

Abstract
Experiments have shown that actin is structurally polymorphic, but knowledge of the details of molecular level heterogeneity in both the dynamics of a single subunit and the interactions between subunits is still lacking. Here, using atomistic molecular dynamics simulations of the actin filament, we identify domains of atoms that move in a correlated fashion, quantify interactions between these domains using coarse-grained (CG) analysis methods, and perform CG simulations to explore the importance of filament heterogeneity. The persistence length and torsional stiffness calculated from molecular dynamics simulation data agree with experimental values. We additionally observe that distinct actin conformations coexist in actin filaments. The filaments also exhibit random twist angles that are broadly distributed. CG analysis reveals that interactions between equivalent CG pairs vary from one subunit to another. To explore the importance of heterogeneity on filament dynamics, we perform CG simulations using different methods of parameterization to show that only by including heterogeneous interactions can we reproduce the twist angles and related properties. Free energy calculations further suggest that in general the actin filament is best represented as a set of subunits with differing CG sites and interactions, and the incorporating heterogeneity into the CG interactions is more important than including that in the CG sites. Our work therefore presents a systematic method to explore molecular level detail in this large and complex biopolymer.
Introduction
Actin monomers (G-actin) assemble into double helical filaments (F-actin), which form mesh-like structures that provide mechanical support to cells and determine their shape (1). Actin filaments also play important roles in cell mobility, division, adhesion, endocytosis, and intracellular transportation (1). A variety of mechanisms regulate the conformation and mechanical properties of actin filaments and networks, including >100 actin-binding proteins and ligands (1–5). The nucleotide at the core of each actin subunit can affect filament properties: ATP-bound filaments have higher bending (6,7) and torsional rigidities than ADP-bound filaments (8). The inherent coupling between actin subunit conformation and large length scale properties of actin filaments and networks makes it critical to explore actin conformations that could facilitate different functions in cellular processes.
Both experimental and computational studies on G- and F-actin have shown that actin exists in multiple conformations and exhibits a range of filament properties (9–29). As an example, subdomain 2 of actin, particularly the D-loop region, appears to be highly conformationally mobile. A comparison of early x-ray structures of G-actin (13,15) showed that the D-loop folds into an α-helix in the ADP state but does not form a well-defined secondary structure in the ATP state. Rould et al (17) suggested that the helical D-loop in ADP actin was a result of specific crystal packing interactions between actin subunits. Meanwhile, computational studies have explored the stability of the D-loop (16,19,21,22). In some molecular dynamics (MD) simulations, the D-loop unfolds from a helical to a coiled conformation (19) in G-actin. This transition was explored in a more systematic manner using the metadynamics simulations (22). The most recent experimental conclusion is that in actin the D-loop can adopt multiple configurations, but these configurations have not been clearly correlated to a particular nucleotide state. Rather, ATP- and ADP-bound G-actin is believed to differ primarily in two loops: the β-hairpin loop containing Ser-14 and the sensor loop with the methylated His-73 (1).
F-actin also exists in different conformational states, which can coexist in the same filament. A recent cryo-electron microscopy study (25) identified six distinct structural modes of actin, which differed in the contacts between neighboring subunits, and in most cases in the conformation of the D-loop. These differences in contacts may cause variations in twist angle along the filament, explaining experiments that show that the twist angle of each actin subunit along the actin filament is broadly distributed despite the longitudinal rise remaining roughly the same (10). Orlova and Egelman (12) hypothesized that the energy landscape of the twist angle per subunit was not a simple harmonic form, but may have many local minima. Taken together, these results suggest that each subunit could be trapped at some specific twist angle for long periods of time in one of several structurally polymorphic states and thus the actin filament consists of an ensemble of different subunit states.
MD simulations have complemented the experimental research into actin filament structure and properties. One area of interest is the hypothesis that different conformations favor specific intersubunit interactions, which in turn affect macroscopic properties. Chu and Voth (16) investigated how the persistence length of actin filaments varied with changes in D-loop conformation, finding that ATP-bound, unfolded D-loop filaments had twice the persistence length of those that were ADP bound and had a folded D-loop. Pfaendtner et al. (26) refined this study using improved filament structures (23) and found that the unfolding of the D-loop increased the persistence length regardless of the state of the bound nucleotide. More recently, a study comparing different F-actin models showed the spontaneous development of heterogeneous subunit conformations and intersubunit interactions that were consistent with the modes observed in the cryo-electron microscopy studies mentioned previously (29). These studies again show how local conformational changes can propagate to affect macroscopic filament properties.
In this work we apply systematic coarse-graining (CG) methods to quantify the differences between subunits in filaments composed of multiple configurational states and explore how allowing different interactions between different pairs of actin subunits affects the properties of a filament. All-atom MD simulations of actin filaments either ATP or ADP bound reveal a range of actin subunit configurations and the filament properties calculated from these simulations are consistent with previous experimental results. Two CG methods allowed us to quantify the differences between subunits. Essential dynamics coarse-graining (ED-CG) (30) identifies domains in which atom motions were highly correlated (dynamic domains) and the heterogeneous elastic network model (HeteroENM) algorithm (31) quantifies how strongly the domains interact with one another. The results show that both the domain composition and the interaction strengths vary between subunits within the same filament. One of the advantages of CG analysis is that it does not rely on differences in single residue interactions but rather captures information about more collective differences in structure and dynamics. Based on this analysis, three sets of CG models were built to determine how heterogeneity between subunits impacted filament properties. The results of these simulations show that only CG actin filaments that incorporate heterogeneous interactions correctly reproduce the persistence length, twist angle, and torsional stiffness. Our work thus reveals that filament properties are strongly affected by structural polymorphism, and demonstrates a systematic method to explore the dynamics of this complex biological polymer.
Methods
Atomistic MD simulations of actin filaments
Both ATP- and ADP-bound filament systems were setup based on PDB entry 2ZWH (23), with Mg2+ placed at the high-affinity cationic binding site and explicitly incorporating the first solvation shell of waters around the cation (28). Filaments containing 13 subunits were created as described previously (16,26,28). The filament was solvated in explicit TIP3P water molecules (32) and potassium and chloride ions were added to a final concentration of 0.18 M using the solvate and autoionize tools in VMD (33), with a minimum 8 Å of solvent between the protein and the boundaries in the x- and y-directions. The filament was aligned to periodically repeat along the z-direction, interacting with itself at the box edges.
Simulations were performed using NAMD (34) and the CHARM 22/27 force field with CMAP correction (35). Electrostatic interactions were calculated using the particle mesh Ewald sum method (36) with a cutoff of 12 Å. An integration time step of 2 fs was used while constraining all intramolecular hydrogen-containing bonds with the SHAKE (37) algorithm. Each system was energy minimized, heated, and equilibrated for 100 ps in the canonical ensemble while harmonically restraining the protein backbone, nucleotide, and active site water oxygen atoms. The NAMD barostat was applied to maintain a pressure of 1 atm and the system was equilibrated for an additional 200 ps. Constraints were released stepwise over 100 ps before starting the production run. The temperature was maintained throughout at 310 K using a Langevin thermostat with damping coefficient of 0.5 ps−1; in total, 94 and 105 ns data were generated for the ATP- and ADP-bound systems, respectively. Unless otherwise specified, the final 10 ns of data were used for CG analysis.
The ED-CG method identifies domains with dynamically correlated atoms
Each subunit was coarse-grained into 6–20 beads using the ED-CG method (25). This method systematically assigns atoms into different CG sites according to the correlations of atomic motions, and has previously been applied to large protein systems, such as the ribosome, to explore dynamically correlated domains (38). The theory underlying this method is described in the Supporting Material.
Two kinds of CG filaments were created using the ED-CG method. In the heterogeneous CG filament, unique CG sites for each subunit were obtained by applying the ED-CG analysis to the trajectory of each individual subunit. In the uniform CG filament representation, the same CG sites were used for all subunits; these were obtained by analyzing the pseudotrajectory consisting of the concatenation of the 13 individual subunit trajectories.
Comparing the similarity of CG sites
A simple algorithm was devised to quantify the similarity of CG sites in different subunits. For a pair of CG sites, overlapping residues are defined as those residues that are in both CG sites. A CG site in subunit B is defined as being analogous to a site in subunit A if, of all the sites in B, it contains the maximum number of overlapping residues. To calculate the similarity of A to B, the sum of the number of overlapping residues in the analogous CG site in B for each site in A is determined, and normalized by dividing by the total number of residues in a subunit.
CG interactions are obtained using HeteroENM
To extract the effective interaction strength between each CG pair, the HeteroENM method (31) was used. In this method, all interactions are modeled using harmonic springs, and the stiffness of each spring is iteratively adjusted until the fluctuations of the CG model as predicted from normal mode analysis match the fluctuations observed during an all-atom MD simulation. This method is more fully described in the Supporting Material.
For the heterogeneous CG filament system (set 1), each subunit was individually coarse-grained and parameterized. In addition, two more uniform models were developed—sets 2 and 3. In set 2, each subunit was represented by the same set of CG sites but the interactions between CG sites were allowed to differ. In set 3, both the sites and the interactions between specific CG sites were required to be identical in all subunits.
CG simulations
A 0.2 μs trajectory was generated for each CG set using Langevin dynamics with a 2 ps inverse friction constant (to damp harmonic oscillations in the absence of solvent) coupled to the Nóse-Hoover thermostat at 310 K and 2 fs integration time step, using GROMACS 4 (39). Systems equilibrated quickly (<10 ns). The final 0.1 μs of these trajectories were used for analysis.
Calculating the twist angle per subunit, persistence length, and torsional stiffness from MD and CG simulations
To calculate experimentally measurable properties from simulation trajectories, we linearized the actin filament and determined the twist angle of each subunit as described in the Supporting Material. The persistence length was calculated as described in (18). The torsional stiffness (C) was derived based on the equipartition theorem, , where Δβ is the standard deviation of the twist angle between two subunits with their COM separated by distance h.
Free energy of CG systems
The free energy of the coarse-grained systems was calculated analytically from the enthalpy and entropy derived using normal mode analysis (40). These energy terms are exact for the present CG model because the CG systems are coupled networks of harmonic oscillators; the derivation is provided in the Supporting Material.
Note: All error estimates are given as the standard deviation unless otherwise noted.
Results and Discussion
Properties of actin filaments extracted from MD simulations are consistent with experimental results
We calculated several properties of actin filaments from MD simulation data to demonstrate that the MD simulations correctly captured important long length scale filament features. These results, summarized in Table 1, are consistent with experimental measurements. The half twist length and twist angle per subunit do not distinguish between ATP- and ADP-bound systems because the geometries of the two systems are similar. However, the nucleotide is known to affect mechanical properties including bending rigidity (K), persistence length (Lp = K/kBT), and torsional stiffness. On the basis of our simulation data, Lp was calculated to be 12.0 ± 2.2 μm for ATP-bound actin and 9.9 ± 2.1 μm for ADP-bound actin. These results agree well with the experimental measurements, 13.5 ± 1 μm and 9 ± 1 μm (6,7), which reflect that the ADP binding softens the actin filament bending mode, as noted in previous studies (16,26). The torsional stiffnesses, 0.65 × 10−26Nm2/rad and 0.47 × 10−26Nm2/rad for ATP- and ADP-bound filaments, respectively, were also comparable to experimental values (41–45).
Table 1.
MD simulation results versus experimental results
| MD simulation results |
Experimental results |
|||
|---|---|---|---|---|
| ATP-bound | ADP-bound | ATP-bound | ADP-bound | |
| Half twist (Å) | 359.52 (0.59) | 358.24 (0.52) | 358–358.8∗ | |
| Twist per subunit (°) | 166.09 (1.76) | 165.84 (2.00) | 166.15–167.14∗ | |
| Persistence length (μm) | 12.0 (2.2) | 9.9 (2.1) | 13.5 (1.0)†§ | 9.0 (1.0)† |
| Torsional stiffness (E-26Nm2/rad) | 0.65 (0.19) | 0.47 (0.10) | — | 0.14–8.0‡ |
A variable twist angle is coupled to polymorphism in actin structure
From our MD simulation data, the twist angle distribution was quantified and is shown in Fig. 1. In both the ATP- and ADP-bound filaments, the twist angles are broadly distributed from 160° to 172°, consistent with previous experimental results (9,10), even though periodic boundary conditions were imposed. Enforcing these boundary conditions means that the twist angle is constrained to ∼ −166.2°. The broad distribution of twist angles along the filament is perhaps surprising under these conditions and is in itself a strong indication that subunits are behaving heterogeneously in the filament. The twist angle does show some nucleotide dependence: in the ATP-bound filament, the twist angle is twice as likely to be between 163° and 166° than it is between 166°and 169°, whereas in the ADP-bound filament it is more likely to be between 165° and 167° than it is between either 163° and 165° or 167° and 171°. Neither distribution curve is unimodal, supporting the hypothesis that there are multiple local energy minima (12).
Figure 1.
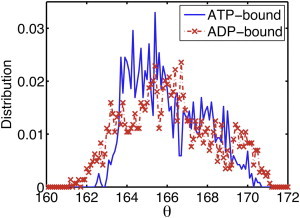
(Color online) Twist angle per subunit is broadly distributed (160°–172°) in both ATP-bound (blue curve) and ADP-bound (red curve) actin filaments. The bin size is 0.1°.
MD simulations also enable us to directly visualize configurations of each subunit. At this level of detail, the D-loops are found to be highly dynamic. The heterogeneity of this region is evidenced in Fig. S1 in the Supporting Material, which shows the final configurations of different subunit pairs in the ADP-bound filament. Although the D-loop was initially unfolded in all the subunits, by the end of simulation the D-loop conformation varies significantly between subunits. The D-loop in one of the 13 subunits became folded ∼55 ns into the simulation and remained folded to the end of the simulation as shown in the bottom subunit of Fig. S1 a. A folded-to-unfolded D-loop transition has been previously reported in the MD simulation of G-actin (19), but this is the first report, to our knowledge, of the reverse transition in either G- or F-actin. The probability of observing a folding event is quite low; the fact that we have observed this transition in a relatively short simulation window suggests that in the filament the barrier to folding is very low, and that the folded state is favored under certain local filament conditions.
In conjunction with the variations in D-loop structure, subunits also differ in the N-terminal configuration and in the contacts they form with their longitudinal neighbors, as shown in Fig. S1 and further discussed in (29). This is consistent with experimental observations (25) suggesting that changes in the D-loop conformation are coupled to differences in longitudinal contacts; this range of contact types would be expected to induce a broad range of twist angles, such as that shown in Fig. 1. Similar results are seen in the ATP-bound filament (data not shown).
CG analysis reveals variation in dynamic domains and in interactions between these domains in different subunits within an actin filament
Simply observing differences in morphology between subunits does not provide information about their collective motions. To reveal differences in the internal correlated dynamics of individual subunits, we applied the ED-CG method (30) to our MD simulation data. This method systematically divides atoms into dynamic domains based upon the identification of which atoms show correlated movements during the slow modes of motion obtained from principle component analysis (46). The results show that the atoms assigned to each dynamic domain vary from one subunit to another. To illustrate this, Fig. 2 shows the ED-CG map for a 6-site per subunit CG model with each site colored differently. In panel 2 a, which shows the CG site mapping for the ATP-bound filament, for example, the dark blue domain shows that S-loop (residues 11–16) is grouped with N-terminal (residues 1–5) in four subunits (subunit index 2, 4, 8, 11). The light blue domains reveal that the H-loop (residues 70–78) is in the same group as G-loop (residues 154–161) in two subunits (7th and 12th). In orange, the V-stretch (residues 227–237) and H-plug (residues 263–273) are grouped together in two subunits (6th and 13th). Similar variations in CG grouping are found in the ADP-bound filament (Fig. 2 b). Four typical different 6 CG sites subunit configurations are plotted in Fig. 2 c, and the complete list could be found in Fig. S2 for the ATP- and ADP-bound filament. It should be noted that because of the limited timescales that are accessible to MD, we can only observe this configurational heterogeneity by comparing different subunits within the filaments. As has been shown experimentally (47), transitions between states within a particular subunit are expected to occur on significantly longer timescales. One of the long-range objectives in characterizing the CG dynamics of the actin system is to create realistic CG models that we will be able to simulate on the timescale of seconds or longer.
Figure 2.
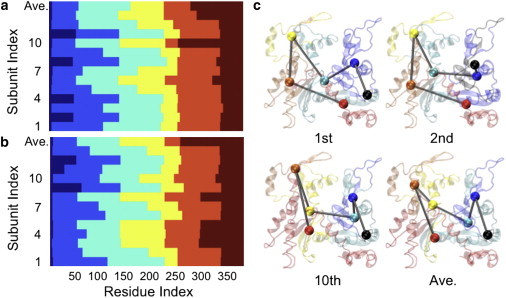
(Color online) Dynamic domains identified using the ED-CG method (NCG = 6) differ from one subunit to another, indicating heterogeneous collective motions. Each color represents one dynamic domain: (a) for the ATP-bound filament, (b) for the ADP-bound filament, and (c) examples of the 6 CG site per subunit mapping of ATP-bound actin; colors are consistent with those used in panel a.
To quantify the differences in conformation and examine the effect of increasing the resolution of the CG model (number of CG sites) on heterogeneity between subunits, we calculated the similarity index, as described in the Methods section. Fig. 3, a and b, show the average similarity between subunits as a function of the number of CG sites in the model. As the model resolution increases, the similarity between subunits increases to a plateau of ∼75–80%. In Fig. 3, c and d, the similarity matrices for the 6 and 16 CG site models of the ADP filament are shown. It is interesting to note that the highly similar subunits are not necessarily conserved as the number of CG sites increases. For example, in the 6-site CG model, subunit 7 is similar to subunit 1, whereas in the 16-site CG model this similarity is lost despite the overall increase in the average similarity. This suggests that different dynamic motions are being captured in the different CG representations.
Figure 3.
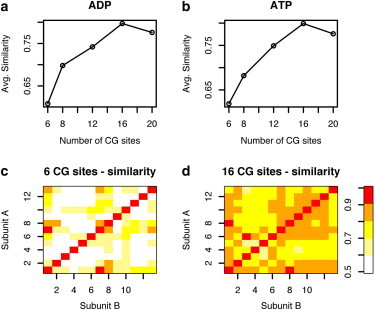
(Color online) Similarity in CG sites between actin subunits increases as the resolution of the model is increased.
The effective interactions between pairs of CG sites were determined using the HeteroENM method. Meaningful comparison between the interactions was complicated by the fact that the definition of CG sites in the filament was not identical. A more reasonable standard would be to compare the interactions between the same groups of atoms in different subunits. To accomplish this, a uniform mapping from the atomistic to the CG representation was needed. To determine the optimal uniform mapping, the ED-CG method was applied to the concatenation of the trajectories of all the individual subunits within the filament. Using this mapping, the CG pair interactions were then determined using the HeteroENM method. Fig. 4 illustrates the variation in CG interactions for different subunits using the CG spring constants, kij, for an intuitive minimalist CG map of four sites (one for each subdomain, see reference (16)). We show the 4-site data both for simplicity in comparing interactions and to illustrate the unique role of CG site 2 (which includes the D-loop), although the same trend applies for CG models with more sites. The six intrasubunit interactions are plotted using different colors and symbols, and the intersubunit interactions are plotted in gray dots. Variation in the kij values can be seen in the vertical variation of points of the same color. For example, k34 (red circles) varies from 4 to 16 kcal/mol Å2 for equivalent CG pairs in different subunits. Further examination of the intrasubunit interactions (data listed in Table S1) shows that the three interactions involving CG site 2 have a much higher relative variation in kij. CG site 2 appears to be highly dynamic because its interactions vary more between subunits than any other CG site. This is probably at least partially due to the conformational mobility of the D-loop segment. These results are consistent with very recently published work using a different CG methodology that also identified variations in the interactions of subdomain 2 (48).
Figure 4.
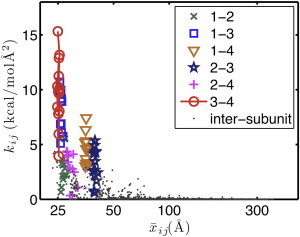
(Color online) CG effective interaction strength, kij, decreases with CG site pair distance. Intrasubunit interactions are plotted in different symbols and colors, whereas intersubunit interactions are plotted using gray dots.
The strength of the kij value of the intersubunit interactions decreases rapidly as the distance between CG sites increases, implying that interactions between nearby subunits are the most important. CG simulations using only the interactions with the six nearest subunits reproduced the results obtained using no cutoff criteria, confirming that relatively local interactions determine filament behavior.
Only CG simulations that incorporate heterogeneous interactions yield the correct filament properties
Using CG analysis methods, we have identified heterogeneity in both the identity of the dynamic domains and in the interactions between them in actin filaments. To examine the importance of this variation, three sets of CG systems were constructed based on the results from ED-CG and HeteroENM methods. Set 1 contains different CG sites in each subunit and different interactions between CG sites for each subunit. This CG mapping is obtained by applying the ED-CG algorithm to the whole 13-subunit filament without requiring that the subunits be coarse-grained identically. Sets 2 and 3 use the same CG sites in every subunit (i.e., a CG mapping is obtained by requiring the ED-CG method to give the same CG sites in each subunit while using averaged data from the whole filament to determine the average value and the standard deviation of the distances between CG sites). In set 2 the CG interactions between equivalent CG pairs vary between subunits, whereas in set 3 they are the same for all subunits within the filament.
We quantified the persistence length and torsional stiffness of actin filaments from simulations with the three CG models detailed previously, and compared them to atomistic MD results. As shown in Figs. 5 and 6, only sets 1 and 2 reproduce the correct filament properties. The calculated persistence lengths for ATP- and ADP-bound actin filaments are also plotted in Fig. 5 a and Fig. S3 a as a function of the number of CG sites used in the model. The results from sets 1 (red) and 2 (green) agree well with MD simulation results (black), whereas the results from set 3 (blue curve) are significantly larger and increase with the number of CG sites. In Fig. 5 b and Fig. S3 b, the torsional stiffness is plotted as a function of the number of CG sites. Similar to what was seen in the Lp plots, sets 1 (red) and 2 (green) reproduce the atomistic value (black) reasonably well for both filaments, whereas in set 3 (blue) the torsional stiffness increases with the number of CG sites. For NCG <10 in the ATP-bound filament, set 3 yields a smaller torsional stiffness, whereas for NCG >10 it yields a larger torsional stiffness and seems to stabilize as NCG >15. In the ADP-bound filament, when NCG <8, the torsional stiffness values are comparable with the atomistic MD one, but for NCG ≥8, it becomes larger and stabilizes at NCG >15. This indicates that for a small number of CG sites, a model with uniform effective interactions could still represent the elasticity of actin filaments reasonably well (in agreement with our previous CG simulation results (18)), whereas for a larger number of CG sites it fails to do so. When the number of CG beads increases, the corresponding intra- and intersubunit CG interactions become more diverse and heterogeneous, and so CG systems with uniform effective interactions fail to reproduce the correct physical filament properties. This result is especially important because it shows that a biomolecular complex such as actin that has been overly coarse-grained can give results for a few properties that may appear quite good when in fact the information content (and presumably its predictive capability) of that CG model is relatively minimal.
Figure 5.
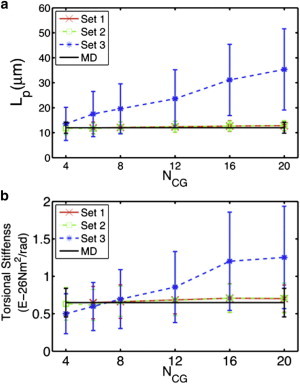
(Color online) Incorporating heterogeneity into CG models better reproduces (a) the persistence length and (b) torsional stiffness observed in MD simulations.
Figure 6.
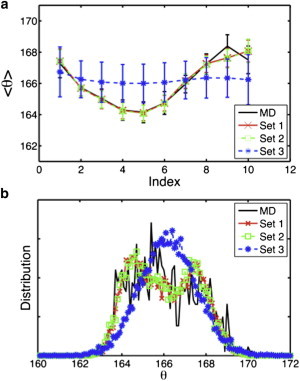
(Color online) Incorporating heterogeneity into CG models better represents (a) the twist per subunit and (b) its distribution observed in MD simulations.
To examine how well the CG model captures the inherent heterogeneity of the actin filament, the twist angle per subunit and twist angle distribution are plotted in Fig. 6, a and b, respectively, for ATP-bound CG filaments, and Fig. S4 for ADP-bound CG filaments. All data shown here are from CG simulations with NCG = 20; this example shows the most variance between the different CG models. Fig. 6 a and Fig. S4 a show that the twist angle of CG set 1 (red curve) and set 2 (green curve) remain variable, in agreement with MD results (black curve), and the distribution is broad (Fig. 6 b and Fig. S4 b), whereas the distribution from set 3 is not as variable, nor as broad, and it forms a smooth Gaussian-like distribution (blue curve). The fact that the random variable twist angle could only be reproduced with heterogeneous interactions (set 1 and set 2) also indicates that structural polymorphism and the associated differences in the interactions between subunits may be a key factor in generating a variable twist angle along the filament.
As long as heterogeneous interactions are permitted, additionally adding heterogeneity in the identity of CG sites appears to have only a minimal effect on the free energy of the model
Because sets 1 and 2 yielded similar results for all of the filament properties discussed previously, it is worthwhile to further compare these two CG systems to evaluate their relative merits. The free energy was calculated for both ATP- and ADP-bound CG filaments (data summarized in Table 2). We found that for the ATP-bound filament the free energy of set 1 is consistently slightly smaller than that of set 2 for NCG = 6 to 20, indicating that set 1 may better represent the ATP-bound all-atom system, but that the effect is not great. For the ADP-bound filament, the free energy of set 1 is very close to that of set 2 when taking the error limit into consideration for NCG = 6 to 12. For NCG = 16 and 20, the free energy of set 1 is slightly smaller than that of set 2. This indicates that sets 1 and 2 are equally good at representing the ADP-bound all-atom system for NCG <16, and for larger NCG values set 1 represents the system slightly better. This is interesting because, as shown in Fig. 3, when the number of CG sites is large the beads become more similar when comparing the same beads between subunits. What this implies is that even though the beads in set 1 are very similar at NCG = 16 and 20, the small differences between them make some impact on the free energy calculated by this method. Despite these differences in free energy, for the purpose of calculating the long-range properties of the system included in this work the simple CG representation in set 2 appears quite good. Furthermore, the free energy difference analysis indicates that in general the heterogeneity of CG interactions (used in set 1 and set 2) is more important than that of CG sites (incorporated only in set 1).
Table 2.
Normalized free energy difference between set 1 and set 2 (G1-G2)/NCG (kcal/mol)
| NCG | 6 | 8 | 12 | 16 | 20 |
|---|---|---|---|---|---|
| ATP-bound | −0.63 (0.35) | −0.76 (0.36) | −1.21 (0.32) | −0.55 (0.36) | −0.25 (0.34) |
| ADP-bound | −0.59 (0.93) | −0.62 (0.70) | 0.02 (0.46) | −0.48 (0.39) | −0.44 (0.36) |
Conclusions and Perspectives
Our work has explored the influence of heterogeneity on actin filament properties. We first carefully constructed atomistic filament systems, performed MD simulations, and obtained results consistent with experiments, including comparable mechanical properties, random variable twist angles, and the coexistence of subunits of different modes. Two mechanical properties, persistence length and torsional stiffness, were determined from MD simulations, which showed that ATP-bound filaments are more rigid than ADP-bound filaments based on either persistence length or torsional stiffness. These values are comparable to experimental measurements (6,7,41–45), and agree with previous MD simulations (16,26). We have quantified the subunit twist angle for both filament systems and observe that it varies significantly along the filament and is broadly distributed, consistent with experimental observations (9,10). These results support the hypothesis that the subunit twist angle exists in a rough energy landscape and can occupy multiple minima in actin filaments (12).
MD simulations enable us to visualize the molecular details of the system in addition to providing information about larger scale properties. In the ADP-bound filament, one of the D-loops in the ADP-bound filament spontaneously folded from an unfolded conformation and the conformations of the other D-loops differed between subunits. This direct observation of actin subunits with different structures coexisting in the same filament segment is in agreement with experimental results on structure polymorphism in actin (25). The folded-to-unfolded transition has been previously observed in G-actin (19), whereas the reverse transition has not. Getting a protein to fold enough times to be statistically significant within the timescales that can be sampled by MD without some type of biasing force is, at this point, an unrealistic goal even for small systems. This is why many studies of protein folding will start from a folded state and heat the system to promote unfolding. In the actin filament this problem is further complicated by the fact that we have only relatively low-resolution structural data on the contacts formed between the D-loop and the adjacent subunit. It is entirely possible that many of the unfolding events that have been observed in previous studies are because the filament environment around the D-loop is not optimal. For these reasons, the fact that we observe even one of the loops folding is remarkable. Our group has previously applied an enhanced sampling technique, metadynamics, to study the folding of the D-loop and shown that there is a local minimum for a folded D-loop in the filament environment (22). The observation of the folding of the D-loop in the current work strengthens these previous findings because it occurred in an unbiased simulation.
The spontaneous folding suggests that the free energies of subunits in different modes are comparable and that the barrier to transition between states is not very high. These different conformations may also provide distinct binding surfaces for different actin-binding proteins (1–5). The variations in contact interactions between pairs of subunits observed in MD simulations suggest that actin subunit polymorphism may be coupled to the variations in twist angle along the filament. Different subunit conformations result in distinct longitudinal contacts, which affect the twist angle per subunit, and conversely, changing the twist angle of subunits induces different intersubunit interactions, which then affect the conformations of the neighboring subunits. Future research into the correlation between the conformations of neighboring subunits may provide insight into the mechanism by which actin-binding proteins allosterically affect filament conformation and properties.
We further applied CG analysis methods to explore the heterogeneity observed in the atomistic MD simulations. CG analysis has previously been used to analyze different F-actin models and to identify the key interactions in large biomolecular complexes such as the ribosome (38). The motion of subunits in the filament can be represented based on CG sites, reducing the complexity of the data. In this work, we identified dynamic domains based on correlated motions at the atomic scale using the ED-CG method and found that these domains are different between subunits. This result may at first be unexpected, because each subunit is chemically identical. However, the different local conformations observed in our MD simulations, including different D-loop configurations, can reasonably be expected to form distinct intersubunit interactions, and therefore exhibit different local dynamics. This hypothesis has been further verified by showing that qualitatively different interdomain effective interactions are obtained using HeteroENM.
With the aid of CG simulations, the effects of the heterogeneity in the interdomain interactions have also been explored. The comparison of three sets of CG simulation results suggests that only CG systems with heterogeneous effective interactions can represent the actin filament correctly. Interestingly, we found that including heterogeneity becomes more important as the number of CG sites increases. When NCG is small, CG averages out the heterogeneity in the effective interactions kij, making these values roughly the same among the subunits. When NCG is larger, the various kij values capture the interactions at a finer level and reveal key difference in kij values. Finally, free energy calculations suggest that in general the CG system consisting of subunits with heterogeneous CG beads and heterogeneous effective interactions is slightly better at representing both the ATP- and ADP-bound CG filaments than a model that only includes heterogenous interactions, because each subunit is represented by a group of CG beads tailored specifically to its own dynamics. However, allowing heterogeneity in the CG interactions is significantly more important than allowing that of CG sites, because the model with uniform CG beads but heterogeneous effective interactions across the filament can reasonably represent the filament properties.
In summary, we have examined the structural polymorphism in actin filaments at the molecular level and quantified the heterogeneity in the dynamic domains and in the interactions between these domains using CG analysis. The importance of the variation in effective interactions between the CG domains was also explored. The heterogeneity is focused to be an essential feature of the actin filament and a necessary element to include in a CG model that reproduces the properties of the filament.
Acknowledgments
We specifically acknowledge the assistance of Greg Cross and Lorenzo Pesce in the Computation Institute. We also thank Shulu Feng, Zhen Cao, and Lanyuan Lu for helpful discussions throughout this project.
This material is based upon work supported by the National Science Foundation through the Center for Multiscale Theory and Simulation (grant CHE-1136709). Two computer resources provided computational time: The National Science Foundation XSEDE resources at the Pittsburgh Supercomputing Center and the National Institute for Computational Sciences, and the Computation Institute and the Biological Sciences Division of the University of Chicago and Argonne National Laboratory, under National Institutes of Health (NIH) grant S10 RR029030-01.
Supporting Material
References
- 1.Dominguez R., Holmes K.C. Actin structure and function. Annu. Rev. Biophys. 2011;40:169–186. doi: 10.1146/annurev-biophys-042910-155359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hild G., Bugyi B., Nyitrai M. Conformational dynamics of actin: effectors and implications for biological function. Cytoskeleton (Hoboken) 2010;67:609–629. doi: 10.1002/cm.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oda T., Maéda Y. Multiple conformations of F-actin. Structure. 2010;18:761–767. doi: 10.1016/j.str.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Drewes G., Faulstich H. Cooperative effects on filament stability in actin modified at the C-terminus by substitution or truncation. Eur. J. Biochem. 1993;212:247–253. doi: 10.1111/j.1432-1033.1993.tb17656.x. [DOI] [PubMed] [Google Scholar]
- 5.Prochniewicz E., Katayama E., Thomas D.D. Cooperativity in F-actin: chemical modifications of actin monomers affect the functional interactions of myosin with unmodified monomers in the same actin filament. Biophys. J. 1993;65:113–123. doi: 10.1016/S0006-3495(93)81057-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Isambert H., Venier P., Carlier M.F. Flexibility of actin filaments derived from thermal fluctuations. Effect of bound nucleotide, phalloidin, and muscle regulatory proteins. J. Biol. Chem. 1995;270:11437–11444. doi: 10.1074/jbc.270.19.11437. [DOI] [PubMed] [Google Scholar]
- 7.McCullough B.R., Blanchoin L., De la Cruz E.M. Cofilin increases the bending flexibility of actin filaments: implications for severing and cell mechanics. J. Mol. Biol. 2008;381:550–558. doi: 10.1016/j.jmb.2008.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rebello C.A., Ludescher R.D. Influence of tightly bound Mg(2+) and Ca(2+), nucleotides, and phalloidin on the microsecond torsional flexibility of F-actin. Biochemistry. 1998;37:14529–14538. doi: 10.1021/bi981240i. [DOI] [PubMed] [Google Scholar]
- 9.Hanson J. Axial period of actin filaments. Nature. 1967;213:353–356. [Google Scholar]
- 10.Egelman E.H., Francis N., DeRosier D.J. F-actin is a helix with a random variable twist. Nature. 1982;298:131–135. doi: 10.1038/298131a0. [DOI] [PubMed] [Google Scholar]
- 11.Kabsch W., Mannherz H.G., Holmes K.C. Atomic structure of the actin:DNase I complex. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- 12.Orlova A., Egelman E.H. F-actin retains a memory of angular order. Biophys. J. 2000;78:2180–2185. doi: 10.1016/S0006-3495(00)76765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otterbein L.R., Graceffa P., Dominguez R. The crystal structure of uncomplexed actin in the ADP state. Science. 2001;293:708–711. doi: 10.1126/science.1059700. [DOI] [PubMed] [Google Scholar]
- 14.Galkin V.E., VanLoock M.S., Egelman E.H. A new internal mode in F-actin helps explain the remarkable evolutionary conservation of actin’s sequence and structure. Curr. Biol. 2002;12:570–575. doi: 10.1016/s0960-9822(02)00742-x. [DOI] [PubMed] [Google Scholar]
- 15.Graceffa P., Dominguez R. Crystal structure of monomeric actin in the ATP state. Structural basis of nucleotide-dependent actin dynamics. J. Biol. Chem. 2003;278:34172–34180. doi: 10.1074/jbc.M303689200. [DOI] [PubMed] [Google Scholar]
- 16.Chu J.W., Voth G.A. Allostery of actin filaments: molecular dynamics simulations and coarse-grained analysis. Proc. Natl. Acad. Sci. USA. 2005;102:13111–13116. doi: 10.1073/pnas.0503732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rould M.A., Wan Q., Trybus K.M. Crystal structures of expressed non-polymerizable monomeric actin in the ADP and ATP states. J. Biol. Chem. 2006;281:31909–31919. doi: 10.1074/jbc.M601973200. [DOI] [PubMed] [Google Scholar]
- 18.Chu J.W., Voth G.A. Coarse-grained modeling of the actin filament derived from atomistic-scale simulations. Biophys. J. 2006;90:1572–1582. doi: 10.1529/biophysj.105.073924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng X.G., Diraviyam K., Sept D. Nucleotide effects on the structure and dynamics of actin. Biophys. J. 2007;93:1277–1283. doi: 10.1529/biophysj.107.109215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sawaya M.R., Kudryashov D.S., Yeates T.O. Multiple crystal structures of actin dimers and their implications for interactions in the actin filament. Acta Crystallogr. D Biol. Crystallogr. 2008;64:454–465. doi: 10.1107/S0907444908003351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Splettstoesser T., Noé F., Smith J.C. Nucleotide-dependence of G-actin conformation from multiple molecular dynamics simulations and observation of a putatively polymerization-competent superclosed state. Proteins. 2009;76:353–364. doi: 10.1002/prot.22350. [DOI] [PubMed] [Google Scholar]
- 22.Pfaendtner J., Branduardi D., Voth G.A. Nucleotide-dependent conformational states of actin. Proc. Natl. Acad. Sci. USA. 2009;106:12723–12728. doi: 10.1073/pnas.0902092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda T., Iwasa M., Narita A. The nature of the globular- to fibrous-actin transition. Nature. 2009;457:441–445. doi: 10.1038/nature07685. [DOI] [PubMed] [Google Scholar]
- 24.Wang H., Robinson R.C., Burtnick L.D. The structure of native G-actin. Cytoskeleton (Hoboken) 2010;67:456–465. doi: 10.1002/cm.20458. [DOI] [PubMed] [Google Scholar]
- 25.Galkin V.E., Orlova A., Egelman E.H. Structural polymorphism in F-actin. Nat. Struct. Mol. Biol. 2010;17:1318–1323. doi: 10.1038/nsmb.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfaendtner J., Lyman E., Voth G.A. Structure and dynamics of the actin filament. J. Mol. Biol. 2010;396:252–263. doi: 10.1016/j.jmb.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujii T., Iwane A.H., Namba K. Direct visualization of secondary structures of F-actin by electron cryomicroscopy. Nature. 2010;467:724–728. doi: 10.1038/nature09372. [DOI] [PubMed] [Google Scholar]
- 28.Saunders M.G., Voth G.A. Water molecules in the nucleotide binding cleft of actin: effects on subunit conformation and implications for ATP hydrolysis. J. Mol. Biol. 2011;413:279–291. doi: 10.1016/j.jmb.2011.07.068. [DOI] [PubMed] [Google Scholar]
- 29.Saunders M.G., Voth G.A. Comparison between actin filament models: coarse-graining reveals essential differences. Structure. 2012;20:641–653. doi: 10.1016/j.str.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z.Y., Lu L.Y., Voth G.A. A systematic methodology for defining coarse-grained sites in large biomolecules. Biophys. J. 2008;95:5073–5083. doi: 10.1529/biophysj.108.139626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyman E., Pfaendtner J., Voth G.A. Systematic multiscale parameterization of heterogeneous elastic network models of proteins. Biophys. J. 2008;95:4183–4192. doi: 10.1529/biophysj.108.139733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 33.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 34.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 36.Darden T., York D., Pedersen L. Particle mesh Ewald—an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 37.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical-integration of Cartesian equations of motion of a system with constraints: molecular-dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 38.Zhang Z.Y., Sanbonmatsu K.Y., Voth G.A. Key intermolecular interactions in the E. coli 70S ribosome revealed by coarse-grained analysis. J. Am. Chem. Soc. 2011;133:16828–16838. doi: 10.1021/ja2028487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 40.McQuarrie D.A. Harper & Row; New York: 1973. Statistical Thermodynamics. [Google Scholar]
- 41.Yasuda R., Miyata H., Kinosita K., Jr. Direct measurement of the torsional rigidity of single actin filaments. J. Mol. Biol. 1996;263:227–236. doi: 10.1006/jmbi.1996.0571. [DOI] [PubMed] [Google Scholar]
- 42.Tsuda Y., Yasutake H., Yanagida T. Torsional rigidity of single actin filaments and actin-actin bond breaking force under torsion measured directly by in vitro micromanipulation. Proc. Natl. Acad. Sci. USA. 1996;93:12937–12942. doi: 10.1073/pnas.93.23.12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshimura H., Nishio T., Ikegami A. Torsional motion of eosin-labeled F-actin as detected in the time-resolved anisotropy decay of the probe in the sub-millisecond time range. J. Mol. Biol. 1984;179:453–467. doi: 10.1016/0022-2836(84)90075-5. [DOI] [PubMed] [Google Scholar]
- 44.Prochniewicz E., Zhang Q.N., Thomas D.D. Microsecond rotational dynamics of actin: spectroscopic detection and theoretical simulation. J. Mol. Biol. 1996;255:446–457. doi: 10.1006/jmbi.1996.0037. [DOI] [PubMed] [Google Scholar]
- 45.Prochniewicz E., Janson N., De la Cruz E.M. Cofilin increases the torsional flexibility and dynamics of actin filaments. J. Mol. Biol. 2005;353:990–1000. doi: 10.1016/j.jmb.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 46.Amadei A., Linssen A.B.M., Berendsen H.J.C. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 47.Kozuka J., Yokota H., Yanagida T. Dynamic polymorphism of single actin molecules in the actin filament. Nat. Chem. Biol. 2006;2:83–86. doi: 10.1038/nchembio763. [DOI] [PubMed] [Google Scholar]
- 48.Deriu M.A., Bidone T.C., Morbiducci U. Biomechanics of actin filaments: a computational multi-level study. J. Biomech. 2011;44:630–636. doi: 10.1016/j.jbiomech.2010.11.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.