

Abstract
Hantaviruses, similarly to other negative-strand segmented RNA viruses, initiate the synthesis of translation-competent capped mRNAs by a unique cap-snatching mechanism. Hantavirus nucleocapsid protein (N) binds to host mRNA caps and requires four nucleotides adjacent to the 5′ cap for high-affinity binding. N protects the 5′ caps of cellular transcripts from degradation by the cellular decapping machinery. The rescued 5′ capped mRNA fragments are stored in cellular P bodies by N, which are later efficiently used as primers by the hantaviral RNA-dependent RNA polymerase (RdRp) for transcription initiation. We showed that N also protects the host mRNA caps in P-body-deficient cells. However, the rescued caps were not effectively used by the hantavirus RdRp during transcription initiation, suggesting that caps stored in cellular P bodies by N are preferred for cap snatching. We examined the characteristics of the 5′ terminus of a capped test mRNA to delineate the minimum requirements for a capped transcript to serve as an efficient cap donor during hantavirus cap snatching. We showed that hantavirus RdRp preferentially snatches caps from the nonsense mRNAs compared to mRNAs engaged in translation. Hantavirus RdRp preferentially cleaves the cap donor mRNA at a G residue located 14 nucleotides downstream of the 5′ cap. The sequence complementarity between the 3′ terminus of viral genomic RNA and the nucleotides located in the vicinity of the cleavage site of the cap donor mRNA favors cap snatching. Our results show that hantavirus RdRp snatches caps from viral mRNAs. However, the negligible cap-donating efficiency of wild-type mRNAs in comparison to nonsense mRNAs suggests that viral mRNAs will not be efficiently used for cap snatching during viral infection due to their continuous engagement in protein synthesis. Our results suggest that efficiency of an mRNA to donate caps for viral mRNA synthesis is primarily regulated at the translational level.
INTRODUCTION
Hantaviruses, members of the Bunyaviridae family, are transmitted to humans through aerosolized excreta of infected rodent hosts. Their infection causes hantavirus cardiopulmonary syndrome (HCP) and hemorrhagic fever with renal syndrome (HFRS) (62, 63), with mortalities of 50% and 15%, respectively. The spherical hantavirus particles harbor three negative-sense genomic RNA segments, S, M, and L, within a lipid bilayer (64). The mRNAs derived from S, L, and M segments encode viral nucleocapsid protein (N), RNA-dependent RNA polymerase (RdRp), and glycoprotein precursor (GPC), respectively. The GPC precursor is cleaved into two glycoproteins, Gn and Gc. The characteristic feature of the hantaviral genome is the partially complementary sequence at the 5′ and 3′ termini of each of the three genome segments that undergo base pairing and form panhandle structures (48, 53, 57). N is a multifunctional protein playing a vital role in multiple processes of the virus replication cycle and has been found to undergo trimerization both in vivo and in vitro (2, 3, 8, 13, 28, 32, 41–44, 54, 66, 67). N specifically encapsidates the three viral genomic RNAs into nucleocapsids which are packaged into virions.
The sequence of L segment RNA has been determined for about 20 viruses in the Bunyaviridae family, including nine hantaviruses. Except for the tospovirus and nairovirus, the RdRps of all other bunyaviruses are of a similar molecular mass (∼250 kDa). The requirement of both RdRp and N for replication/transcription of the viral genome has been demonstrated for both hantaviruses and other bunyaviruses (1, 8, 21, 39). Using a green fluorescent protein (GFP) fusion protein, the localization of Tula hantavirus RdRp has been found to be perinuclear. The punctate expression pattern of the L-enhanced GFP (EGFP) fusion protein has led to the suggestion that RdRp is membrane associated (34, 37). The 5′ and 3′ termini of the hantaviral genome contain untranslated regions (UTRs) of various lengths. Assays in which reporter genes have been flanked by these UTRs have shown that promoters for viral RdRp are located in these critical UTR sequences. In the orthobunyavirus Bunyamwera, base pairing of 5′ and 3′ termini of the viral genomic RNA was found to be required for the synthesis of RNA by viral RdRp (6). Studies on the characterization of the influenza A virus promoter have suggested a corkscrew-like secondary structure formed by the base pairing of partially complementary 5′ and 3′ ends of the viral genome (17).
The RdRp from segmented negative-sense RNA viruses requires a capped RNA primer to initiate the transcription (12, 15, 18, 55, 68). The capped RNA primer is generated from the 5′ terminus of host cell mRNA by the “cap-snatching” mechanism, which has been well characterized for the influenza virus (7, 11, 26, 33, 59). Although the knowledge about the sequence, length, and structure of the 5′ mRNA terminus that donates the primer is rather limited, most common cap donor mRNAs are cleaved 15 nucleotides downstream of the cap, with a variation of 10 to 20 nucleotides (7, 9, 15, 18, 22, 31, 51, 52, 65). The use of capped primers following a “prime and realign” mechanism has been suggested for the Bunyaviridae transcription initiation (24). Transcription termination signals have been identified in Hantaan and Sin Nombre virus (SNV) mRNAs (27). SNV S and L segment mRNAs are not polyadenylated; however, M segment-derived mRNA is polyadenylated and synthesis is terminated at a (U)8 polyadenylation transcription termination signal (27).
A cap-snatching mechanism similar to that in influenza virus has been proposed for all minus-strand segmented RNA viruses, including the bunyaviruses and arenaviruses, although their RdRps are structurally different and they replicate at different locations inside the host cell. Unlike the situation in influenza virus, the RdRp of bunyaviruses and arenaviruses is encoded by one rather than three genes. Recent studies have suggested that RdRp from bunyaviruses and arenaviruses harbors the endonuclease domain at the N terminus, and its endonuclease activity has been demonstrated (47, 61). Moreover, influenza viruses carry out cap snatching and transcription in the nucleus of infected cells, whereas bunyavirus and arenavirus transcription and genome replication are cytoplasmic. Unlike influenza virus, the viruses carrying out cap snatching in the cytoplasm have to compete with the cellular RNA degradation machinery, which actively removes caps and degrades cellular transcripts after the completion of translation.
The eukaryotic mRNA degradation machinery follows two general decay pathways, both of which begin with the shortening of the 3′ poly(A) tail by a process known as deadenylation (50). Following deadenylation, mRNAs can be degraded by a 3′-to-5′ exosome under the control of peptides of the SKI complex. Alternatively, after deadenylation, mRNAs can be decapped by the Dcp1/Dcp2 decapping enzymes, followed by 5′-to-3′ degradation by exonuclease XRN1 (23, 40, 49). Decapping and XRN1-dependent 5′-to-3′ decay form the predominant pathway for the degradation of cellular mRNAs. Moreover, the components of this pathway, including decapping enzymes Dcp1/Dcp2, exonuclease XRN1, and other peptides that function in mRNA degradation and regulation, are located in discrete cytoplasmic foci termed processing bodies (P bodies) (23, 40, 49).
In addition to these two mRNA degradation pathways, eukaryotic cells also use elegant mRNA surveillance or quality control mechanisms to ensure the translation of error-free mRNAs. Among these, the most prevalent and well-characterized mechanism is the nonsense-mediated mRNA decay (NMD) pathway, which recognizes and degrades mRNAs containing premature translation termination codons (PTCs) (29). Premature translation termination leads to the assembly of the surveillance complex on mRNA, which triggers NMD. The surveillance complex is composed of the UPF1 to three proteins and four additional NMD effectors (SMG1 and SMG5 to 7) (4, 16, 35). Assembly of the surveillance complex recruits the decapping enzymes and XRN1, but it can also accelerate the deadenylation and 3′-to-5′ degradation by the exosome and the SKI complex (14, 46). The enzymes that function in general mRNA decay also function in NMD, and the mRNA molecules containing PTCs are targeted to P bodies for rapid decay (4, 16, 35).
We have recently found that SNV N protein resides in cellular P bodies and also binds specifically to the mRNA 5′ caps (42, 43, 45). This specific interaction prevented the 5′ caps of cellular mRNAs from degradation by the cellular decapping machinery. The rescued 5′-capped oligoribonucleotides were stored in P bodies by N and were later used as primers by the hantavirus RdRp (42). We reported that 5′-capped mRNA oligonucleotides sequestered in P bodies by N were at least 180 nucleotides in length (42). The mechanism generating the shorter primers of appropriate length and specificity is still unknown. In this paper, we examine the characteristics of the 5′ terminus of a capped test mRNA to delineate the minimum requirements for a capped transcript to serve as an efficient cap donor during cap-snatching mechanism of transcription initiation by the Sin Nombre virus RdRp.
MATERIALS AND METHODS
Oligonucleotides, enzymes, and other reagents.
PCR primers were from Integrated DNA technologies. All restriction enzymes were from New England BioLabs. Platinum PCR Supermix was from Invitrogen. Phusion high-fidelity DNA polymerase was from NEB. RNA purification reagents were from Qiagen, and reverse transcription reagents were from Invitrogen. Power Sybr green PCR master mix was from Applied Biosystems. TA cloning reagents were from Invitrogen. All other chemicals were purchased from Sigma. The reagents for 5′ rapid amplification of cDNA ends (RACE) were purchased from Roche Applied Science.
Constructs.
The plasmid pCDNAGFP expressing green fluorescent protein (GFP) was PCR amplified from plasmid pEGFP-C1 (Clontech) using a forward primer, 5′-GATTATGCTAGCATGGGGTCTCATGGCGAGGA-3′, and a reverse primer, 5′-GTATTCTCGAGTTATCTAGATCCGGTGGATCCC-3′ (boldface and italics indicate restriction sites). The PCR product was gel purified, digested with NheI and XhoI restriction enzymes, and cloned between the same restriction sites in pCDNA3.1+ vector (Invitrogen). The plasmid pCDNAGFPns, which does not express GFP due to two substitution mutations, was cloned in pCDNA3.1+ using the forward primer GATTATGCTAGCATGGGGTGATCATGGCGAGGA-3′ and the reverse primer described above. The plasmid pCDNAGFPnsG0, expressing a GFP mRNA harboring five substitution mutations at the 5′ untranslated region (UTR) of mRNA, was constructed by generating a PCR product from pCDNAGFPns plasmid using forward primer F1, AGTGTATCATATGCCAAGTAC, and reverse primer R1, 5′-GGATAAGGGAGTAAGGAGTGGGTTGTGTA. Similarly, another PCR product was generated from pCDNAGFPns plasmid using forward primer F2, 5′-TACACAACCCACTCCTTACTCCCTTATCC-3′, and reverse primer R2, 5′-TCTAGACTCGAGTTACTTGTACAGCT-3′. The two PCR products were gel purified and mixed together. The mixture was used as the template and a third PCR product was generated using forward primer F1 and reverse primer R2. This final PCR product was again gel purified, digested with NdeI and XhoI, and cloned between the same restriction sites in pCDNA3.1+ backbone. Using this cloning strategy, the mutations were incorporated through forward primer F2 and reverse primer R1. The same strategy was used for the construction of other plasmids (see Fig. 2A and Fig. 3A). In all these constructs the same forward F1 and reverse R2 primers were used. However, the sequences of reverse R1 and forward F2 primers were different, depending upon the type of mutation (Table 1).
Fig 2.
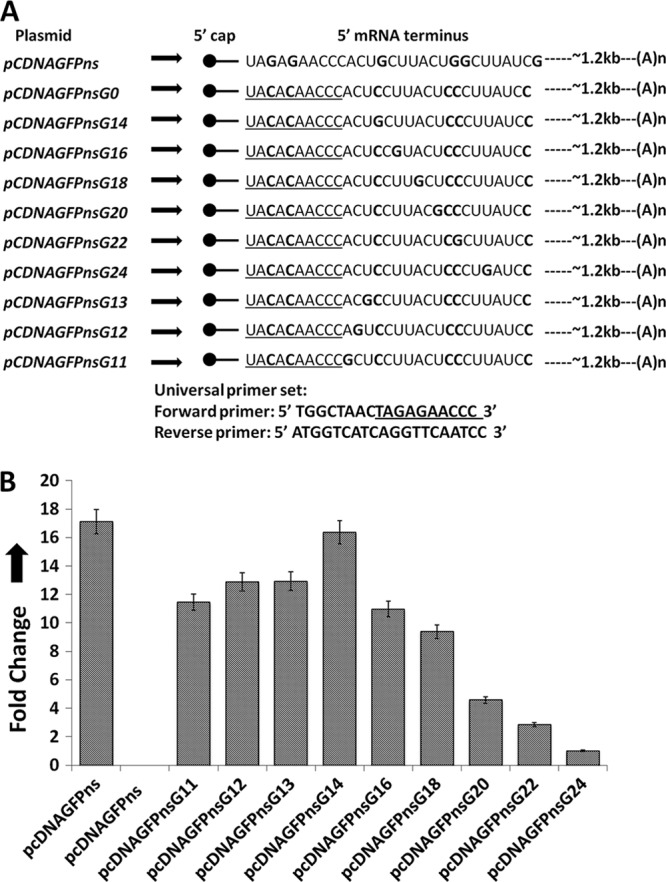
Characteristics of a preferred cap donor mRNA for hantavirus cap snatching. (A) To determine the characteristics of the 5′ mRNA termini that are required for cap snatching, we generated multiple constructs, shown on the left side. The mRNA encoded by each plasmid is shown by an arrow. The bold letters show the mutations in the 5′ mRNA terminus. The universal primer set used in the real-time PCR studies is shown at the bottom. (B) Vero E6 cells were transfected with the plasmids shown in panel A, followed by SNV infection. Total mRNA was purified and reverse transcribed using a primer specific to the S-segment mRNA. Using the universal primer set, the cDNA was used in real-time PCR analysis to quantify the caps obtained by the S-segment mRNA from the mutant mRNAs expressed from the transfected plasmid of interest (see Materials and Methods for details).
Fig 3.

Role of nucleotide complementarity in hantavirus cap snatching. (A) To determine whether nucleotide complementarity at the 3′ termini of viral genomic RNA and the capped primer have a role in cap snatching, we generated five additional plasmids, shown on the left side. The mRNAs expressed from these plasmids are shown by arrows. The bold letters show the mutations in the 5′ mRNA terminus. The complementary nucleotides between the test mRNA and the viral genomic RNA are shown. (B) Vero E6 cells were transfected with the plasmids shown in panel A, followed by SNV infection. Total mRNA was purified and reverse transcribed using a primer specific to the S-segment mRNA. Using the universal primer set, the cDNA was used in real-time PCR analysis to quantify the caps obtained by the S-segment mRNA from the mutant mRNAs expressed from the transfected plasmids shown in panel A (see Materials and Methods for details).
Table 1.
Primers used for the construction of plasmidsa
| Plasmid | Primer for PCR1 |
Primers for PCR2 |
||
|---|---|---|---|---|
| Name | Sequence | Name | Sequence | |
| pCDNAGFPnsG0 | F1 | 5′AGTGTATCATATGCCAAGTAC | F2 | 5′TACACAACCCACTCCTTACTCCCTTATCC |
| R1 | 5′GGATAAGGGAGTAAGGAGTGGGTTGTGTA | R2 | 5′TCTAGACTCGAGTTACTTGTACAGCT | |
| pCDNAGFPnsG14 | R1 | 5′GGATAAGGGAGTAAGCAGTGGGTTGTGTA | F2 | 5′TACACAACCCACTGCTTACTCCCTTATCC |
| pCDNAGFPnsG16 | R1 | 5′GGATAAGGGAGTACGGAGTGGGTTGTGTA | F2 | 5′TACACAACCCACTCCGTACTCCCTTATCC |
| pCDNAGFPnsG18 | R1 | 5′GGATAAGGGAGCAAGGAGTGGGTTGTGT | F2 | 5′TACACAACCCACTCCTTGCTCCCTTATCC |
| pCDNAGFPnsG20 | R1 | 5′GGATAAGGGCGTAAGGAGTGGGTTGTGTA | F2 | 5′TACACAACCCACTCCTTACGCCCTTATCC |
| pCDNAGFPnsG22 | R1 | 5′GGATAAGCGAGTAAGGAGTGGGTTGTGTA | F2 | 5′TACACAACCCACTCCTTACTCGCTTATCC |
| pCDNAGFPnsG24 | R1 | 5′GGATACGGGAGTAAGGAGTGGGTTGTGTA | F2 | 5′TACACAACCCACTCCTTACTCCCGTATCC |
| pCDNAGFPnsG13 | R1 | 5′GGATAAGGGAGTAAGGCGTGGGTTGTGTA | F2 | 5′TACACAACCCACGCCTTACTCCCTTATCC |
| pCDNAGFPnsG12 | R1 | 5′GGATAAGGGAGTAAGGACTGGGTTGTGTA | F2 | 5′TACACAACCCAGTCCTTACTCCCTTATCC |
| pCDNAGFPnsG11 | R1 | 5′GGATAAGGGAGTAAGGAGCGGGTTGTGTA | F2 | 5′TACACAACCCGCTCCTTACTCCCTTATCC |
| pCDNAGFPns(i) | R1 | 5′GGATAAGGGAGTAAGCTGTGGGTTGTGTA | F2 | 5′TACACAACCCACAGCTTACTCCCTTATCC |
| pCDNAGFPns(ii) | R1 | 5′GGATAAGGGAGTAAGCTATGGGTTGTGTA | F2 | 5′TACACAACCCATAGCTTACTCCCTTATCC |
| pCDNAGFPns(iii) | R1 | 5′GGATAAGGGAGTCTACAGTGGGTTGTGTA | F2 | 5′TACACAACCCACTGTAGACTCCCTTATCC |
| pCDNAGFPns(iv) | R1 | 5′GGATAAGGGCTACTACAGTGGGTTGTGTA | F2 | 5′TACACAACCCACTGTAGTAGCCCTTATCC |
| pCDNAGFPns(v) | R1 | 5′GGATAACTACTACTACAGTGGGTTGTGT | F2 | 5′TACACAACCCACTCTAGTAGTAGTTATCC |
As discussed in Materials and Methods, two PCR products (PCR1 and PCR2) were generated from plasmid pcDNAGFPns using the primer sets F1 and R1 and F2 and R2, respectively. The two PCR products were mixed and used as a template for a third PCR along with primers F1 and R2. This third PCR product was digested with NdeI and XhoI restriction enzymes and cloned between the same sites in plasmid pCDNA3.1+ to generate plasmid pCDNAGFPnsG0. The same strategy was used for the construction of the remaining plasmids. The sequences of the F1 and R2 primers were the same. However, the sequences of the R1 and F2 primers were changed depending upon the type of mutation to be incorporated in the plasmid of interest.
Reverse transcription and real-time PCR.
Vero E6 cells in six-well plates were transfected with the plasmid of interest, using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Eight hours posttransfection, cells were infected with Sin Nombre virus (strain 77734, a gift from Brian Hjelle, University of New Mexico) at a multiplicity of infection (MOI) of 1.0. Cells were lysed 48 h postinfection, and total RNA was purified using the RNeasy kit (Qiagen), including treatment with RNase-free DNase I (Qiagen), following the manufacturer's protocol. Two micrograms of total RNA from each well was reverse transcribed using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen) and a primer specific to the viral S-segment mRNA (5′-ACTAAAGCCAATCACACCCATGACA-3′) in a total volume of 20 μl. Two microliters of the resulting cDNA was used in 20-μl real-time PCRs. The relative quantification method was used for real-time PCR using an ABI Prism 7700 sequence detection system following the manufacturer's instructions (Applied Biosystems). Fold change in mRNA levels and standard deviation were calculated by the relative quantification method, which is described in detail in the ABI instruction manual (http://www3.appliedbiosystems.com/cms/groups/mcb_support/documents/generaldocuments/cms_040980.pdf). We used a universal primer set with a forward primer (5′-TGGCTAACTACACAACCC-3′) and a reverse primer (5′-ATGGTCATCAGGTTCAATCC) to amplify 290 nucleotides from the 5′ terminus of viral S-segment mRNA. This universal primer set was used in all real-time PCRs reported in this paper, unless otherwise stated. The forward primer is complementary to the 5′ terminus of GFP mRNA, which is expressed from the transfected plasmid, and the reverse primer is complementary to the open reading frame of viral S-segment mRNA. This primer set will generate products only if the viral S-segment mRNA has snatched a cap from the GFP mRNA (discussed in more detail in Results). Amplification of β-actin mRNA as an “internal control” was carried out using a forward primer, 5′-CCATCATGAAGTGTGACGTGG, and a reverse primer, 5′-GTCCGCCTAGAAGCATTTGCG, as previously reported (42). To ensure the amplicon specificity of each primer set, the PCR products were subjected to melting curve analysis followed by sequential agarose gel electrophoresis. The efficiency for amplification of the target (5′ terminus of the viral S-segment mRNA) and the internal control gene (β-actin) was examined using serial dilutions of cDNA. The mean difference between threshold cycle number values was calculated for each cDNA dilution. The mean difference values corresponding to each dilution were plotted and fit to a straight line with a slope of <0.1. After this validation test, the levels of S-segment mRNA which have snatched caps from the test mRNA expressed from the transfected plasmid in Vero E6 cells were calculated following normalization to the β-actin mRNA levels and expressed as relative units. Intrinsic steady-state levels of GFP reporter mRNA in transfected cells were monitored by real-time PCR analysis using a forward primer, 5′-CACATGAAGCAGCACGACTT-3′, and a reverse primer, 5′-AGTTCACCTTGATGCCGTTC-3′. This primer set is specific to the GFP open reading frame.
TA cloning.
During cap snatching, hantaviruses typically cleave the host cell mRNA at a G residue located 8 to 17 nucleotides downstream of the terminal cap (24). To determine whether the caps derived from the mRNAs expressed from transfected plasmids (pCDNAGFP/pCDNAGFPns) exhibit these hallmarks of correct cap snatching, we sequenced the cap-viral UTR junctions of viral S-segment mRNAs, which have obtained their caps from either GFP mRNA or nonsense GFP mRNA. Vero E6 cells were transfected with either plasmid pCDNAGFP or plasmid pCDNAGFPns, followed by viral infection 4 h posttransfection. Cells were lysed 48 h postinfection, and total RNA was purified and reverse transcribed using a primer specific to the S-segment mRNA, as described above. The cDNA was PCR amplified using a primer set shown in Fig. 1A. The PCR product was cloned using a TA cloning kit (Invitrogen) by following the manufacturer's instructions. Plasmid DNA was purified from 20 randomly selected clones and sequenced in the region corresponding to the cap-UTR junction, as previously reported (42).
Fig 1.

Hantavirus cap-snatching assay. (A) A diagrammatic representation of the cap-snatching assay. Step 1, Vero E6 cells were transfected with cap donor plasmid (pCDNAGFPns or pCDNAGFP) followed by SNV infection 4 h posttransfection (step 2). Step 3, cells were lysed 48 h postinfection, and total RNA was purified as described in Materials and Methods. Twenty-five nanograms of the purified RNA was reverse transcribed using a primer specific to the S-segment-derived mRNA. The cDNA was PCR amplified using a forward primer specific to the 5′ terminus of GFP mRNA (purple) and a reverse primer specific to the N gene (black). See Materials and Methods for details. (B) As expected, the PCR product was generated only from the cells which were transfected with either pCDNAGFPns or pCDNAGFP plasmid, followed by viral infection. (C) The Vero E6 cells were transfected with cap donor plasmid pCDNAGFPns or pCDNAGFP, followed by SNV infection 4 h posttransfection, as described for panel A. Cells were lysed 48 h postinfection, and total RNA was purified. Intrinsic mRNA levels expressed from cap donor plasmids pCDNAGFPns and pCDNAGFP were quantified by real-time PCR analysis using a primer set specific to the GFP open reading frame, as described in Materials and Methods. (D) Similarly, the cap-donating potential of the transcripts expressed from cap donor plasmids pCDNAGFPns and pCDNAGFP was determined by real-time PCR using a primer set shown in panel A. (E) The PCR product from panel B was cloned in a TA cloning vector (Invitrogen), and plasmid DNA from 20 random clones was sequenced to examine the cap-UTR junction. As shown at the bottom, the capped primers were terminated at the 3′ G residue. (F) The GFP mRNA and nonsense GFP mRNA were synthesized by in vitro T7 transcription, as described in Materials and Methods. To distinguish their 5′ UTRs, two nucleotides in the 5′ UTR of nonsense GFP mRNA were mutated (bold and underlined). Vero E6 cells were cotransfected with GFP mRNA and nonsense GFP mRNA, followed by virus infection. The cap-donating potential of these two transcripts was examined by 5′RACE, as described in Materials and Methods.
siRNA knockdown.
To substantiate the role of P bodies in hantavirus cap snatching, two essential P-body components, GW-182 and Ge-1, were downregulated by small interfering RNA (siRNA) transfection. The GW-182, Ge-1, and control siRNAs were purchased from IDT. The sequences for the GW-182 siRNAs were 5′-GGAAUGUUACAAGACAAACGAAUGG and 5′-CCAUUCGUUUGUCUUGUAACAUUCCUA-3′. The sequences of the Ge-1 siRNAs were 5′-GGAUGUUAGCCAGAUCAAGCAGGGC-3′ and 5′-GCCCUGCUUGAUCUGGCUAACAUCCAC-3′. Both GW-182 and Ge-1 siRNAs were transfected at a final concentration of 50 nM each into monolayers of Huh-7 cells (a gift from Yu-Jui-Yvonne Wan, KUMC) seeded in six-well plates using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Control siRNA was similarly transfected into control wells. Twelve hours after transfection, GW-182 and Ge-1 siRNA or control siRNA was retransfected together with 4 μg of pCDNAGFPns plasmid. The effect of siRNA knockdown on the expression levels of GW-182 and Ge-1 proteins was verified 24 h after first transfection by Western blot analysis, using either anti-GW182 or anti-Ge-1 antibodies (Santa Cruz). Cells were infected with SNV at an MOI of 1.0, 24 h after first transfection. Cells were lysed 36 h postinfection, and total RNA was purified and reverse transcribed using a primer specific to the S-segment mRNA, as mentioned in the “Reverse transcription and real-time PCR” section above. The effect of siRNA knockdown on hantavirus cap snatching was monitored by quantitative estimation of caps snatched from the GFPns mRNA by the SNV RdRp, using real-time PCR analysis, as discussed in the above “Reverse transcription and real-time PCR” section.
To determine the effect of a P body on the protection of mRNA caps by SNV N protein, monolayers of Huh-7 cells were either mock transfected or transfected with both GW-182 and Ge-1 siRNAs at a final concentration of 50 nM each. Twelve hours posttransfection, cells were retransfected with GW182 and Ge-1 siRNAs along with 4 μg each of plasmids pCDNAGFPns and pCDNA-SNVN. Cells were lysed 24 h after plasmid transfection, and total RNA was purified. Two micrograms of total RNA was reverse transcribed using random primers. The cDNA generated from both mock- and siRNA-transfected cells was used in real-time PCR analysis to quantitatively estimate the 5′ terminus of GFPns mRNA, using a forward primer, 5′-TAGAGAACCCACTGCTTACTGGC-3′, and a reverse primer 5′-CAGATGAACTTCAGGGTCAG-3′.
5′ RACE.
5′ RACE was performed using a 5′/3′ RACE kit (catalog no. 03353621001; Roche Applied Science) following the manufacturer's instruction. Briefly, Vero E6 cells seeded in six-well plates were transfected with 4 μg of either pCDNAGFP or pCDNAGFPns plasmid or cotransfected with 2 μg of each GFP mRNA and nonsense GFP mRNA and synthesized by in vitro T7 transcription. Eight hours posttransfection, cells were infected with Sin Nombre virus at an MOI of 10. Cells were lysed 48 h postinfection, and total RNA was purified using the RNeasy kit (Qiagen). Two micrograms of purified total RNA was reverse transcribed using a primer, 5′-ACTAAAGCCAATCACACCCATGACA-3′, complementary to the S-segment mRNA from 696 to 720 nucleotides. The resulting cDNA was purified using a PCR cleanup kit (Qiagen), and a homopolymeric (dA) tail was added at the 3′ end of the cDNA, using terminal transferase provided in the kit. The (dA)-tailed cDNA was then used to generate a PCR product with the forward oligo(dT)-anchor primer 5′-GACCACGCGTATCGATGTCGACTTTTTTTTTTTTTTTTV-3′ (V = A, C, or T) and a reverse primer, 5′-GCGAAACTTAGAATGTAGAGTCCGATG-3′. The reverse primer was complementary to the S-segment mRNA from 405 to 431 nucleotides. Finally, the resulting PCR product was used as a template to generate a short PCR product using the anchor primer (5′-GACCACGCGTATCGATGTCGAC-3′) containing a MluI site and a reverse primer (5′-ATTATATAGCGGCCGCATGGTCATCAGGTTCAATCC-3′) containing a NotI site. The reverse primer was complementary to the S-segment mRNA from 290 to 309 nucleotides. The final PCR product was digested with MluI and NotI and cloned in pcDNA 3.1+ vector between the same restriction sites. The plasmid DNA isolated from 20 colonies was sequenced to read the cap-UTR junction of the S-segment mRNA.
Staining and microscopy.
Adherent Huh-7 cells were grown on sterilized glass coverslips in a six-well plate. Cells were transfected with either GW182 siRNA (IDT) or Ge-1 siRNA (IDT) using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer's instructions. Similarly, the control siRNA was transfected into the control well. After 24 h, cells were fixed at −20°C for 5 min using acetone and then permeabilized with 0.5% Triton X-100 at room temperature for 15 min. Cells were then incubated for 1 h at room temperature with rabbit anti-Dcp2 antibody at a dilution of 1:100 in phosphate-buffered saline (PBS) containing 2% fetal calf serum (FCS). After washing with PBS, cells were incubated for 1 h at room temperature with fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG antibody at a 1,000-fold dilution in PBS containing 2% FCS. Fluorescent images were recorded by a Nikon Eclipse 80i upright microscope.
Synthesis of mRNA and 5′ capping.
The mRNA synthesis was carried out using the Ribomax T7 transcription kit (Promega) as previously reported (39–42). Briefly, the gene of interest was PCR amplified using a forward primer containing a flanking T7 promoter and an appropriate reverse primer. The PCR product was gel purified and used as the template in a 50-μl transcription reaction. Following synthesis, template DNA was degraded with DNase I, and RNA was purified by RNAeasy (Qiagen) and stored in 10-μl aliquots at −70°C. The resulting mRNA was 5′ capped using the ScriptCap m7G capping system (Cell Script Catnumber C-SCCE0610), according to the manufacturer's instructions. Briefly, 50 μg of purified mRNA was added to the reaction mix containing 50 mM Tris HCl, pH 8.0, 6 mM KCl, 1.25 mM MgCl2, 1 mM GTP, 100 μM S-adenosyl-methionine (SAM), and 1 unit of the capping enzyme in a final volume of 100 μl. The reaction mixture was incubated at 37°C for 30 min, followed by purification of capped mRNA using the RNeasy kit (Qiagen). The purified capped mRNA was used to transfect Huh-7 cells.
Using this approach, we also synthesized GFP mRNA and nonsense GFP mRNA, which were cotransfected to Huh-7 cells to examine whether hantaviruses preferentially snatch caps from nonsense transcripts. Briefly, the GFP ORF was PCR amplified from pCDNAGFP plasmid using a forward primer, 5′-CTAGCTAATACGACTCACTATAGTAGAGAACCCACTGCTTACTGGCTTATCG-3′, and a reverse primer, 5′-CCATAGAGCCCACCGCATCCCC-3′. Similarly a forward primer, 5′-CTAGCTAATACGACTCACTATAGTAGAGAACCACCTGCTTACTGGCTTATCG-3′, and the above-described reverse primer were used to generate another PCR product from plasmid pCDNAGFPns. Both the PCR products were gel purified and used as the templates to generate two transcripts, as described above. The two transcripts were 5′ capped using the ScriptCap m7G capping system, as described above. The 3′ tailing of purified capped mRNAs was carried out using a Cell Script A-Plus poly(A) polymerase tailing kit, following the manufacturer's instructions. Briefly, 50 μg of purified capped GFP mRNA or nonsense GFP mRNA were mixed with 1× tailing buffer (50 mM Tris-HCl, pH 8.0, 250 mM NaCl, and 10 mM MgCl2) containing 10 mM ATP and 8 units of Cellscript A-plus poly(A) polymerase in a total volume of 50 μl. The reaction mixture was incubated for 30 min at 37°C, and the tailed mRNA was purified by the RNeasy kit.
RESULTS
SNV preferentially snatches caps from mRNAs containing premature termination codons.
We have previously reported that hantavirus N protein efficiently protects the 5′ caps of cellular nonsense mRNAs in comparison to mRNAs, which encode proteins (42). The protected caps were abundantly found in cellular P bodies, which were later efficiently used by the RdRp for transcription initiation (42). To further confirm this observation, we cloned GFP in the pCDNA3.1+ vector, which expresses GFP mRNA having 5′ and 3′ UTRs of 71 and 372 nucleotides in length, respectively (Fig. 1E). In addition, we incorporated two extra nucleotides in the open reading frame (ORF) of the GFP expression construct, which generated a premature termination codon two amino acids downstream of the start codon (Fig. 1E). We transfected Vero E6 cells with these GFP constructs expressing either GFP mRNA or nonsense GFP mRNA, followed by infection with SNV 8 h posttransfection. We used our previously established cap-snatching assay to quantitatively estimate the caps snatched from either GFP mRNA or GFP nonsense mRNA by the viral RdRp. Briefly, 48 h postinfection, cells were lysed and total RNA was purified and reverse transcribed using a primer specific to the S-segment mRNA (Fig. 1A; also see Materials and Methods). The cDNA was PCR amplified using a forward primer specific to the 5′ terminus of GFP mRNA and a reverse primer specific to the N protein open reading frame (ORF) (Fig. 1A). This PCR strategy was designed to specifically identify the S-segment mRNAs, which have obtained their 5′ caps from either GFP mRNA or GFP nonsense mRNA. As expected, this PCR strategy generated the PCR product of appropriate size only from SNV-infected cells that were previously transfected with the constructs expressing either GFP mRNA or GFP nonsense mRNA (Fig. 1B). A comparatively intense band from cells expressing GFP nonsense mRNA suggests that RdRp preferentially snatches caps from PTC-containing mRNAs. To rule out the possibility that the difference in the band intensities shown in Fig. 1B was not due to a difference in the intrinsic steady-state levels of GFP mRNA and GFP nonsense mRNA in host cells, we repeated the above-described experiment and quantitatively estimated the expression levels of these two transcripts using real-time PCR. As shown in Fig. 1C, the intrinsic steady-state levels of GFP mRNA were 3-fold higher than those of GFP nonsense mRNA, consistent with preferential degradation of PTC-containing mRNA by host NMD machinery. To further confirm the observation made in Fig. 1B that SNV RdRp preferentially snatches caps from PTC-containing mRNAs, we used our previously established real-time PCR-based cap-snatching method to quantitatively estimate the caps snatched by the S-segment mRNA from either GFP mRNA or GFP nonsense mRNA. Consistent with the observations made in Fig. 1C, we observed that although intrinsic steady-state levels of GFP nonsense mRNA were lower than those of GFP mRNA, the PTC-containing GFP mRNA served as a better cap donor than GFP mRNA (Fig. 1D).
In addition, we used 5′ RACE to examine the 5′ terminus of S-segment mRNA and to further confirm that SNV RdRp preferentially uses the PTC-containing mRNAs for cap snatching. We expressed either GFP mRNA or GFP nonsense mRNA in virus-infected Vero E6 cells and examined the 5′ terminus of S-segment mRNA by 5′ RACE to further confirm that GFP nonsense mRNA serves as a preferential cap donor in comparison to GFP mRNA. The PCR product corresponding to virus-infected cells expressing either GFP mRNA or GFP nonsense mRNA was cloned, and plasmid DNA isolated from 20 colonies was sequenced (see Materials and Methods for details). Interestingly, 19 of 20 colonies were positive for cap snatching from GFP nonsense mRNA, suggesting that SNV RdRp snatched 95% of the caps from GFP nonsense mRNA in virus-infected cells expressing this transcript. In comparison, 1 of 20 colonies was positive for cap snatching from GFP mRNA, suggesting that only 5% of caps were snatched from GFP mRNA in virus-infected cells expressing this transcript (data not shown). Thus, both 5′ RACE and real-time PCR analysis suggest that the cap-donating potential of nonsense GFP mRNA is ∼25-fold higher than that of GFP mRNA.
To further strengthen this observation, we synthesized the GFP mRNA and nonsense GFP mRNA by in vitro T7 transcription, as described in Materials and Methods. Both the mRNAs were capped at the 5′ terminus and polyadenylated at the 3′ terminus (see Materials and Methods). To differentiate the 5′ UTRs of GFP mRNA and nonsense GFP mRNA, we mutated the 10th and 11th residues downstream of the terminal cap in the 5′ terminus of GFP mRNA from CA to AC (Fig. 1F). We cotransfected Vero E6 cells with GFP mRNA and nonsense GFP mRNA, followed by viral infection. Total RNA was purified from infected cells, and 5′ RACE was again used to examine the 5′ terminus of viral S-segment mRNA, as described above. An examination of 15 colonies revealed that 13 out of 15 colonies were positive for cap snatching from nonsense GFP mRNA. One colony each was positive for cap snatching from GFP mRNA and cellular mRNA. Taken together, these observations strongly establish that hantaviruses preferentially snatch caps from nonsense mRNAs.
It has been previously reported that hantavirus RdRp preferentially cleaves the host cell mRNA at a G residue during cap snatching (24, 42). To confirm these hallmarks of correct cap snatching, the PCR products from Fig. 1B were cloned in a TA cloning vector (Invitrogen), and plasmid DNAs from 20 random clones were sequenced to examine the cap-UTR junction. Although the assay is not quantitative, it is evident from Fig. 1E that capped mRNAs are cleaved at G residues, with a preference for the 14th G residue downstream of the 5′ cap. In addition, this observation further confirms the specificity of the PCR-based cap-snatching assay. Similar observations were made from the 5′ RACE experiment (Fig. 1F).
SNV preferentially cleaves the host mRNA at a G residue 14 nucleotides downstream of the terminal cap.
Since PTC-containing GFP mRNA was preferentially used for cap snatching, this mRNA was further examined to demonstrate the characteristics of the 5′ mRNA termini that are prerequisite for cap snatching. Both wild-type and PTC-containing GFP mRNAs contain five G residues in the first 30-nucleotide region of the 5′ UTR (Fig. 1E). Using site-directed mutagenesis, we first mutated these five G nucleotides to C residues (pCDNAGFPnsG0) and asked whether the resulting mutant mRNA was still an efficient cap donor for the hantavirus cap snatching (Fig. 2A). We used a real-time PCR analysis to quantitatively estimate the caps obtained by the S-segment mRNA from the test mRNAs expressed from the transfected plasmids of interest (see Materials and Methods). As shown by the Fig. 2B, the lack of G residues in the 5′ terminus of the test mRNA drastically decreased its cap-donating potential to an undetectable level. This observation further confirms that G residues in the 5′ terminus of an mRNA are required for efficient cap snatching. From plasmid pCDNAGFPnsG0 (Fig. 2A), we generated multiple constructs to further examine the requirements of a capped mRNA to serve as an efficient cap donor during cap snatching (Fig. 2A). We incorporated a single G residue at either the 14th (pCDNAGFPnsG14), 16th (pCDNAGFPnsG16), 18th (pCDNAGFPnsG18), 20th (pCDNAGFPnsG20), 22nd (pCDNAGFPnsG22), 24th (pCDNAGFPnsG24), 13th (pCDNAGFPnsG13), 12th (pCDNAGFPnsG12), or 11th position (pCDNAGFPnsG11) from the 5′ cap (Fig. 2A). Vero E6 cells were transfected with these mutant plasmids, followed by the viral infection. Using the universal primer set (Fig. 2A, bottom), the real-time PCR analysis showed that incorporation of a single G residue at the 14th position from the 5′ cap dramatically enhanced the ability of the test mRNA to donate caps during cap snatching (Fig. 2B). However, the displacement of the 14th G residue further downstream or upstream had a negative effect on the cap-donating ability of the test mRNA (Fig. 2B), suggesting that the presence of a G residue at the 14th position from the 5′ cap improves the chances of an mRNA to serve as an efficient cap donor during hantavirus transcription initiation.
Sequence complementarity between the 3′ termini of the capped primer and viral genomic RNA favors cap snatching.
To determine whether the complementarity between the nucleotides at the 3′ terminus of viral genomic RNA and the nucleotides located in the vicinity of the high-priority cleavage site (14th G residue) of the cap donor mRNA improves the efficiency for cap donation, we generated five additional mutants (Fig. 3A). The mutants pCDNAGFPns(i) and pCDNAGFPns(ii) express mRNAs having either two or three nucleotides at positions 13 to 14 or 12 to 14, respectively, complementary to the 3′ terminus of the viral genomic RNA (Fig. 3A). Similarly, three other mutants, pCDNAGFPns(iii), pCDNAGFPns(iv), and pCDNAGFPns(v), which have either three, six, or nine nucleotides complementary to the 3′ terminus of viral genomic RNA were generated (Fig. 3A). The mutant plasmids were transfected into Vero E6 cells, followed by viral infection, and caps obtained by the S-segment mRNA from the corresponding mutant mRNAs were quantified by real-time PCR, using the universal primer set. It is evident from Fig. 3B that a gradual increase in the nucleotide complementarity significantly enhances preferential usage of caps from the test mRNA during cap snatching. Although it has been previously reported that cleavage of capped mRNAs by the endonuclease subunit of influenza virus RdRp occurs independent of the 3′ terminus of viral genomic RNA (60), the recent findings suggest that influenza virus transcriptase also prefers capped primers with 3′ nucleotides more complementary to the 3′ terminus of viral genomic RNA (25).
SNV snatches caps from its own mRNA.
SNV preferentially snatched caps from the transcripts containing 5′ nucleotides complementary to the 3′ terminus of the viral genomic RNA (Fig. 3B). Caps were snatched with remarkable efficiency from the GFP nonsense transcript having a nine-nucleotide-long triplet repeat sequence (UAGUAGUAG) in the 5′ UTR, which is complementary to the 3′ terminus of viral genomic RNA (Fig. 3B). All hantaviral mRNAs contain this triplet repeat sequence at the same location, raising a question of whether hantaviruses snatch caps from their own mRNAs during infection. To test this hypothesis, we cloned the gene encoding the hantavirus glycoprotein precursor (GPC) along with 5′ and 3′ UTRs in the ptriEX1.1 vector. A PCR product was generated from the resulting plasmid using a forward primer containing a flanking 5′ T7 promoter preceded by the 14 nucleotides of the 5′ UTR of the GFP mRNA and a reverse primer containing a flanking 5′ poly(A) tail of 50 nucleotides in length. The resulting PCR was used as the template in a T7 transcription reaction for the synthesis of GPC mRNA (see Materials and Methods for details). The purified mRNA was capped at the 5′ terminus, as described in Materials and Methods. This capping method incorporates cap 1 structure, predominantly found in all higher eukaryotic mRNA (5). The cap 1 structure was found to be required for priming the transcription of influenza virus mRNA (10). The synthesis of GPC mRNA was performed in such a way that the 5′ 14 nucleotides of the GPC transcript matched all other transcripts used in this study (Fig. 4A). In addition, the synthesized GPC mRNA also matched the transcript generated by the viral RdRp. Similarly, we synthesized the GPCns mRNA containing a PTC 15 amino acids downstream of the initiating methionine (Fig. 4A). For comparison, we also synthesized GFP mRNA and GFP nonsense mRNA using T7 RNA polymerase (Fig. 4A). Vero E6 cells were transfected with these mRNAs, and the expression of GPC and GFP was monitored by Western blot analysis. Unlike nonsense mRNAs, the GFP and GPC expression was observed in cells transfected with GFP mRNA and GPC mRNA, respectively (Fig. 4B, inset). The cap-donating potential of these four transcripts (Fig. 4A) was quantified by a real-time PCR, using the primer set shown in Fig. 1A. As shown in Fig. 4B, SNV snatched caps from both mRNAs encoding either GFP or viral GPC protein. However, both GFP and GPC mRNAs harboring PTCs served as efficient cap donors in comparison to their respective wild-type mRNAs. Moreover, it is noticeable from Fig. 4B that in comparison to GFP nonsense mRNA, SNV preferentially snatched caps from its own mRNA having a PTC.
Fig 4.

SNV snatches caps from its own mRNA. (A) To determine whether hantaviruses snatch caps form their own mRNAs, we synthesized SNV glycoprotein precursor (GPC) mRNA and GPC nonsense mRNA by T7 transcription reaction, as described in Materials and Methods. The two mRNAs are the same in sequence except that nonsense GPC mRNA has two additional CC residues in the GPC ORF four amino acids from the initiating methionine. This additional mutation generated a stop codon 15 amino acids downstream of the initiating methionine. For comparison we synthesized GFP mRNA and nonsense GFP mRNA using a T7 transcription reaction, as described in Materials and Methods. GFP mRNA and nonsense GFP mRNA are the same in sequence except that nonsense GFP mRNA has a PTC two amino acids from the initiating methionine. It must be noted that the 5′-terminal 15 nucleotides of all these four mRNAs are the same in sequence. (B) Vero E6 cells were transfected with the mRNAs shown in panel A, followed by SNV infection. Caps snatched by SNV RdRp from these four transcripts were quantified by real-time PCR analysis using the universal primer set shown in Fig. 2A (see Materials and Methods for details). To ensure that transfected mRNA were properly engaged in translation, Vero E6 cells were transfected with the mRNA shown in panel A. Cells were lysed 36 h posttransfection, and cell lysates were examined by Western blot analysis (inset) using either anti-GFP (i) or anti-His tag (ii) antibody. The GPC contained a C-terminal His tag and was examined by ant-His tag antibody.
Since GPC generates two transmembrane proteins, Gn and Gc, the translation of GPC mRNA occurs on the endoplasmic reticulum (ER). The GFP is a soluble cytoplasmic protein; it is likely that GFP mRNA is translated by the cytoplasmic ribosomes. Thus, the possibility that hantaviruses preferentially snatch caps from nonsense mRNAs associated with ER cannot be ruled out.
Role of cellular P bodies in hantavirus cap snatching.
We have previously reported that capped host mRNA oligoribonucleotides bound with SNV N protein were abundantly found in cellular P bodies which were later efficiently used as primers by the hantavirus RdRp for transcription initiation (42). It is possible that N independently migrates to P bodies and selectively associates with the 5′ caps of those cellular transcripts that are transported to P bodies for degradation. Alternatively, it is equally likely that N independently associates with the mRNA 5′ caps in the cytoplasm and migrates to P bodies along with bound mRNAs, which are targeted to P bodies for degradation. To test these two possibilities and to demonstrate the general role of cellular P bodies in hantavirus cap snatching, we used siRNAs to downregulate GW182 and Ge-1 proteins in Huh-7 cells. Downregulation of Gw182 and Ge-1 was confirmed by Western blot analysis (Fig. 5A and B). Both Gw182 and Ge-1 are critical P-body components whose downregulation has been reported to cause P-body loss in cells (38, 69). The decapping enzyme Dcp2 is a P-body resident and a commonly used marker to examine the P-body formation in cells. Using an anti-Dcp2 antibody, the P-body formation in wild-type and siRNA-downregulated Huh-7 cells was examined by fluorescence microscopy. As evident from Fig. 5C, the downregulation of GW182 and Ge-1 causes P-body loss in Huh-7 cells.
Fig 5.

Role of cellular P bodies in hantavirus cap snatching. (A) Downregulation of GW182 by siRNA. To determine whether GW182 was downregulated after siRNA treatment, cells transfected with either siRNA or control siRNA were lysed and GW182 expression was monitored by Western blot analysis using anti-GW182 antibody. To confirm that downregulation of GW182 observed by Western blot analysis was not a loading error, equal volumes of cell lysate were examined for the expression of β-actin by Western blot analysis (bottom bands). (B) Similar to what is shown in panel A, the downregulation of Ge-1 by siRNA was confirmed by Western blot analysis using anti-Ge-1 antibody. (C) Downregulation of GW182 and Ge-1 by siRNA causes the downregulation of cellular P bodies. Huh-7 cells were transfected with 150 nM either control siRNA, GW-182 siRNA, or Ge-1 siRNA. Twenty-four hours posttransfection, cells were fixed with formaldehyde and stained with anti-Dcp2 antibody. Cells were visualized under fluorescence microscope using a FITC-conjugated secondary antibody. Bar, 10 μm. (D) Wild-type or P-body-downregulated Huh-7 cells were cotransfected with pCDNAGFPns and pCDNA-SNVN plasmids, expressing the cap donor test mRNA and SNV N protein, respectively. Thirty-six hours posttransfection, cells were lysed and total RNA was purified and reverse transcribed using random primers. The effect of N upon the stability of the test mRNA was quantified by real-time PCR analysis using a forward primer, 5′-TAGAGAACCCACTGCTTACTGGC-3′, and a reverse primer, 5′-CAGATGAACTTCAGGGTCAG-3′, to amplify 241 nucleotides from the 5′ mRNA terminus. (E) Huh-7 cells transfected with either siRNA or control siRNA along the cap donor plasmid pCDNAGFPns were infected with SNV. Caps snatched by the SNV from the test mRNA were quantified by real-time PCR analysis using the primer set shown in Fig. 1A.
We next asked whether SNV N can bind to the 5′ caps and equally protect the 5′ terminus of the nonsense mRNA in wild-type and P-body-deficient Huh-7 cells. If N selectively protects the 5′ caps of cellular mRNAs in P bodies, we do not expect such protection in Gw182-deficient cells. We cotransfected the wild-type and P-body-deficient Huh-7 cells with a pCDNAGFPns construct expressing nonsense GFP mRNA along with a plasmid expressing SNV N protein. At 36 h posttransfection, cells were lysed and total RNA was purified and reverse transcribed using random primers, as previously reported (42). The 5′ terminus of GFP nonsense mRNA was quantitatively estimated by real-time PCR analysis using a primer set targeted to amplify 180 nucleotides at the 5′ terminus of the mRNA, as previously reported (42). As shown in Fig. 5D, the loss of P-body machinery resulted in the protection of the 5′ mRNA terminus independent of N protein expression. This protection was likely due to the inefficient decapping machinery in P-body-downregulated cells (38, 69). Interestingly, we observed a remarkable protection of the 5′ mRNA terminus by SNV N in both wild-type and P-body-downregulated cells. This observation suggests that SNV N likely binds to the capped host cell mRNAs in the cytoplasm outside the P bodies. To delineate whether P bodies play a role in hantavirus cap snatching and viral mRNA synthesis, we transfected the Huh-7 cells with the cap donor plasmid pCDNAGFPns, followed by the viral infection. The cap-snatching efficiency by SNV RdRp in P-body-deficient Huh-7 cells was compared with that of wild-type Huh-7 cells having intact P-body machinery. Since the protection of the 5′ mRNA terminus by N protein in P-body-deficient cells was around 2-fold greater than that in the wild-type Huh-7 cells (Fig. 5D), we expected corresponding increases in cap snatching in P-body-deficient cells. Interestingly, we observed that cap snatching, and hence viral mRNA synthesis, was not improved in P-body-deficient cells compared to that in wild-type cells (Fig. 5E). This observation suggests that N increases the RNA half-life with no mechanism inferred.
DISCUSSION
The viral genome of negative-sense RNA viruses is encapsidated into viral nucleocapsids, which serve as templates for RdRp during transcription and replication of the viral genome (56, 58). The negative-strand segmented RNA viruses of the Orthomyxoviridae (e.g., influenza A, B, and C and Thogoto viruses), Bunyaviridae (e.g., La Crosse, hanta, Rift Valley fever, and Crimean-Congo hemorrhagic fever viruses) and Arenaviridae (e.g., Lassa virus) families synthesize the translation-competent capped mRNAs by the cap-snatching mechanism. This mechanism has been well studied for the influenza virus, having an RdRp composed of three subunits, PA, PB1, and PB2. The PB1 subunit contains a conserved polymerase domain, which carries out RNA elongation during RNA synthesis (55). The PB2 subunit binds to the 5′ caps of host pre-mRNAs (26, 36), and the PA subunit has the endonuclease domain, which cleaves the capped pre-mRNAs 10 to 13 nucleotides downstream of the 5′ cap (19, 70). The capped oligoribonucleotides are used as primers by the viral RdRp to initiate the transcription. The presence of an endonuclease domain in the RdRps of several other viruses, including SNV, has recently been proposed (47, 61). However, it is not yet clear whether, similarly to that in influenza viruses, the entire cap-snatching process in these viruses is carried out solely by the RdRp or other host factors or whether viral proteins also play a role. Sequence analyses of many viral mRNA 5′ termini have revealed a nucleotide preference at the 3′ end of the capped primer, which has been assumed to reflect the sequence preference for cleavage by the viral endonuclease during cap snatching. For example, in the case of Dugbe virus endonucleolytic cleavage is assumed to take place after a C residue (31), whereas for Bunyamwera virus a strong preference for cleavage after a U residue has been proposed (30). In tomato spotted wilt virus, preference for an A residue has been confirmed (20). However, it has been reported that influenza virus RdRp effectively uses only CA-terminated capped fragments as primers for viral mRNA synthesis in vitro. Consistent with our previous observation (42), we have found that primers used by the SNV RdRp are terminated at G residues. Taking these observations into consideration, it is likely that RdRps have a preference for the endonucleolytic cleavage at certain nucleotides in the mRNA sequence. However, it is also possible that RdRps randomly cleave the capped mRNAs and that the resulting capped fragments with appropriate terminal nucleotides are selected as primers. The selection may depend upon the appropriate location of complementary nucleotides at the 3′ terminus of the vRNA template.
Previous studies have reported that most viruses use 15-nucleotide-long capped primers with a variation of 10 to 20 nucleotides for transcription initiation (7, 9, 15, 18, 22, 31, 51, 52, 65). However, viruses of the Arenaviridae and the nairovirus genus use relatively shorter primers, varying in length from 1 to 4 and 5 to 16 nucleotides, respectively (24, 31, 59). We observed that SNV has a strong preference for 14-nucleotide-long primers containing a 3′-terminal G residue. It is still a mystery why the RdRps from different viruses use capped primers of various lengths for the transcription initiation. A possible role of the length of a capped primer in transcription initiation has been suggested in the cap-snatching model (Fig. 6).
Fig 6.
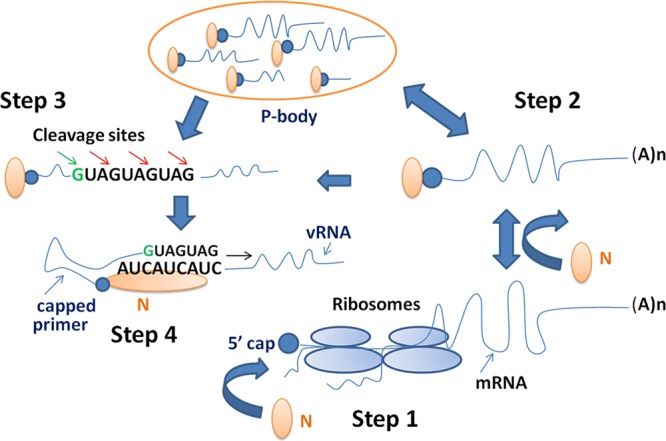
Hantavirus cap-snatching model. (Step 1) An mRNA engaged in translation is not used for cap snatching. N protein can bind to the mRNA cap before translation or after the completion of translation. (Step 2) N-associated mRNAs are targeted to P bodies for degradation. N protects the mRNA caps from degradation in P bodies. The rescued 5′-capped mRNA oligoribonucleotides are stored in P bodies by N and are efficiently used for cap snatching outside P bodies. Alternatively, N-associated mRNAs outside the P bodies can be directly used in cap snatching with less efficiency. (Step 3) The endonuclease activity of hantavirus RdRp or a possible cellular endonuclease preferentially cleaves the capped oligonucleotides at G residues. (Step 4) N protein with a capped primer at the cap binding site simultaneously binds the 3′ terminus of the viral RNA genome and facilitates the annealing of the capped primer with the 3′ terminus of the vRNA template. The capped primer with 3′ nucleotides more complementary to the 3′ terminus of vRNA genome is preferred for transcription initiation.
In hantaviruses, the capped primer containing a 3′ G residue has been proposed to undergo base pairing with one of the C residues at the 3′ terminus of the vRNA template during transcription initiation (24). It has been suggested that RdRp elongates the annealed primer during transcription initiation using a “prime and realign” mechanism (24, 42). However, it is interesting to imagine how a single G-C base pairing between the RNA primer and 3′ terminus of the vRNA template stabilizes the primer and favors its annealing. We have previously reported that hantavirus N protein stabilizes a capped primer at the 3′ terminus of the vRNA template (45). We showed that N protein binds the mRNA caps and has distinct cap and RNA binding sites (42, 43). N protein with a capped primer loaded at its cap binding site simultaneously binds the 3′ terminus of the vRNA template with specificity and facilitates the annealing of the capped primer with the template, which favors transcription. To further address the annealing of the capped primer with the vRNA template during the cap-snatching mechanism of transcription initiation, we asked whether the complementarity between the 3′ termini of the primer and vRNA template favors cap snatching. We incorporated two nucleotides preceding the high-priority cleavage site (14th G) in the 3′ terminus of test mRNA which were complementary to the 3′ terminus of the genomic RNA (Fig. 3A). Similarly, either three, six, or nine nucleotides complementary to the 3′ terminus of the vRNA template were incorporated in the test mRNA, preceding the 14th G residue (Fig. 3A). These complementary nucleotides contained G residues, which can also serve as low-priority cleavage sites for the RdRp. An examination by a very sensitive real-time PCR method demonstrated that these test mRNAs served as efficient cap donors during cap snatching. These observations support the recent findings that although the cleavage of capped mRNAs by the endonuclease subunit of influenza RdRp occurs independent of the 3′ terminus of viral genomic RNA (60), the influenza virus transcriptase prefers capped primers with 3′ nucleotides more complementary to the 3′ terminus of viral genomic RNA (25). We propose that for an mRNA to be an efficient cap donor during cap snatching, it must contain a high-priority cleavage site at an appropriate length from the 5′ terminus. For example, an mRNA having a G residue at the 14th position will be the preferred cap donor for hantavirus cap snatching. In addition, the nucleotide complementarity between the 3′ termini of the capped primer and vRNA template favors the annealing of the primer with the vRNA template.
Although the basic mechanism for the generation of capped primers of appropriate length and specificity might be similar between the RdRps of influenza virus and SNV, the basic difference in their cap-snatching mechanism would be due to their replications at different locations in the host cell. The cellular mRNAs are engaged in translation in the cell cytoplasm and are targeted to P bodies for degradation after the completion of translation. Thus, unlike influenza virus, whose replication takes place in the nucleus, the viruses replicating in the cytoplasm, such as SNV and Rift Valley Fever virus, have to effectively compete with the host decapping machinery to protect the mRNA caps from degradation. We have previously reported that hantavirus N protein actively protects the host mRNA caps from degradation (42). N binds to the host mRNA caps and likely blocks the binding of the decapping enzyme DCP2 at the mRNA 5′ cap by competitive inhibition, which would result in the protection of host mRNA caps from degradation. The rescued 5′ capped RNA oligoribonucleotides were sequestered in the cellular P bodies by N and were later efficiently used as primers by the RdRp. These observations raised the questions of whether N binds to the mRNA caps in the cytoplasm or inside the P bodies and whether P bodies have any role in the cap-snatching process. Protection of host mRNA caps in P-body-downregulated cells support the idea that N likely binds the mRNA caps inside the cytoplasm. N protein is likely transported to the P bodies along with the mRNAs, which are targeted for degradation after the completion of translation. Although N rescued the mRNA caps in P-body-deficient cells, the rescued caps were not used with significant efficiency in comparison to cells having intact P-body machinery. These observations suggest that P bodies might play a role in virus replication by providing the capped primers with high efficiency for the transcription initiation. Alternatively, a possible role of a P-body component in hantavirus cap snatching cannot be ruled out. Further experimentation is required to demonstrate the exact role of cellular P bodies in the cap-snatching process. A critical observation that PTC-containing mRNAs were remarkably efficient cap donors in comparison to the wild-type mRNAs suggests that efficiency of an mRNA to donate caps for viral mRNA synthesis is regulated primarily at the translation process. An mRNA engaged in translation may not be an efficient cap donor even if it contains a potential G residue at the 14th position or contains a sequence more complementary to the 3′ terminus of the viral RNA template. We propose that hantaviruses obtain most of their caps from the mRNAs, which abort translation due to PTCs or other translation defects.
The interesting observation that hantaviruses can snatch caps from their own mRNAs is consistent with the positive role of the nucleotide complementarity between the 3′ termini of the capped primer and the vRNA template in cap snatching. However, the cap-donating efficiency of wild-type mRNAs is negligible in comparison to that of the nonsense mRNAs; it is unlikely that viral mRNAs will be efficiently used for cap snatching during viral infection due to their continued engagement in the translation. We found that viral mRNAs containing a PTC were remarkably used for cap snatching with high efficiency. Hantavirus RdRp lacks proofreading activity. The promiscuous nature of polymerization by RdRps due to the lack of proofreading ability is thought to be the primary source of evolution in RNA viruses, which provides them an ability to replicate in different hosts and produce more pathogenic strains. An error rate of approximately 1 mutation/replication/genome for hantaviruses has been estimated. Although this mutation rate is negligible, the nonsense mRNAs generated due to the lack of proofreading activity of the RdRp will be actively recycled for cap snatching. We suggest that apart from helping in the evolutionary strategies of the virus, the lack of proofreading activity of the RdRp plays a role in cap snatching. A model depicting the role of N protein and cellular P bodies in hantavirus cap snatching is shown in Fig. 6.
ACKNOWLEDGMENTS
This work was supported by research grants 5R21AI083672-02 and 5R01AI095236-02 from the NIH.
We thank Nilshad Salim for reading the manuscript.
Footnotes
Published ahead of print 11 July 2012
REFERENCES
- 1. Accardi L, et al. 2001. Activity of Toscana and Rift Valley fever virus transcription complexes on heterologous templates. J. Gen. Virol. 82:781–785 [DOI] [PubMed] [Google Scholar]
- 2. Alfadhli A, et al. 2001. Hantavirus nucleocapsid protein oligomerization. J. Virol. 75:2019–2023 doi:10.1128/JVI.75.4.2019-2023.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alfadhli A, Steel E, Finlay L, Bachinger HP, Barklis E. 2002. Hantavirus nucleocapsid protein coiled-coil domains. J. Biol. Chem. 277:27103–27108 [DOI] [PubMed] [Google Scholar]
- 4. Amrani N, Sachs MS, Jacobson A. 2006. Early nonsense: mRNA decay solves a translational problem. Nat. Rev. Mol. Cell Biol. 7:415–425 [DOI] [PubMed] [Google Scholar]
- 5. Banerjee AK. 1980. 5′-terminal cap structure in eucaryotic messenger ribonucleic acids. Microbiol. Rev. 44:175–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barr JN, Wertz GW. 2004. Bunyamwera bunyavirus RNA synthesis requires cooperation of 3′- and 5′-terminal sequences. J. Virol. 78:1129–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bishop DH, Gay ME, Matsuoko Y. 1983. Nonviral heterogeneous sequences are present at the 5′ ends of one species of snowshoe hare bunyavirus S complementary RNA. Nucleic Acids Res. 11:6409–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blakqori G, Kochs G, Haller O, Weber F. 2003. Functional L polymerase of La Crosse virus allows in vivo reconstitution of recombinant nucleocapsids. J. Gen. Virol. 84:1207–1214 [DOI] [PubMed] [Google Scholar]
- 9. Bouloy M, Pardigon N, Vialat P, Gerbaud S, Girard M. 1990. Characterization of the 5′ and 3′ ends of viral messenger RNAs isolated from BHK21 cells infected with Germiston virus (Bunyavirus). Virology 175:50–58 [DOI] [PubMed] [Google Scholar]
- 10. Bouloy M, Plotch SJ, Krug RM. 1980. Both the 7-methyl and the 2′-O-methyl groups in the cap of mRNA strongly influence its ability to act as primer for influenza virus RNA transcription. Proc. Natl. Acad. Sci. U. S. A. 77:3952–3956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bouloy M, Plotch SJ, Krug RM. 1978. Globin mRNAs are primers for the transcription of influenza viral RNA in vitro. Proc. Natl. Acad. Sci. U. S. A. 75:4886–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Braam J, Ulmanen I, Krug RM. 1983. Molecular model of a eucaryotic transcription complex: functions and movements of influenza P proteins during capped RNA-primed transcription. Cell 34:609–618 [DOI] [PubMed] [Google Scholar]
- 13. Bridgen A, Elliott RM. 1996. Rescue of a segmented negative-strand RNA virus entirely from cloned complementary DNAs. Proc. Natl. Acad. Sci. U. S. A. 93:15400–15404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cao D, Parker R. 2003. Computational modeling and experimental analysis of nonsense-mediated decay in yeast. Cell 113:533–545 [DOI] [PubMed] [Google Scholar]
- 15. Caton AJ, Robertson JS. 1980. Structure of the host-derived sequences present at the 5′ ends of influenza virus mRNA. Nucleic Acids Res. 8:2591–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Conti E, Izaurralde E. 2005. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr. Opin. Cell Biol. 17:316–325 [DOI] [PubMed] [Google Scholar]
- 17. Crow M, Deng T, Addley M, Brownlee GG. 2004. Mutational analysis of the influenza virus cRNA promoter and identification of nucleotides critical for replication. J. Virol. 78:6263–6270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dhar R, Chanock RM, Lai CJ. 1980. Nonviral oligonucleotides at the 5′ terminus of cytoplasmic influenza viral mRNA deduced from cloned complete genomic sequences. Cell 21:495–500 [DOI] [PubMed] [Google Scholar]
- 19. Dias A, et al. 2009. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914–918 [DOI] [PubMed] [Google Scholar]
- 20. Duijsings D, Kormelink R, Goldbach R. 2001. In vivo analysis of the TSWV cap-snatching mechanism: single base complementarity and primer length requirements. EMBO J. 20:2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dunn EF, Pritlove DC, Jin H, Elliott RM. 1995. Transcription of a recombinant bunyavirus RNA template by transiently expressed bunyavirus proteins. Virology 211:133–143 [DOI] [PubMed] [Google Scholar]
- 22. Eshita Y, Ericson B, Romanowski V, Bishop DH. 1985. Analyses of the mRNA transcription processes of snowshoe hare bunyavirus S and M RNA species. J. Virol. 55:681–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eulalio A, Behm-Ansmant I, Izaurralde E. 2007. P bodies: at the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 8:9–22 [DOI] [PubMed] [Google Scholar]
- 24. Garcin D, et al. 1995. The 5′ ends of Hantaan virus (Bunyaviridae) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. J. Virol. 69:5754–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geerts-Dimitriadou C, Zwart MP, Goldbach R, Kormelink R. 2011. Base-pairing promotes leader selection to prime in vitro influenza genome transcription. Virology 409:17–26 [DOI] [PubMed] [Google Scholar]
- 26. Guilligay D, et al. 2008. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 15:500–506 [DOI] [PubMed] [Google Scholar]
- 27. Hutchinson KL, Peters CJ, Nichol ST. 1996. Sin Nombre virus mRNA synthesis. Virology 224:139–149 [DOI] [PubMed] [Google Scholar]
- 28. Ikegami T, Peters CJ, Makino S. 2005. Rift valley fever virus nonstructural protein NSs promotes viral RNA replication and transcription in a minigenome system. J. Virol. 79:5606–5615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Isken O, Maquat LE. 2007. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 21:1833–1856 [DOI] [PubMed] [Google Scholar]
- 30. Jin H, Elliott RM. 1993. Characterization of Bunyamwera virus S RNA that is transcribed and replicated by the L protein expressed from recombinant vaccinia virus. J. Virol. 67:1396–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jin H, Elliott RM. 1993. Non-viral sequences at the 5′ ends of Dugbe nairovirus S mRNAs. J. Gen. Virol. 74(Part 10):2293–2297 [DOI] [PubMed] [Google Scholar]
- 32. Kohl A, et al. 2004. A Bunyamwera virus minireplicon system in mosquito cells. J. Virol. 78:5679–5685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krug RM. 1981. Priming of influenza viral RNA transcription by capped heterologous RNAs. Curr. Top. Microbiol. Immunol. 93:125–149 [DOI] [PubMed] [Google Scholar]
- 34. Kukkonen SK, Vaheri A, Plyusnin A. 2004. Tula hantavirus L protein is a 250 kDa perinuclear membrane-associated protein. J. Gen. Virol. 85:1181–1189 [DOI] [PubMed] [Google Scholar]
- 35. Lejeune F, Maquat LE. 2005. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr. Opin. Cell Biol. 17:309–315 [DOI] [PubMed] [Google Scholar]
- 36. Li ML, Rao P, Krug RM. 2001. The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J. 20:2078–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li XD, et al. 2004. Tula hantavirus infection of Vero E6 cells induces apoptosis involving caspase 8 activation. J. Gen. Virol. 85:3261–3268 [DOI] [PubMed] [Google Scholar]
- 38. Liu J, et al. 2005. A role for the P-body component GW182 in microRNA function. Nat. Cell Biol. 7:1261–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lopez N, Muller R, Prehaud C, Bouloy M. 1995. The L protein of Rift Valley fever virus can rescue viral ribonucleoproteins and transcribe synthetic genome-like RNA molecules. J. Virol. 69:3972–3979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meyer S, Temme C, Wahle E. 2004. Messenger RNA turnover in eukaryotes: pathways and enzymes. Crit. Rev. Biochem. Mol. Biol. 39:197–216 [DOI] [PubMed] [Google Scholar]
- 41. Mir MA, Brown B, Hjelle B, Duran WA, Panganiban AT. 2006. Hantavirus N protein exhibits genus-specific recognition of the viral RNA panhandle. J. Virol. 80:11283–11292 doi:10.1128/JVI.00820-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mir MA, Duran WA, Hjelle BL, Ye C, Panganiban AT. 2008. Storage of cellular 5′ mRNA caps in P bodies for viral cap-snatching. Proc. Natl. Acad. Sci. U. S. A. 105:19294–19299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mir MA, Panganiban AT. 2008. A protein that replaces the entire cellular eIF4F complex. EMBO J. 27:3129–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mir MA, Panganiban AT. 2006. The bunyavirus nucleocapsid protein is an RNA chaperone: possible roles in viral RNA panhandle formation and genome replication. RNA 12:272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mir MA, Sheema S, Haseeb A, Haque A. 2010. Hantavirus nucleocapsid protein has distinct m7G cap- and RNA-binding sites. J. Biol. Chem. 285:11357–11368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mitchell P, Tollervey D. 2003. An NMD pathway in yeast involving accelerated deadenylation and exosome-mediated 3′→5′ degradation. Mol. Cell 11:1405–1413 [DOI] [PubMed] [Google Scholar]
- 47. Morin B, et al. 2010. The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathog. 6:e1001038 doi:10.1371/journal.ppat.1001038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Obijeski JF, Bishop DH, Murphy FA, Palmer EL. 1976. Structural proteins of La Crosse virus. J. Virol. 19:985–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Parker R, Sheth U. 2007. P bodies and the control of mRNA translation and degradation. Mol. Cell 25:635–646 [DOI] [PubMed] [Google Scholar]
- 50. Parker R, Song H. 2004. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 11:121–127 [DOI] [PubMed] [Google Scholar]
- 51. Patterson JL, Holloway B, Kolakofsky D. 1984. La Crosse virions contain a primer-stimulated RNA polymerase and a methylated cap-dependent endonuclease. J. Virol. 52:215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Patterson JL, Kolakofsky D. 1984. Characterization of La Crosse virus small-genome transcripts. J. Virol. 49:680–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pettersson RF, von Bonsdorff CH. 1975. Ribonucleoproteins of Uukuniemi virus are circular. J. Virol. 15:386–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pinschewer DD, Perez M, de la Torre JC. 2003. Role of the virus nucleoprotein in the regulation of lymphocytic choriomeningitis virus transcription and RNA replication. J. Virol. 77:3882–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Plotch SJ, Bouloy M, Ulmanen I, Krug RM. 1981. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23:847–858 [DOI] [PubMed] [Google Scholar]
- 56. Raju R, Kolakofsky D. 1986. Inhibitors of protein synthesis inhibit both La Crosse virus S-mRNA and S genome syntheses in vivo. Virus Res. 5:1–9 [DOI] [PubMed] [Google Scholar]
- 57. Raju R, Kolakofsky D. 1989. The ends of La Crosse virus genome and antigenome RNAs within nucleocapsids are base paired. J. Virol. 63:122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Raju R, Kolakofsky D. 1987. Unusual transcripts in La Crosse virus-infected cells and the site for nucleocapsid assembly. J. Virol. 61:667–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Raju R, et al. 1990. Nontemplated bases at the 5′ ends of Tacaribe virus mRNAs. Virology 174:53–59 [DOI] [PubMed] [Google Scholar]
- 60. Rao P, Yuan W, Krug RM. 2003. Crucial role of CA cleavage sites in the cap-snatching mechanism for initiating viral mRNA synthesis. EMBO J. 22:1188–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Reguera J, Weber F, Cusack S. 2010. Bunyaviridae RNA polymerases (L-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 6:e1001101 doi:10.1371/journal.ppat.1001101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schmaljohn CS. 1996. Molecular biology of hantaviruses. Plenum Press, New York, NY [Google Scholar]
- 63. Schmaljohn CS, Hooper JW. 2001. Bunyaviridae: the viruses and their replication, p 1581–1602 In Knipe DM, et al. (ed), Fields virology, vol 2 Lippincott, Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 64. Schmaljohn CS, Jonsson CB. 2001. Replication of hantaviruses, p 15–32 In Schmaljohn CS, Nichol ST. (ed), Hantaviruses. Springer-Verlag, Berlin, Germany: [DOI] [PubMed] [Google Scholar]
- 65. Simons JF, Pettersson RF. 1991. Host-derived 5′ ends and overlapping complementary 3′ ends of the two mRNAs transcribed from the ambisense S segment of Uukuniemi virus. J. Virol. 65:4741–4748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Taylor SL, Frias-Staheli N, Garcia-Sastre A, Schmaljohn CS. 2009. Hantaan virus nucleocapsid protein binds to importin alpha proteins and inhibits tumor necrosis factor alpha-induced activation of nuclear factor kappa B. J. Virol. 83:1271–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Taylor SL, Krempel RL, Schmaljohn CS. 2009. Inhibition of TNF-alpha-induced activation of NF-kappaB by hantavirus nucleocapsid proteins. Ann. N. Y. Acad. Sci. 1171(Suppl 1):E86–E93 [DOI] [PubMed] [Google Scholar]
- 68. Ulmanen I, Broni BA, Krug RM. 1981. Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl. Acad. Sci. U. S. A. 78:7355–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang Z, et al. 2004. GW182 is critical for the stability of GW bodies expressed during the cell cycle and cell proliferation. J. Cell Sci. 117:5567–5578 [DOI] [PubMed] [Google Scholar]
- 70. Yuan P, et al. 2009. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458:909–913 [DOI] [PubMed] [Google Scholar]