying this isoform as a negative regulator. We speculate that rapid egress of nascent virions, even in this overproduction setting, leaves the steady-state levels of intracellular virion proteins essentially unchanged.
Though the mechanism is not immediately clear, the striking increase (∼5-fold higher over 48 h) in virion production following knockdown of ERK1 supports a growing body of literature suggesting that ERK1 may have an inhibitory effect on ERK2 signaling. Both in vivo and in vitro studies have demonstrated that removal of ERK1 leads to a gain-of-function phenotype where downstream biological responses are either improved or enhanced. Mazzucchelli et al., for example, reported that though the ERK2 levels in the brain of ERK1 null mice did not increase, the activation of ERK2 was enhanced (43). Similarly, our data indicate that ERK1 knockdown in RRV-infected RhF leads to a modest increase in ERK2 activation and that, as we have demonstrated, this activation, in general, is necessary for virion production. Vantaggiato et al. showed that ERK1 could exert its inhibitory effects by competing with ERK2 for MEK, the upstream activator, such that ERK1 knockdown would lead to an increased association of MEK with ERK2, thereby increasing its activation (71). In the context of infection, it is plausible that the downstream targets of pERK2 could also include upregulation of viral and cellular genes involved in egress and release. Therefore, an increase in ERK2 activation could also contribute to the marked overproduction of virus we observed under siERK1 conditions. As a whole, the data reveal that ERK1 plays a negative regulatory role in RRV production. This is the first example (of which we are aware) of a demonstration of such a role for a virus.
In summary, this report is the first to assign distinct functional roles to ERK1 and ERK2 in viral infection. ERK1 negatively regulates RRV production, whereas activation of ERK, and most likely ERK2, promotes and is critical for viral gene expression. Though levels of ERK1 were only slightly lower than those of ERK2 at 48 h p.i., recent data from our laboratory indicate that they are significantly lower than those of ERK2 at much earlier times postinfection (Woodson and Kedes, unpublished). A slow rise in ERK1 levels over the course of productive infection might serve to orchestrate optimal viral production and release. Low ERK1 levels in the earliest stages of productive infection would favor robust lytic viral gene transcription, while rising ERK1 levels during later stages may dampen this transcription in favor of virion assembly and release. The preferential activation of ERK2 and the maintenance of intracellular pools of pERK2 despite ERK2 knockdown in infected cells suggest that the importance of pERK2 in productive RRV infection extends beyond support of viral gene expression. Finally, the strong bias toward the incorporation of pERK2 in preference to pERK1 suggests that minimal levels of pERK2 may be essential for either the structural integrity of the virion or, perhaps, the optimization of infection in vivo. The results, taken in their entirety, point to separate and distinct functions for ERK1 and ERK2 in RRV infection. We anticipate that RRV is not unique in this regard and that other viruses, including other herpesviruses like KSHV, may likewise exploit the functional differences between these two ERK isoforms to optimally modulate infection in host cells.
Supplementary Material
ACKNOWLEDGMENTS
The research was supported by the NIH (grants R01CA088768, 1R01DE022291, and 5RC2CA148038 [to DHK] and F31 NRSA 5F31CA138100 [to ENW]).
We thank Nicholas E. Sherman at the W. M. Keck Biomedical Mass Spectrometry Laboratory for helpful discussions regarding MS analyses, Daniel Gioeli for helpful scientific discussions, and Geneva T. Dodson for help with statistical analyses. We also thank Scott Wong (Oregon National Primate Center) for providing us with the RRV MCP antibody. Finally, we thank Melissa Anderson and Thomas Ellison for critical reading of the manuscript and providing technical advice.
Footnotes
Published ahead of print 27 June 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Adang LA, Parsons CH, Kedes DH. 2006. Asynchronous progression through the lytic cascade and variations in intracellular viral loads revealed by high-throughput single-cell analysis of Kaposi's sarcoma-associated herpesvirus infection. J. Virol. 80:10073–10082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander L, et al. 2000. The primary sequence of rhesus monkey rhadinovirus isolate 26-95: sequence similarities to Kaposi's sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J. Virol. 74:3388–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrade AA, et al. 2004. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 381:437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barber SA, et al. 2002. Visna virus-induced activation of MAPK is required for virus replication and correlates with virus-induced neuropathology. J. Virol. 76:817–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bechtel JT, Winant RC, Ganem D. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:4952–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. 1998. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr. Biol. 8:1049–1057 [DOI] [PubMed] [Google Scholar]
- 7. Bermudez O, Pages G, Gimond C. 2010. The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am. J. Physiol. Cell Physiol. 299:C189–C202 [DOI] [PubMed] [Google Scholar]
- 8. Cai Y, Liu Y, Zhang X. 2007. Suppression of coronavirus replication by inhibition of the MEK signaling pathway. J. Virol. 81:446–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cartier C, et al. 1997. Association of ERK2 mitogen-activated protein kinase with human immunodeficiency virus particles. J. Virol. 71:4832–4837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 11. Chang Y, et al. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 12. Chen J, Stinski MF. 2002. Role of regulatory elements and the MAPK/ERK or p38 MAPK pathways for activation of human cytomegalovirus gene expression. J. Virol. 76:4873–4885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cohen A, Brodie C, Sarid R. 2006. An essential role of ERK signalling in TPA-induced reactivation of Kaposi's sarcoma-associated herpesvirus. J. Gen. Virol. 87:795–802 [DOI] [PubMed] [Google Scholar]
- 14. Damania B, et al. 2004. Comparison of the Rta/Orf50 transactivator proteins of gamma-2-herpesviruses. J. Virol. 78:5491–5499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Desrosiers RC, et al. 1997. A herpesvirus of rhesus monkeys related to the human Kaposi's sarcoma-associated herpesvirus. J. Virol. 71:9764–9769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DeWire SM, McVoy MA, Damania B. 2002. Kinetics of expression of rhesus monkey rhadinovirus (RRV) and identification and characterization of a polycistronic transcript encoding the RRV Orf50/Rta, RRV R8, and R8.1 genes. J. Virol. 76:9819–9831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DeWire SM, Money ES, Krall SP, Damania B. 2003. Rhesus monkey rhadinovirus (RRV): construction of a RRV-GFP recombinant virus and development of assays to assess viral replication. Virology 312:122–134 [DOI] [PubMed] [Google Scholar]
- 18. Diao L, et al. 2005. Activation of c-Jun N-terminal kinase (JNK) pathway by HSV-1 immediate early protein ICP0. Exp. Cell Res. 308:196–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dittmer DP, et al. 2005. Whole-genome transcription profiling of rhesus monkey rhadinovirus. J. Virol. 79:8637–8650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duffy C, et al. 2006. Characterization of a UL49-null mutant: VP22 of herpes simplex virus type 1 facilitates viral spread in cultured cells and the mouse cornea. J. Virol. 80:8664–8675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dupin N, et al. 2000. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 95:1406–1412 [PubMed] [Google Scholar]
- 22. Dupin N, et al. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Favata MF, et al. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273:18623–18632 [DOI] [PubMed] [Google Scholar]
- 24. Ford PW, et al. 2006. Raf/MEK/ERK signalling triggers reactivation of Kaposi's sarcoma-associated herpesvirus latency. J. Gen. Virol. 87:1139–1144 [DOI] [PubMed] [Google Scholar]
- 25. Gupta P, et al. 2011. Mechanism of host cell MAPK/ERK-2 incorporation into lentivirus particles: characterization of the interaction between MAPK/ERK-2 and proline-rich-domain containing capsid region of structural protein Gag. J. Mol. Biol. 410:681–697 [DOI] [PubMed] [Google Scholar]
- 26. Hemonnot B, et al. 2004. The host cell MAP kinase ERK-2 regulates viral assembly and release by phosphorylating the p6gag protein of HIV-1. J. Biol. Chem. 279:32426–32434 [DOI] [PubMed] [Google Scholar]
- 27. Hui EK. 2002. Virion-associated protein kinases. Cell. Mol. Life Sci. 59:920–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jacqué JM, et al. 1998. Modulation of HIV-1 infectivity by MAPK, a virion-associated kinase. EMBO J. 17:2607–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Järviluoma A, Ojala PM. 2006. Cell signaling pathways engaged by KSHV. Biochim. Biophys. Acta 1766:140–158 [DOI] [PubMed] [Google Scholar]
- 30. Johnson RA, Huong SM, Huang ES. 2000. Activation of the mitogen-activated protein kinase p38 by human cytomegalovirus infection through two distinct pathways: a novel mechanism for activation of p38. J. Virol. 74:1158–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kalejta RF. 2008. Tegument proteins of human cytomegalovirus. Microbiol. Mol. Biol. Rev. 72:249–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krens SF, Corredor-Adamez M, He S, Snaar-Jagalska BE, Spaink HP. 2008. ERK1 and ERK2 MAPK are key regulators of distinct gene sets in zebrafish embryogenesis. BMC Genomics 9:196 doi:10.1186/1471-2164-9-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krens SF, et al. 2008. Distinct functions for ERK1 and ERK2 in cell migration processes during zebrafish gastrulation. Dev. Biol. 319:370–383 [DOI] [PubMed] [Google Scholar]
- 34. Krishnan HH, et al. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krishnan HH, Sharma-Walia N, Streblow DN, Naranatt PP, Chandran B. 2006. Focal adhesion kinase is critical for entry of Kaposi's sarcoma-associated herpesvirus into target cells. J. Virol. 80:1167–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuang E, Tang Q, Maul GG, Zhu F. 2008. Activation of p90 ribosomal S6 kinase by ORF45 of Kaposi's sarcoma-associated herpesvirus and its role in viral lytic replication. J. Virol. 82:1838–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuang E, Wu F, Zhu F. 2009. Mechanism of sustained activation of ribosomal S6 kinase (RSK) and ERK by kaposi sarcoma-associated herpesvirus ORF45: multiprotein complexes retain active phosphorylated ERK AND RSK and protect them from dephosphorylation. J. Biol. Chem. 284:13958–13968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lefloch R, Pouyssegur J, Lenormand P. 2008. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol. Cell. Biol. 28:511–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lloyd AC. 2006. Distinct functions for ERKs? J. Biol. 5:13 doi:10.1186/jbiol46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lyman MG, Randall JA, Calton CM, Banfield BW. 2006. Localization of ERK/MAP kinase is regulated by the alphaherpesvirus tegument protein Us2. J. Virol. 80:7159–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marchi M, et al. 2008. The N-terminal domain of ERK1 accounts for the functional differences with ERK2. PLoS One 3:e3873 doi:10.1371/journal.pone.0003873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marzluff WF, Jr, Murphy EC, Jr, Huang RC. 1973. Transcription of ribonucleic acid in isolated mouse myeloma nuclei. Biochemistry 12:3440–3446 [DOI] [PubMed] [Google Scholar]
- 43. Mazzucchelli C, et al. 2002. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34:807–820 [DOI] [PubMed] [Google Scholar]
- 44. Memar OM, Rady PL, Tyring SK. 1995. Human herpesvirus-8: detection of novel herpesvirus-like DNA sequences in Kaposi's sarcoma and other lesions. J. Mol. Med. (Berl.) 73:603–609 [DOI] [PubMed] [Google Scholar]
- 45. Miller G, et al. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Monick M, Staber J, Thomas K, Hunninghake G. 2001. Respiratory syncytial virus infection results in activation of multiple protein kinase C isoforms leading to activation of mitogen-activated protein kinase. J. Immunol. 166:2681–2687 [DOI] [PubMed] [Google Scholar]
- 47. Naranatt PP, Akula SM, Zien CA, Krishnan HH, Chandran B. 2003. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity. J. Virol. 77:1524–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Naranatt PP, et al. 2004. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 64:72–84 [DOI] [PubMed] [Google Scholar]
- 49. Newcomb WW, et al. 2000. Isolation of herpes simplex virus procapsids from cells infected with a protease-deficient mutant virus. J. Virol. 74:1663–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. O'Connor CM, Damania B, Kedes DH. 2003. De novo infection with rhesus monkey rhadinovirus leads to the accumulation of multiple intranuclear capsid species during lytic replication but favors the release of genome-containing virions. J. Virol. 77:13439–13447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O'Connor CM, Kedes DH. 2006. Mass spectrometric analyses of purified rhesus monkey rhadinovirus reveal 33 virion-associated proteins. J. Virol. 80:1574–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. O'Connor CM, Kedes DH. 2007. Rhesus monkey rhadinovirus: a model for the study of KSHV. Curr. Top. Microbiol. Immunol. 312:43–69 [DOI] [PubMed] [Google Scholar]
- 53. Ott DE. 1997. Cellular proteins in HIV virions. Rev. Med. Virol. 7:167–180 [DOI] [PubMed] [Google Scholar]
- 54. Pagès G, et al. 1999. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 286:1374–1377 [DOI] [PubMed] [Google Scholar]
- 55. Pagès G, Pouyssegur J. 2004. Study of MAPK signaling using knockout mice. Methods Mol. Biol. 250:155–166 [DOI] [PubMed] [Google Scholar]
- 56. Pan H, Xie J, Ye F, Gao SJ. 2006. Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 80:5371–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pleschka S, et al. 2001. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 3:301–305 [DOI] [PubMed] [Google Scholar]
- 58. Rady PL, et al. 1995. Herpesvirus-like DNA sequences in classic Kaposi's sarcomas. J. Med. Virol. 47:179–183 [DOI] [PubMed] [Google Scholar]
- 59. Rady PL, et al. 1995. Herpesvirus-like DNA sequences in non-Kaposi's sarcoma skin lesions of transplant patients. Lancet 345:1339–1340 [DOI] [PubMed] [Google Scholar]
- 60. Rahaus M, Desloges N, Wolff MH. 2006. Varicella-zoster virus influences the activities of components and targets of the ERK signalling pathway. J. Gen. Virol. 87:749–758 [DOI] [PubMed] [Google Scholar]
- 61. Reeves MB, Breidenstein A, Compton T. 2012. Human cytomegalovirus activation of ERK and myeloid cell leukemia-1 protein correlates with survival of latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 109:588–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rodems SM, Spector DH. 1998. Extracellular signal-regulated kinase activity is sustained early during human cytomegalovirus infection. J. Virol. 72:9173–9180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Roux PP, Blenis J. 2004. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 68:320–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rubinfeld H, Seger R. 2005. The ERK cascade: a prototype of MAPK signaling. Mol. Biotechnol. 31:151–174 [DOI] [PubMed] [Google Scholar]
- 65. Sadagopan S, et al. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 81:3949–3968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schümann M, Dobbelstein M. 2006. Adenovirus-induced extracellular signal-regulated kinase phosphorylation during the late phase of infection enhances viral protein levels and virus progeny. Cancer Res. 66:1282–1288 [DOI] [PubMed] [Google Scholar]
- 67. Searles RP, Bergquam EP, Axthelm MK, Wong SW. 1999. Sequence and genomic analysis of a rhesus macaque rhadinovirus with similarity to Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 73:3040–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Seger R, Krebs EG. 1995. The MAPK signaling cascade. FASEB J. 9:726–735 [PubMed] [Google Scholar]
- 69. Sharma-Walia N, et al. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 79:10308–10329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Staudt MR, Dittmer DP. 2007. The Rta/Orf50 transactivator proteins of the gamma-herpesviridae. Curr. Top. Microbiol. Immunol. 312:71–100 [DOI] [PubMed] [Google Scholar]
- 71. Vantaggiato C, et al. 2006. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J. Biol. 5:14 doi:10.1186/jbiol38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang SE, et al. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 78:4248–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Winkler M, Stamminger T. 1996. A specific subform of the human cytomegalovirus transactivator protein pUL69 is contained within the tegument of virus particles. J. Virol. 70:8984–8987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xie J, Ajibade AO, Ye F, Kuhne K, Gao SJ. 2008. Reactivation of Kaposi's sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 371:139–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xie J, Pan H, Yoo S, Gao SJ. 2005. Kaposi's sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J. Virol. 79:15027–15037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang X, Gabuzda D. 1999. Regulation of human immunodeficiency virus type 1 infectivity by the ERK mitogen-activated protein kinase signaling pathway. J. Virol. 73:3460–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yao Y, et al. 2003. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc. Natl. Acad. Sci. U. S. A. 100:12759–12764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yoon S, Seger R. 2006. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24:21–44 [DOI] [PubMed] [Google Scholar]
- 79. Yu F, et al. 2007. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog. 3:e44 doi:10.1371/journal.ppat.0030044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yu XK, et al. 2003. Three-dimensional structures of the A, B, and C capsids of rhesus monkey rhadinovirus: insights into gammaherpesvirus capsid assembly, maturation, and DNA packaging. J. Virol. 77:13182–13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhu FX, Chong JM, Wu L, Yuan Y. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:800–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.