

A majority of Trypanosoma brucei proteins have unknown functions, a consequence of its independent evolutionary history within the order Kinetoplastida that allowed for the emergence of several unique biological properties. Among these is RNA editing, needed for expression of mitochondrial-encoded genes. The recently discovered mitochondrial RNA binding complex 1 (MRB1) is composed of proteins with several functions in processing organellar RNA. The authors characterize two MRB1 subunits, referred to herein as MRB8170 and MRB4160, and show that they are both novel RNA binding proteins, possibly representing a new class of these proteins.
Keywords: RNA editing, RNA binding protein, ribonuclear protein (RNP), mitochondria, trypanosome
Abstract
A majority of Trypanosoma brucei proteins have unknown functions, a consequence of its independent evolutionary history within the order Kinetoplastida that allowed for the emergence of several unique biological properties. Among these is RNA editing, needed for expression of mitochondrial-encoded genes. The recently discovered mitochondrial RNA binding complex 1 (MRB1) is composed of proteins with several functions in processing organellar RNA. We characterize two MRB1 subunits, referred to herein as MRB8170 and MRB4160, which are paralogs arisen from a large chromosome duplication occurring only in T. brucei. As with many other MRB1 proteins, both have no recognizable domains, motifs, or orthologs outside the order. We show that they are both novel RNA binding proteins, possibly representing a new class of these proteins. They associate with a similar subset of MRB1 subunits but not directly with each other. We generated cell lines that either individually or simultaneously target the mRNAs encoding both proteins using RNAi. Their dual silencing results in a differential effect on moderately and pan-edited RNAs, suggesting a possible functional separation of the two proteins. Cell growth persists upon RNAi silencing of each protein individually in contrast to the dual knockdown. Yet, their apparent redundancy in terms of cell viability is at odds with the finding that only one of these knockdowns results in the general degradation of pan-edited RNAs. While MRB8170 and MRB4160 share a considerable degree of conservation, our results suggest that their recent sequence divergence has led to them influencing mitochondrial mRNAs to differing degrees.
INTRODUCTION
Trypanosoma brucei subspecies are the etiological agents of several neglected human and veterinary diseases that mainly impact sub-Saharan Africa, as epitomized by sleeping sickness and nagana. Our understanding of the underlying biology of this flagellate has certainly been advanced by the release of its genome, along with those of its parasitic cohorts Trypanosoma cruzi and Leishmania major. However, there is much fertile ground for future research because more than half of the predicted T. brucei proteins are of unknown function (El-Sayed et al. 2005).
The unfamiliar nature of the T. brucei proteome is not surprising given that it belongs to the order Kinetoplastida, a clade that has several unusual biological features due to its long and independent evolutionary history (Philippe et al. 2000). Most notable of these is the kinetoplast (k) DNA that ultimately lends its name to the whole order (Shlomai 2004; Lukeš et al. 2005). This DNA, usually arranged in the form of a network, represents the mitochondrial (mt) genome, which is made up of two types of circular molecules. In T. brucei, thousands of ∼1 kb-long minicircles, heterogeneous in sequence, constitute the bulk of kDNA, while the dozens of ∼23 kb-long maxicircles resemble the mt genomes of other aerobic eukaryotes because they contain rRNA and protein-coding genes, which predominantly encode subunits of the respiratory complexes (Shlomai 2004; Lukeš et al. 2005).
The complicated affair that is maxicircle gene expression is also a distinguishing feature of kinetoplastid flagellates. Twelve out of the 18 protein-coding mt genes in T. brucei are encrypted, and their mRNAs require for their maturation a post-transcriptional process called kinetoplastid (k) RNA editing (Simpson et al. 2004; Lukeš et al. 2005; Stuart et al. 2005). Uridine (U) residues are inserted into or deleted from preordained sites of the edited mRNA to render a meaningful open reading frame (ORF). The extent of this process varies from the so-called pan-edited mRNAs that require the insertions of hundreds and deletions of dozens of U's throughout the molecule to minimally edited mRNAs that bear short regions where U insertions occur. Numerous 50- to 70-bp-long noncoding guide (g) RNAs encoded predominantly in the minicircles act true to their name by directing the kRNA editing machinery to the appropriate sites of U-insertion/deletion on the mRNA. For pan-edited mRNAs, a series of gRNA:mRNA annealing events decodes the ORFs from a 3′-to-5′ direction (Maslov and Simpson 1992).
Many proteins, usually as subunits of macromolecular complexes, make up the kRNA editing machinery. Among the best characterized of these components is the RNA editing core complex (RECC), also known as the 20S editosome (Simpson et al. 2010). This complex encompasses the cascade of enzymatic activities for U-insertion/deletion events: An endonuclease cleaves the mRNA at the editing site dictated by the gRNA, which also provides information to RECC for how many U's are inserted or deleted here by a terminal uridylyltransferase (TUTase) or exonuclease, respectively; in the last step, the processed site is sealed within the mRNA by an RNA ligase. These activities are being explored as potential drug targets for treating these pathogens in their mammalian hosts (Durrant et al. 2010; Moshiri et al. 2011).
RECC actually occurs in three forms that, while containing overlapping subunits, are distinguished by others that confer specificity to a particular type of editing event (Panigrahi et al. 2006; Carnes et al. 2008, 2011). Notably, each RECC bears one of three types of endonucleases that are able to exclusively cleave U-insertion or U-deletion editing sites, plus one that is created by a unique cis-acting gRNA element within the 3′-UTR of an edited mRNA (Golden and Hajduk 2005). As implied by the name RECC, other proteins play a role in kRNA editing as well as other aspects of kinetoplastid mtRNA metabolism. The heterotetrameric complex that is formed by the two mtRNA binding proteins MRP1/2 is believed to facilitate gRNA:mRNA duplex formation (Schumacher et al. 2006; Zíková et al. 2008), as is the RBP16 protein (Ammerman et al. 2008). RNAi-mediated depletion of either of these entities affects only a subset of edited mRNAs (Pelletier and Read 2003; Vondrušková et al. 2005), while their simultaneous down-regulation leads to a more pronounced reduction of the overall panel of these transcripts (Fisk et al. 2009). RNA editing helicase (REH) 1 comes into play after editing of a given site is completed by unwinding the utilized gRNA from its cognate mRNA, a particularly important task in maturation of pan-edited mRNAs that require many of these small molecules (Li et al. 2011). TbRGG1 is a poly(U) binding protein that appears to have a role in the stability of mRNAs that have completed editing (Vanhamme et al. 1998; Hashimi et al. 2008). An enzyme called RNA editing TUTase 1 (RET1) is responsible for the post-transcriptional addition of a short 3′-poly(U) tail onto the trans-acting gRNAs, an essential feature of these molecules (Aphasizhev et al. 2003).
The gRNAs are not the only species in the mt transcriptome that is modulated by RET1, because polyuridylylation is a widespread processing event in the organelle (Ryan and Read 2005; Aphasizheva and Aphasizhev 2010). The mRNAs occur in two populations distinguished by 3′-extensions that are composed of short poly(A) (∼20 nt) or long A/U tails (∼200 nt) (Bhat et al. 1992; Militello and Read 1999; Etheridge et al. 2008). A mt poly(A) polymerase (KPAP1) is responsible for the former structure while KPAP1 and RET1 act in concert with two pentatricopeptide repeat (PPR) proteins to produce the latter, a feature that marks mature mRNAs for translation (Ryan and Read 2005; Etheridge et al. 2008; Aphasizheva et al. 2011). In contrast to such a plethora of polyuridylylation events, a novel RNase D exoribonuclease targets the gRNA poly(U) tail, perhaps serving to fine-tune this heterogeneous population of molecules (Zimmer et al. 2011). A close homolog of the RECC endonucleases has been recently shown to process polycistronic minicircle transcripts to yield functional gRNAs of the appropriate size and has thus been dubbed mtRNA precursor-processing endonuclease 1 (mRPN1) (Madina et al. 2011).
Contributing to kRNA metabolism is the recently discovered mtRNA binding complex 1 (MRB1), whose subunits appear to have diverse functions (Hashimi et al. 2008; Panigrahi et al. 2008; Weng et al. 2008). MRB1 was so named because among its constituent proteins are those having motifs that clearly indicate a role in mtRNA metabolism. However, most MRB1 subunits lack these features or orthologs outside of the kinetoplastid flagellates, obscuring their specific roles in kRNA processing/maintenance. A further impediment to understanding MRB1 is that reported isolations from different groups contain both overlapping as well as differing proteins (summarized in Ammerman et al. 2011). However, recent work based on a pairwise yeast two-hybrid screen of 31 putative MRB1 subunits has defined the architecture of MRB1 (Ammerman et al. 2012). It is composed of a core, which persisted in all reported isolations, of the following proteins: the two gRNA-associated proteins GAP1 and 2 (also known as GRBC), MRB3010, MRB5390, MRB8620, and MRB11870 (Ammerman et al. 2012). Furthermore, there are peripheral subcomplexes that interact with this core in an RNA-enhanced or -dependent manner, possibly reflecting a dynamic assembly onto kRNAs undergoing processing.
Adding to the difficulty in pinpointing the role of MRB1 is the variety of apparent functions of its studied subunits. The two paralogous GAPs partner with each other in a way reminiscent of the aforementioned MRP1/2 complex and promote the stability of these short RNAs (Weng et al. 2008; Hashimi et al. 2009). The REH2 RNA helicase, which has been identified in some MRB1 isolates, also appears to have a still-undefined role during the lifetime of the gRNAs that also affects their stability (Hashimi et al. 2009; Hernandez et al. 2010). Ablation of the core MRB3010 by RNAi indicates that it facilitates an early step in RNA editing while not directly affecting gRNAs (Ammerman et al. 2011). TbRGG2, a peripheral MRB1 protein outside of the core, participates in the progression of multi-round editing required by pan-edited transcripts in an evocative fashion of the previously mentioned REH1 (Fisk et al. 2008; Acestor et al. 2009; Ammerman et al. 2010). A Nudix hydrolase (also called MERS1) has a role in the stability of potentially all mRNAs (Weng et al. 2008; Hashimi et al. 2009).
In this study, we investigate the functions of two MRB proteins called MRB8170 and MRB4160 that are paralogs sharing striking sequence similarity on the DNA level, a result of a duplication event that occurred during or after the speciation of T. brucei (Jackson 2007). MRB8170 and MRB4160 have no known motifs or orthologs outside the sequenced kinetoplastid flagellates. They have been shown to interact with the pre-gRNA processing endonuclease mRPN1 in an RNA-independent manner, although their direct contact with this enzyme was not verified by yeast two-hybrid analysis (Madina et al. 2011). However, it is known that their association with the MRB1 core is mediated by their interaction with TbRGG2 (Ammerman et al. 2012). We found that each protein associates with a highly similar subset of core and peripheral MRB1 subunits, while showing no direct interaction with each other, thus clarifying their status within the MRB1 complex. Very significantly, MRB8170 and MRB4160 bind RNA and thus represent a novel, kinetoplastid-restricted class of nucleic acid binding proteins. Finally, to elucidate their function within MRB1, we used RNAi-silencing techniques. Single knockdown of MRB8170 or MRB4160 did not affect procyclic cell growth, although mild effects on abundance of some edited and pre-edited transcripts were observed with MRB8170 depletion only. In contrast, tandem MRB8170/4160 RNAi silencing inhibits cell growth, coinciding with a substantial depletion of pan-edited RNAs and a significant, although qualitatively and quantitatively different effect on moderately edited transcripts. Thus, we conclude that while these proteins do have a fair degree of functional redundancy in their critical role in the MRB1 complex, although some suggestions of functional divergence are evident.
RESULTS
The two paralogous MRB8170 and MRB4160 genes share a highly conserved sequence
The MRB8170 and MRB4160 genes are located within regions of chromosomes 8 and 4, respectively, that are products of a large 0.5-Mb duplication event that occurred solely in the T. brucei lineage (Jackson 2007) and is absent from the other Trypanosomatids such as Leishmania species and T. cruzi. These genes are among the 47% of such duplicated coding sequences (CDS) retained in both locations. In the immediate genomic neighborhood of the MRB8170/MRB4160 genes, they can be thought of as being the first in an array of five genes that are found on both chromosomes (Fig. 1A). Using chromosome 4 as a reference, this array is bounded downstream from MRB4160 by Tb927.4.4170, encoding a putative rRNA methyltransferase that has been lost in chromosome 8. This pair of genes is present in a syntenic block on chromosome 8 of Trypanosoma congolense, a species closely related to T. brucei that is the major causative agent of nagana, and thus can be considered the ancestral state of this locus. Interestingly, it is the MRB8170 CDS that shares slightly higher sequence identity to the T. congolense ortholog than MRB4160 (Fig. 1A).
FIGURE 1.

The paralogous MRB8170 and MRB4160 genes. (A) Scheme showing that the kMAP genes are part of an array of five paralogs retained on both chromosomes 4 and 8 arising from a duplication event (bottom two black lines). In addition, a syntenic region of T. congolense chromosome 8 (top gray line) is aligned to the gene array present on T. brucei chromosome 4. CDSs are depicted as boxes with either its name or locus tag given inside or adjacently just above or below the box in red letters. MRB8170 and MRB4160 loci are in bold. Paralogs retained on both T. brucei chromosome 4 and 8 are yellow, only on the former in red and the latter in green. The top number in the shaded region between the duplicated CDSs is the percentage protein sequence identity between them according to a ClustalW alignment performed in the BioEdit program. The bottom number in parentheses is the locus number as given by Jackson (2007), from which this figure is adapted. The numbers in the shaded region between T. congolense chromosome 8 and T. brucei chromosome 4 represent the protein sequence identity of the CDSs from the former with T. brucei chromosomes 8 (top) and 4 (bottom) as calculated previously. The chromosome location at the beginning and end of the array of genes in question is given in gray adjacent to the relevant bounding CDS. (B) Dot-plot comparing the CDSs plus 3′-UTRs of MRB8170 (x-axis) and MRB4160 (y-axis) to reveal regions of nucleotide sequence identity. Nucleotide matches are represented as a black dot. The diagonal represents the identical regions of the two sequences. The small gray square in the lower left-hand corner encloses the diverged region of two sequences. Schemes depicting the relevant regions of MRB8170 and MRB4160 are provided along the axes. The diverged and conserved regions are indicated, while the 3′-UTR is shown as a black line. The regions targeted for the single (sKD) and double (dKD) knockdowns and regions amplified by quantitative real-time PCR primers (Q) are shown.
The translated MRB8170 and MRB4160 genes have a predicted molecular weight of 100 kDa and share 77.3% amino acid sequence identity, taking into account that the ORF of the latter gene actually starts at the first ATG internal to the annotated start codon (Supplemental Fig. 1; Nilsson et al. 2010; Siegel et al. 2010). However, most of the sequence divergence is restricted to a 5′ region comprising 15% of the ORFs (Fig. 1B). The remaining 85% of the two CDSs exhibit high conservation with 90.2% sequence DNA identity, which is similar to the 90.1% identity between the two predicted 3′-untranslated regions (UTRs) (Jackson 2007). The similarity of the two 3′-UTRs implies that MRB8170 and MRB4160 mRNAs have comparable degrees of post-transcriptional regulation (Jackson 2007).
It should be noted that MRB8170 and MRB4160 are not the only two MRB1 proteins whose genes are located in these paralogous loci (Fig. 1A). A subunit designated MRB8180 (Tb927.8.8180) is encoded in the 3′-CDS adjacent to MRB8170, while MRB4150 (Tb927.4.4150) is located directly downstream from MRB4160. Interestingly, MRB4150 and MRB8170 are virtually indistinguishable from each other as they share 99.1% sequence identity, in contrast to the divergence between MRB8170 and MRB4160 (Fig. 1B).
Both MRB8170 and MRB4160 have in vitro RNA binding activity
The amino acid compositions of MRB8170 and MRB4160 confer basic theoretical isoelectric points (pI) 9.48 and 9.64, respectively. This property is suggestive of RNA binding ability, since it signifies a net positive charge of the polypeptide at physiological pH, which would promote interaction with negatively charged nucleic acids. To test whether MRB8170 and MRB4160 are indeed RNA binding proteins, recombinant MRB8170 and MRB4160 were isolated from E. coli via an incorporated GST tag. Surprisingly, MRB4160 exhibited a considerably lower level of overexpression compared with MRB8170 under the same conditions despite sharing 77.3% amino acid identity. These proteins were subsequently incubated with radiolabeled RNA corresponding to 102 nt of the 5′ end of the pre-edited cyB mRNA followed by UV cross-linking to capture any direct interactions. After these reactions were treated with RNase to degrade the unbound polyribonucleotide, they were resolved on a SDS-polyacrylamide gel and assayed for overlapping radioactive RNA and protein signals.
Figure 2A (lane 1) shows that the radiolabeled RNA does not bind bovine serum albumin (BSA), visualized by the Coomassie stain as an ∼67-kDa band (Fig. 2B), which is a constituent of the RNA binding buffer. The motilities of the recombinant MRB8170, p22, and TbRGG2 proteins in the absence of bound RNA are shown in Figure 2B (lanes 2–5). The MRB8170 protein with the 30-kDa GST tag has a predicted molecular weight of ∼125 kDa. The p22 protein with the ∼64-kDa MBP-tag migrates as a band just below the BSA band, which served as a negative control because it does not bind RNA (Fig. 2, lanes 4,6; Sprehe et al. 2010). TbRGG2-GST acted as a positive control for RNA binding (Fisk et al. 2008). This recombinant protein exhibited the same mobility as BSA, which is apparent from the increased intensity of the 75-kDa band (Fig. 2, lanes 4,7). Both MRB8170 and MRB4160 bind RNA, because they meet the criteria of yielding strong radioactive signals (Fig. 2A, lanes 5,8) that correspond with the recombinant proteins in Figure 2B. While we attempted to use an equal amount of recombinant protein in all the reactions, it is clear from the ∼130-kDa band intensity that less of the poorly expressing recombinant MRB4160 was present. The copious amount of the free GST-tag in this isolation compared with the others (Fig. 2B, lane 8) indicates that this contaminant represented a significant portion of the utilized protein.
FIGURE 2.

The MRB8170 and MRB4160 proteins bind RNA. Proteins incubated with the first 102 nt of pre-edited cyB sequence body-labeled with [α-32P]UTP were UV-cross-linked, treated with RNase A, and separated on a 10% SDS-acrylamide gel. (A) Autoradiograph of recombinant proteins binding radiolabeled single-stranded RNA. (B) Coomassie-stained 10% SDS-acrylamide gel. The band indicated as “b” in lane 1 is BSA, a component of the binding buffer. (*) Bands corresponding to recombinant proteins indicated at the top of the lanes. (#) The 25-kDa band in lane 8 that stains the free GST-tag. Protein ladder sizes are indicated on the right of both panels.
MRB8170 has an apparent higher affinity for mRNAs than guide RNAs
Since both MRB8170 and MRB4160 exhibit in vitro RNA binding activities, we decided to address whether there is a preferential binding to mRNA or gRNA. To this end, we used the double filter binding assay (Wong and Lohman 1993; Hall and Kranz 1999) to quantify recombinant MRB8170 binding to radiolabeled pre-edited and edited RPS12 mRNAs (Kao and Read 2005) as well as a oligo(U) tail containing gRNA (gA6[14]) (Koslowsky et al. 1992). A constant concentration of one of these substrate RNAs was incubated with a range of concentrations of MRB8170 and at least a 2000-fold molar excess of torula yeast RNA. The RNA:protein complex was filtered via a nitrocellulose membrane from the unbound RNA, which was subsequently captured by a nylon membrane. The apparent dissociation constant (Kd) of MRB8170 RNA binding was determined by measuring the percent RNA bound for each protein concentration. The aforementioned poor expression of MRB4160 precluded its use in this assay.
As shown in Figure 3, A and B, the apparent Kd ± SE of MRB8170 binding for pre-edited and edited RPS12 mRNA is 15 ± 1 nM and 21 ± 2 nM, respectively. These values are about five times more than the Kd of MRB8170 association with the U-tail bearing gRNA, which is 117 ± 22 nM (Fig. 3C). These results suggest that MRB8170 has an higher affinity for mRNAs than gRNAs.
FIGURE 3.

MRB8170 appears to preferentially bind mRNA in vitro. Binding curves of MRB8170 binding to (A) pre-edited RPS12 mRNA, (B) edited RPS12 mRNA, and (C) gA6[14] gRNA, which possesses a U17 3′-oligomer tail. The range of protein concentrations used is given on the x-axis in nanomolars and the fraction of bound RNA on the y-axis. The apparent Kd determined from the fitted curves is given along with the Bmax of MRB8170 RNA binding and coefficient of determination (R2) of each curve. Whiskers at each data point represent standard deviation over two separate experiments done in triplicate.
MRB8170 is incorporated into a macromolecular complex in RNA-dependent manner
Several MRB1 subunits have been shown to be incorporated into macromolecular complexes (Panigrahi et al. 2008; Acestor et al. 2009; Hashimi et al. 2009; Hernandez et al. 2010; Ammerman et al. 2011). Because MRB8170 and MRB4160 have been found in several MRB1 isolations (Ammerman et al. 2011), we decided to test whether MRB8170, the only paralog against which specific antibodies are available, also associates with a macromolecular complex(es). To this end, mt proteins were separated on a 10%–30% glycerol gradient in the presence or absence of an RNase cocktail. The odd fractions from the gradient were loaded onto a SDS-PAGE gel, and the resolved proteins were detected by immunodecoration with α-MRB8170 polyclonal and α-KREPA1 and α-KREL1 monoclonal antibodies. The latter two antigens served as markers for the sedimentation of the RECC (Fig. 4). A strong α-MRB8170 signal sediments in the denser fractions (15–23), with a peak around 20S, while a weaker signal appears in fractions 7–13 (Fig. 4). This pattern indicates that MRB8170 is primarily incorporated into a large complex(es). This association is abolished upon RNase treatment, as sedimentation shifts to the lighter fractions, with a somewhat equal distribution of signal in fractions 1–15 and a weaker band trailing from here to the bottom of the gradient. Thus, this association of MRB8170 appears to have an RNA component, which is consistent with both its RNA binding activity and the RNase sensitivity of the incorporation of other subunits into MRB1.
FIGURE 4.

MRB8170 is incorporated into a macromolecular complex(es) in an RNase-sensitive manner. Mitochondria isolated from 1010 cells of the T. brucei 29-13 strain were lysed and separated on a 10%–30% glycerol gradient by ultracentrifugation. The gradient was divided into 24 fractions, out of which all odd ones were run on a 10% SDS-acrylamide gel and transferred onto a PVDF membrane that was immunoprobed with the α-MRB8170 antibody. The lysate used in the top panels was treated with RNase inhibitor (RNase-), while that used in the bottom panel was treated with an RNase cocktail that disrupted interactions mediated by both single- and double-stranded RNAs (RNase+).
MRB8170 and MRB4160 interact with a highly similar subset of MRB1 subunits
Next, we set about determining whether the MRB8170-containing complex(es) contains the characteristic MRB1 subunits described from pull-down isolations in other studies (Hashimi et al. 2008; Panigrahi et al. 2008; Weng et al. 2008; Hernandez et al. 2010; Ammerman et al. 2011), as well as ascertaining whether the composition of the likely MRB4160 RNP(s) is equivalent to that of its paralog. T. brucei cell lines inducibly overexpressing either the MRB8170 or MRB4160 ORF bearing a C-terminal TAP tag were generated. This method allowed the affinity purification of MRB8170 or MRB4160 and identification of copurified proteins in the TAP eluates by mass spectroscopy or Western blot analysis with specific antibodies immunodecorating MRB1 subunits.
As depicted in Table 1 and Figure 5A, both proteins pulled down a highly similar subset of MRB1 subunits in the absence of any nuclease treatment. The core proteins comprising MRB11870, MRB3010, MRB5390, GAP1, and 2 (Ammerman et al. 2012) were present in both isolations, while MRB8620 was identified only in the MRB4160 isolation. When the number of unique peptides and their coverage of the identified interacting partners are taken into account for both MRB8170 and MRB4160 TAP experiments, the similarity between the two RNPs is even more striking. The MRB4150/8180 protein is highly represented in both isolations using this criteria, as well as MERS1, which has a role in mRNA stability and has been proposed to be in a discrete complex apart from MRB1 (Table 1; Weng et al. 2008). TbRGG2, the MRB1 protein involved in the 3′-to-5′ progression of pan-edited RNAs, was also observed in both purifications to the same extent as was Tb927.10.11870. Overall, more proteins were distinguished in the MRB4160 TAP isolation as compared with MRB8170, including the RNA binding MRP2 protein (Fig. 5A; Table 1). However, it should be noted that neither the polycistronic gRNA endonuclease mRPN1, which was demonstrated to associate with either MRB8170 or MRB4160 (Madina et al. 2011), nor the REH2 RNA helicase was observed in any of the TAP experiments.
TABLE 1.
TAP-MRB8170- and TAP-MRB4160-associated proteins identified by mass spectrometry ordered according to the number of unique peptides from MRB4160 TAP tag elutions
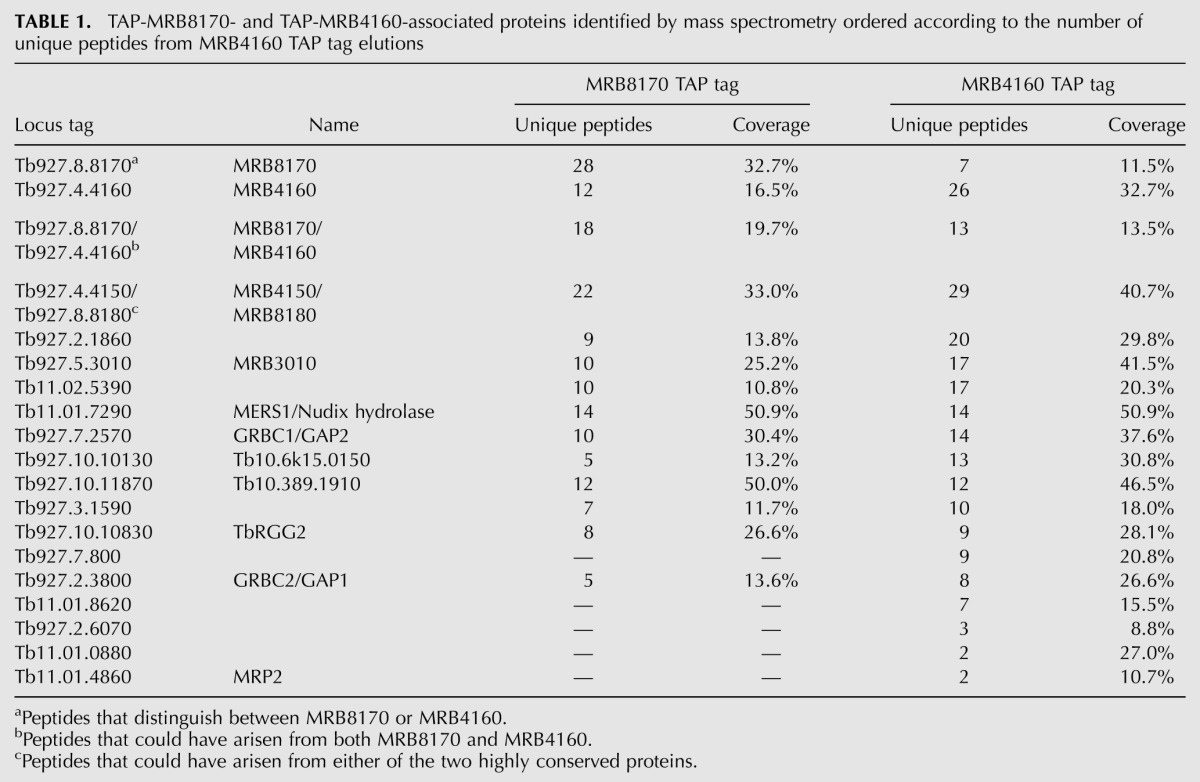
FIGURE 5.

MRB1 proteins interacting with MRB8170 and MRB4160. (A) The proteins identified by mass spectroscopy of the TAP purification of MRB8170 and MRB4160 described in Table 1 are depicted in the scheme introduced by Ammerman and colleagues (Ammerman et al. 2011). The names or gene tags of all proteins described from various MRB1 purifications (Ammerman et al. 2011) are enclosed in the large dashed oval, while the core MRB1 proteins are depicted in the small dotted oval. The proteins associated with either of the TAP-tagged MRB8170 and MRB4160 in the absence of RNase is enclosed by a thick dark line. Proteins that were demonstrated to have RNA-dependent or -enhanced interactions are with double and single underlines, respectively. (B) Immunoblot of TEV eluates from the first column of the TAP purification of MRB8170 and MRB4160, performed in the absence (−) and presence (+) of an RNase cocktail degrading both single- and double-stranded RNAs, The membrane was probed with antibodies recognizing MRB1 proteins GAP1, MRB3010, TbRGG2, and MRB8170. The band recognizing the endogenous and not the larger TAP-tagged MRB8170 protein is shown for this purification. An antibody recognizing the myc-epitope present in the TAP-tag was used as a loading control between the RNase-untreated and -treated purifications.
Since many of these interactions could be mediated by RNA linkers, we decided to compare the presence of some MRB1 subunits against which we have antibody, namely, GAP1, MRB3010, and TbRGG2, in both MRB8170 and MRB4160 TAP isolations performed with or without RNases (Fig. 5B) as previously described (Ammerman et al. 2012). The efficiency of RNase degradation was confirmed by running on an ethidium-bromide-stained gel RNA extracted from an equal volume sample of treated and untreated lysates before application onto the first affinity column (data not shown). While GAP1 interaction with MRB8170 or MRB4160 was abolished due to RNase treatment, interaction with MRB3010 and TbRGG2 was significantly reduced, but not eliminated, in both cases. These results suggest that while MRB8170 and MRB4160 contact MRB3010 and TbRGG2 directly, this interaction is enhanced by the presence of RNA. Furthermore, this observation is consistent with reciprocal experiments that show that MRB8170 is less abundant in MRB3010 or TbRGG2 pull-downs in the presence of ribonucleases (Ammerman et al. 2012).
Finally, we provide here some evidence that while MRB8170 and MRB4160 may interact with a matching subset of MRB1 proteins in a nearly identical manner, each protein may represent a distinct complex(es). Upon mass spectroscopy of either TAP isolation, there is an under-representation of unique peptides attributable to its paralog (Table 1, top two rows); this analysis excludes peptides that can arise from both MRB8170 and MRB4160 (Table 1, third row). Furthermore, interaction of the tagged MRB8170 with its endogenous counterpart persists despite RNase treatment, indicating self-binding, while the interaction of endogenous MRB8170 with tagged MRB4160 is abolished under such conditions (Fig. 5B), indicating that their association is RNA-mediated only and not due to any protein–protein contact.
The RNAi silencing of either MRB8170 or MRB4160 is compensated by the presence of the other to maintain cell growth
To facilitate functional analysis of the MRB8170 and MRB4160, procyclic T. brucei cell lines were generated with the capacity for tetracycline-inducible RNAi silencing of the expression of either protein individually as single knockdowns (sKDs) or both simultaneously as a double knockdown (dKD). We targeted a portion of the diverged 5′ regions of the two ORFs for the sKDs and sequence from the conserved 3′ part for the dKD (Fig. 1B; Supplemental Table 1). To assay the specificity and robustness of RNAi silencing of either MRB8170 or MRB4160 transcripts in these cell lines, qPCR was used using primers annealing to their distinct regions (Fig. 1B; Supplemental Table 1). The dKD equally down-regulated both mRNAs to roughly 57%–66% as compared with their wild-type abundance. In the sKDs, MRB8170 or MRB4160 was reduced to ∼28%–38% of their ordinary levels in their respective KDs, while the other untargeted paralog was not significantly affected (Fig. 6A). This result was confirmed by Western blot analysis with the α-MRB8170 antibody, showing that the protein was undetectable in both the MRB8170 sKD and dKD, while it remained unaffected in the RNAi-induced MRB4160 sKDs (Fig. 6B). The persistence of MRB8170 when MRB4160 is depleted is in contrast to the situation when one of the GAPs or MRPs is silenced, which leads to the reciprocal instability of the other partner protein (Vondrušková et al. 2005; Hashimi et al. 2009).
FIGURE 6.

Growth is inhibited upon simultaneous down-regulation of MRB8170 and MRB4160 but persists when each is individually RNAi-silenced. (A) A bar graph on a linear scale showing the mean relative transcript abundance as determined by qRT-PCR in MRB8170/4160 double- and single-knockdown cell lines treated with the RNAi-induction agent tetracycline in comparison to those grown in the absence of the antibiotic. Data were normalized to cytosolic β-tubulin (black) and 18S rRNA (gray) levels. A value of 1 indicates no change, while a value <1 signifies depletion of the assayed transcript. The range of obtained relative abundances is signified by whiskers around the top of the bars. (B) Western analysis of resolved mitochondrial proteins from the studied cell lines was performed to assay the depletion of MRB8170 in kMAP double and single knockdowns. The mitochondrial protein trCoIV was used as a loading control. (C) The growth rate of cells grown in the presence (black diamond and line) or absence (gray square and line) of tetracycline for the MRB8170/4160 double and single knockdowns over a 14-d course. Cell density (cells/mL) was measured every 24 h, and the cells were diluted to the starting density of 2 × 106 cells/mL every second day.
To ascertain whether the MRB8170 and MRB4160 is essential for the viability of procyclic T. brucei, the growth of the sKD and dKD cell lines in the presence and absence of tetracycline was monitored over the course of 2 wk. Simultaneous ablation of MRB8170 and MRB4160 resulted in slower growth, which was apparent 5 d after RNAi induction and persisted until the end of the time course (Fig. 6C). This retardation of growth was also observed when both MRB8170 and MRB4160 were ablated in the bloodstream stage of the parasite (data not shown). However, no effect on cell growth was observed when only one of these proteins was silenced in the procyclics (Fig. 6C). Thus, while both MRB8170 and MRB4160 together are essential for maintaining normal cell growth, the absence of one of them is compensated by the presence of the other. Based on these data, we decided to perform subsequent experiments on the dKD and sKDs 4 and 5 d after RNAi induction, respectively.
The influence of MRB8170 and MRB4160 ablation on the integrity of MRB1 subunit-containing complexes
Next, we wanted to determine whether the functional redundancy apparent in the sKDs in terms of growth also influences the integrity of MRB1 subunits-containing complexes. The mt proteins from the tetracycline-treated and -untreated dKD and sKD cells were resolved by ultracentrifugation in a 10%–30% glycerol gradient, and the resulting fractions were analyzed as described previously (Fig. 4), using antibodies against the MRB1 subunits GAP1, MRB3010, and TbRGG2. The α-MRB8170 antibody was also used in the analysis of the MRB4160 sKD. In the dKD, a clear shift to lighter fractions was observed in the sedimentation of the core MRB1 subunits GAP1 and MRB3010, indicating that the integrity of macromolecular complexes encompassing these two proteins was compromised. The sedimentation of TbRGG2 was much less affected (Fig. 7A), because only the protein in the two densest fractions was shifted upon simultaneous RNAi silencing.
FIGURE 7.

Simultaneous down-regulation of MRB8170 and MRB4160 leads to disruption of macromolecular complexes containing some MRB1 subunits. Mitochondrial vesicles from the double and single MRB8170/4160 knockdown cell lines were isolated following RNAi induction by tetracycline (Tet+). Parallel samples grown for this time in the absence of the antibiotic were also analyzed (Tet−). The mitochondrial proteins from MRB8160/MRB4160 dKD (A), MRB8170 sKD (B), and MRB4160 sKD (C) were resolved by glycerol gradient sedimentation and analyzed by Western blot as described in Figure 4. The column marked as “mito” contains total mitochondrial lysate. The identity of the antibody used is indicated at the right.
In both of the sKDs, the integrity of the TbRGG2 complex remained unaltered (Fig. 7B,C). The sedimentation of the MRB3010 subunit in the denser fractions was also retained when only one of the MRB8170 and MRB4160 proteins was depleted, although there was a slight shift to lighter fractions in the peak of the antibody signal in the MRB8170 sKD (Fig. 7B). An effect on GAP1 sedimentation to less dense fractions was slightly more apparent in the case of the MRB8170 silencing (Fig. 7B) as compared to that in the MRB4160 sKD (Fig. 7C). Interestingly, the sedimentation of MRB8170 is unchanged when MRB4160 is depleted, consistent with the notion that they are in a distinct complex(es) (Fig. 7C). Thus, it appears that the assembly of MRB1-containing complexes is most affected in the dKD. The integrity of RECC, as represented by the α-KREPA6 antibody signal, is unaffected in all of the KDs.
MRB8170/4160 RNAi silencing does not appreciably alter minicircle-encoded guide RNAs
Because some MRB1 proteins have been demonstrated to affect gRNA stability (Weng et al. 2008; Hashimi et al. 2009; Hernandez et al. 2010) and MRB8170 and MRB4160 were shown to associate with the pre-gRNA processing endonuclease mRPN1 (Madina et al. 2011), we decided to inspect whether MRB8170/4160 RNAi silencing affects the levels of these small transcripts. Guanylyltransferase labeling of gRNAs revealed no changes in the steady-state level or size distribution of these molecules when both proteins were simultaneously down-regulated (Fig. 8A). Further investigation of a panel of four individual gRNAs by Northern analysis in the dKD, using a probe against a cytoplasmic tRNA as a loading control, shows that none are completely degraded, although there appears to be a small but discernible decrease in the molecules directing co3 and ATP synthase subunit 6 (A6) mRNA editing (Fig. 8B). However, the levels of these gRNAs were not greatly affected in either sKD (Fig. 8B). We conclude from these results that MRB8170 and MRB4160 do not play a major role in gRNA biogenesis.
FIGURE 8.

The effect of simultaneous and individual MRB8170/4160 RNAi silencing on minicircle-encoded gRNAs. (A) RNA from the double MRB8170/4160 knockdown grown in the presence (+) and absence (−) of the RNAi-induction agent tetracycline was capped with [α-32P]GTP by the recombinant guanylyltransferase enzyme. The homogeneous population of small gRNA molecules in the samples was resolved as a ladder of bands on a denaturing 7 M urea/8%-acrylamide gel (bottom panel). A cytosolic RNA that is simultaneously labeled by this reaction is shown (top panel) as a loading control. (B) Individual gRNAs directing editing of RPS12, MURF2, co3, and A6 mRNAs (using the abbreviations as defined in Fig. 9) were detected by Northern blot. A probe against a cytoplasmic tRNACys (GCA) was used as a loading control. The “+” and “−” above the lanes indicate the state of tetracycline induction.
Depletion of MRB8170/4160 differentially influences maxicircle-encoded mRNAs
We then turned our attention to whether levels of maxicircle-encoded never-edited and edited mRNAs were impacted in the MRB8170/4160 dKD and sKDs by an established qPCR-based assay (Carnes et al. 2005). The levels of never-edited transcripts were not dramatically affected in any of the RNAi knockdowns (Fig. 9, NE). In fact, the depletion of MRB4160 does not significantly affect any maxicircle transcripts examined (Fig. 9C). In contrast, ablation of MRB8170 exerts a mild effect on many pan-edited and two moderately edited mRNAs. Most of these are depleted in abundance (edited MURF2, A6, and ND7, and both pre-edited and edited co3), while pre-edited cyB is actually more abundant (Fig. 9B; Supplemental Fig. 2). However, the most striking changes in maxicircle edited RNA abundances were obtained upon the dual silencing of MRB8170 and MRB4160 (Fig. 9A). In this case, both pre-edited and edited species of most pan-edited RNAs were clearly less abundant, and to a greater extent than observed in the MRB8170 sKD. Changes in abundance of almost all examined moderately edited RNAs were also observed. Thus it seems that MRB8170 and MRB4160 exhibit a synergistic effect on the stability of pan-edited RNAs, since the dKD affects these molecules more than the sKDs. This synergy in their influence is strikingly demonstrated when nascently edited A6 and RPS12 molecules are assayed by poisoned primer extension (Ammerman et al. 2010). The severity of the down-regulation of edited A6 and RPS12 RNAs decreases steadily from the dKD to the MRB8170 sKD, with the mildest effect in the MRB4160 sKD (Supplemental Fig. 2). Together, these results suggest an apparent functional divergence between the two paralogs since MRB8170 appears to influence mtmRNAs to a higher degree than MRB4160.
FIGURE 9.

The influence of simultaneous and individual MRB8170/4160 depletion on the steady-state levels of maxicircle-encoded mRNAs. qRT-PCR analysis of pre-edited (P), edited (E), and never-edited (NE) mRNAs encoded on maxicircle DNA. Analysis was performed in the presence or absence of the RNAi-induction agent tetracycline for the double (A) and single knockdowns of MRB8170 (B) and MRB4160 1 (C). For each target amplicon, the relative change in RNA abundance due to the depletion of both or one of the MRB8170/4160 proteins as compared with the untreated controls, which were processed in parallel, was determined by using the cytosolic transcripts β-tubulin and 18S rRNA as internal references. The mean relative abundance of each amplicon is plotted as a bar graph on a logarithmic scale on the y-axis, in which levels above and below the middle axis signify an up- or down-regulation, respectively, while no bar indicates that there was no change in steady-state levels of the transcript upon RNAi silencing; whiskers denote the range of obtained relative abundances within the triplicates. The following pre-edited and edited mRNAs were assayed: ATPase subunit 6 (A6), cytochrome oxidase subunits 2 (co2) and 3 (co3), cytochrome reductase subunit b (cyB), maxicircle unknown reading frame 2 (MURF2), NADH dehydrogenase subunit 7 (ND7), and ribosomal protein S12 (RPS12). The following never-edited RNAs were assayed: cytochrome oxidase subunit (co1) and NADH dehydrogenase subunit 4 (ND4).
Interestingly, we noted in the dKD a qualitative difference between pan-edited and moderately edited transcripts. In moderately edited transcripts, the manifested effect was reminiscent of the consequences of the disruption of core RNA editing activities: an accumulation of pre-edited mRNAs and a decrease of fully edited molecules (Carnes et al. 2005; Tarun et al. 2008; Weng et al. 2008; Hashimi et al. 2009; Ammerman et al. 2011). The cytochrome oxidase subunit (co) 2 and cyB transcripts exhibited such a phenotype, while the pre-edited MURF2 mRNA exhibited an increase without the concurrent decrease of the respective edited transcripts (Fig. 9A, moderately edited).
In contrast, pan-edited transcript abundances in the dKD were affected in a fashion that is reminiscent of an effect on RNA stability, namely, that both pre-edited and edited forms of various transcripts are often less abundant (Fig. 9A, pan-edited; Hashimi et al. 2008, 2009). The effects observed here ranged from a similar degree of down-regulation of edited and pre-edited co3, to the marginally affected or unaltered pre-edited NADH dehydrogenase subunit (ND) 7 and RPS12 mRNAs, respectively (Fig. 9A) with coincident depletion of both edited forms. The fully edited ribosomal protein S12 (RPS12) mRNA was further analyzed to determine whether the long A/U appended population, which stabilizes and marks these transcripts for translation (Ryan and Read 2005; Etheridge et al. 2008; Aphasizheva et al. 2011), was preferentially degraded over the short poly(A) forms; both types were equally down-regulated in the dKDs (Supplemental Fig. 3). Thus, it is some feature other than the long A/U tail that results in the differential effect on abundance of fully edited and pre-edited RPS12 resulting from MRB8170 and MRB4160 codepletion. Finally, we note that the steady-state levels of fully processed pan-edited molecules were more reduced (25%–45% relative abundance, average 35%) as compared with those from the moderately edited population (60%–94% relative abundance, average 75%) (Fig. 9A, pan-edited). This phenotype, in addition to all of the other mRNA phenotypes described above, hints at a role of MRB8170 and MRB4160 in mRNA stability and/or processing.
DISCUSSION
The MRB1 complex proteins are among the bewildering number of proteins that play a role in the unique process of U-insertion/deletion RNA editing. As such, not only are many MRB1 subunits exclusive to the order Kinetoplastida, but they also exhibit a variety of RNAi phenotypes, complicating the determination of their roles in mediating this massive processing event and integrating it into the RNA metabolism required for mt gene expression. Here, we expand our knowledge about MRB1 by functional characterization of its subunits MRB8170 and MRB4160. They are paralogs in T. brucei while occurring only as a single locus in other sequenced trypanosomatid lineages. The proteins share a large conserved C terminus while having divergent sequence in a relatively smaller N-terminal region. In this work, we show that although these proteins share many biological properties due to their significant conservation, they also differ in a key aspect.
Both MRB8170 and MRB4160 proteins have basic pIs, a biophysical feature that facilitates their ability to bind RNA in vitro. Because neither of them has any identifiable domains or orthologs outside of kinetoplastids, they represent novel RNA binding proteins that will be an interesting topic of future study. As a start toward this endeavor, we investigated whether MRB8170 has higher affinity in vitro for RNA species that are well represented in the mt transcriptome: gRNAs, pre-edited or edited mRNAs. The results from the filter binding assay used suggest that MRB8170 exhibits ∼5× higher affinity for mRNAs than gRNAs. To ensure that any observed differences in binding are not caused by varying numbers of nucleotides in assayed binding substrates, a molar excess of torula yeast RNA was included during the incubation of the tested substrate RNAs. Not much is known about the structure of mt mRNAs, in contrast to studies demonstrating three-helical structures in mRNA:gRNA duplexes (Reifur and Koslowsky 2008) and the presence of two hairpin loops in gRNAs, including gA6[14] (Schmid et al. 1995). While our data do not indicate which structural features of the mRNAs are exploited by the 100-kDa MRB8170 for binding, one possible structural determinant of the apparent preference for mRNAs is that they may have significant stretches that are single-stranded compared with gRNA. Nevertheless, these data are consistent with the RNAi silencing of MRB8170 influencing pan-edited mRNAs but not gRNAs.
A common feature of MRB8170 and MRB4160 is that they interact with a highly overlapping subset of the MRB1 subunits, as evidenced by the very similar peptide composition profiles of proteins pulled down with either TAP-MRB8170 or TAP-MRB4160. In both cases, interaction with GAP1 is mediated completely by RNA linkers, while that with MRB3010 and TbRGG2 is RNA-enhanced, consistent with a direct contact. This finding agrees with other studies reporting the existence of a complex containing TbRGG2 and either MRB8170 or MRB4160 (Madina et al. 2011; Ammerman et al. 2012), interacting with the core MRB1 proteins via MRB3010 and MRB8620 (Ammerman et al. 2012). Furthermore, when both MRB8170 and MRB4160 are down-regulated simultaneously, the assembly of the three MRB1 subunits GAP1, MRB3010, and TbRGG2 into macromolecular complexes appears to be hindered, albeit to different degrees, further supporting the notion that they interact with each other via RNAs slated for editing, which are also reduced in this background, and/or protein–protein binding.
We also show that MRB8170 and MRB4160 do not interact directly with each other, as evidenced by RNase-treated TAP-MRB4160 failing to pull down MRB8170. Furthermore, the stability of MRB8170 is not dependent on the presence of MRB4160, as observed for the GAP paralogs (Hashimi et al. 2009) and the MRP1/2 proteins (Zíková et al. 2008), and even the integrity of the MRB8170-containing complex(es) is not affected in the MRB4160 sKD. Thus it appears that the MRB8170 and MRB4160 proteins function independently.
Current evidence suggests that proteins within the MRB1 complex undergo numerous transient and dynamic interactions (Hashimi et al. 2008; Panigrahi et al. 2008; Weng et al. 2008; Ammerman et al. 2012). Interestingly, MRB8170 and MRB4160 along with TbRGG2 were recently shown to associate with the gRNA endonuclease mRPN1 (Madina et al. 2011). We did not detect mRPN1 in mass spectrometry of the MRB8170 or MRB4160 TAP purifications described here. Since neither we, nor other laboratories, have detected this endonuclease by mass spectrometry analyses of purified MRB1 subunits, we speculate that mRNP1 is not amenable to such a technique, either due to its intrinsic properties or an association with these MRB1 subunits in substoichiometric amounts. Our data also indicate that the primary role of MRB8170 and MRB4160 is not the processing of gRNAs, because the dKD does not significantly affect the steady-state levels or sizes of these molecules. Rather, our data are more consistent with a role for TbRGG2/MRB8170/4160 in a hand-over mechanism between the biogenesis of gRNAs and their utilization in the RNA editing process as proposed by Madina et al. (2011).
MRB8170 and MRB4160 are among 74 paralogs that have arisen due to a massive duplication of a 0.5-Mb-long region between chromosomes 4 and 8 that occurred after the T. brucei clade branched off from the other trypanosomes. As described by Jackson (2007), the affected loci were either lost from one of the chromosomes only or retained on both, the latter event resulting in paralogs that either remained conserved or diverged in sequence. The CDSs of MRB8180 and MRB4150, which are immediately downstream from the topics of this work, MRB8170 and MRB4160, respectively, are loci that fit into the category of conserved paralogs, sharing 99.1% translated sequence identity. On the other hand, MRB8170 and MRB4160 are considered to be divergent, because their amino acid sequence identity is 77.3%. The question is whether this divergence in sequence has resulted in two functionally divergent proteins. The similarities between MRB8170 and MRB4160 discussed so far seem to argue against this. Their functional redundancy is also implied by our finding that cell growth persists upon RNAi silencing of either MRB8170 or MRB4160 alone, at least under the culture condition used, while their simultaneous down-regulation leads to substantial growth inhibition.
However, MRB8170 and MRB4160 do differ in one key aspect: They influence pan-edited RNAs to differing degrees. When MRB8170 is ablated, pan-edited RNAs appear to be destabilized because their levels are decreased. This is in contrast to the MRB4160 sKD in which neither pan-edited RNAs nor any other classes of maxicircle transcripts are affected, despite equally efficient RNAi. When both proteins are silenced in the dKD, the pan-edited RNAs exhibit a more severe decrease than that observed in the MRB8170 sKD. Together, these results suggest that after their divergence, MRB8170 retained more influence on the stability of pan-edited transcripts than MRB4160, but that both proteins may act synergistically on these transcripts.
The picture is somewhat different with regard to moderately edited RNAs. Here, MRB8170 and MRB4160 display a clearly redundant function. Neither of the sKDs exhibited alterations in the abundances of moderately edited transcripts, while these RNAs were affected in the dKD. Moreover, this redundant MRB8170/4160 function appears to be at the level of RNA editing, since in the dKD moderately edited RNAs were affected in a manner reminiscent of that observed when core editing processes are interfered with: an accumulation of pre-edited mRNAs and a depletion of edited mRNAs. This phenotype may reflect a redundant structural role of MRB8170/4160 in the assembly of partner MRB1 subunits that impact RNA editing, resulting in a downstream effect on moderately edited RNAs. The profound disassembly of the MRB complex in the dKD, in contrast to either sKD, agrees with this scenario.
Although the pan-editing phenomenon has been reported in several kinetoplastids, it seems most prevalent in the Trypanosoma clade, as exemplified by the nine maxicircle genes that require extensive editing in T. brucei as compared with only six such transcripts in Leishmania tarentolae (Arts and Benne 1996; Alfonzo et al. 1997; Speijer 2006). Thus, we propose that this milieu could be conducive to retain both MRB8170 and MRB4160 in the duplicon and perhaps the subsequent divergence of the two paralogs. Since both MRB8170 and MRB4160 function independently of each other, one of them can be modified in the background of the other maintained paralog, as long as any potential innovations fit within the context of the other MRB1 subunits. Indeed, there is evidence for some of the codons being positively selected between the two loci (Jackson 2007). Such a situation could then give rise to a functionally distinct MRB8170 that plays a bigger role in the stability of pan-edited RNAs than its paralog.
MATERIALS AND METHODS
Dot-plot analysis of MRB8170 and MRB4160 ORFs and 3′-UTRs
The MRB8170 and MRB4160 ORFs plus predicted 3′-UTR DNA sequences (Jackson 2007) from the 927 T. brucei strain (available at TriTrypDB: http://tritrypdb.org/tritrypdb/) were entered into the CLC Main Workbench 6.1.1 program (CLC Bio). The included dot-plot function was used to create the similarity matrix using the default window size setting of 9. The output was further adjusted manually in the program to reduce the background noise, which did not affect the signal on the diagonal of the plot. The corrected MRB4160 ORF from recent spliced leader trapping studies (Nilsson et al. 2010; Siegel et al. 2010), which was confirmed by spliced leader-mediated PCR (Supplemental Fig. 1), was used in this analysis, as was the annotated MRB8170 ORF that was also confirmed with this method (data not shown).
Expression and purification of recombinant MRB8170 and MRB4160
The MRB8170 and MRB4160 ORFs were PCR-amplified using Pfx polymerase (Invitrogen) from genomic DNA extracted from procyclic form (PF) T. brucei (strain 29-13) using primers incorporating the restriction sites described in Supplemental Table 1. The resultant PCR products were cloned into the pGBD-C2 vector using the In-Fusion cloning kit (Clontech). The inserts were then shuttled into the expression vector pGEX4T-1 (Novagen) and transformed into the Rosetta strain of Escherichia coli (Novagen). Cells were grown to an OD600 of ∼0.4 at 37°C and induced with 0.1 mM IPTG, while the temperature was reduced to 17°C overnight. GST-tagged MRB8170 and MRB4160 were purified using glutathione-agarose (Invitrogen) and a standard single-step GST purification protocol.
RNA cross-linking assay
To generate substrate RNA, a previously described BamHI-linearized construct containing the 5′ 102 nt of pre-edited cytochrome B (cyB) sequence (Koslowsky et al. 1996) was in vitro transcribed with a MAXIscript kit (Ambion) using unlabeled and [α-32P]-labeled UTP (800 Ci/mmol) at a 1:1 molar ratio in a 20-μL volume. DNase-treated RNA was resuspended in an equal volume of 90% formamide and run on a 8%-acrylamide/7 M urea gel, after which the band of appropriate size was excised. RNA was eluted from the cut gel by constant rotation overnight in elution buffer (10 mM Tris-HCl at pH 8.0, 1 mM EDTA at pH 8, 0.75 M NH4oAc, 0.1% SDS) and subsequently precipitated with isopropanol. Five femtomoles of RNA and 7.5 pmol of protein were incubated in cross-linking buffer (4.5 mM HEPES, 1.6 mM MgCl2, 0.38 mM DTT, 1.13 mM ATP, 3.75 mM creatine phosphate, 0.075 mM EDTA, 7.5 μg /mL torula yeast RNA type VI [Sigma-Aldrich], 4.5% glycerol, 15 μg/mL BSA, 6 U RNaseOUT [Invitrogen]) for 20 min at room temperature, UV cross-linked for 10 min on ice, and treated with 30 μg of RNase A for 15 min at 37°C. These reactions were run on a 10% SDS-acrylamide gel, stained with Coomassie dye, and dried. The radioactive signal on the gel was detected using the Storm PhosphorImager (Molecular Dynamics).
RNA double filter binding assay
The untailed, full-length pre-edited and edited RPS12 mRNAs used in this assay were 221 and 325 nt in length (Kao and Read 2005). The 79-nt gA6[14] substrate gRNA consisted of a previously reported sequence (Koslowsky et al. 1992) with a 17-nt 3′ U tail and 10 nt of 5′ vector-derived sequence. These RNAs were body-labeled as described above and subsequently gel-purified. RNAs were then renatured by incubation for 3 min at 70°C, slow cooling at a rate of 2°C per min to 27°C, at which they remained for 30 min, followed by quenching on ice to allow them to most closely achieve their native secondary state. RNA with an invariant concentration of 30 pM was incubated with different concentrations of protein for 30 min at room temperature in two separate experiments each performed in triplicate. Buffer conditions during binding were 0.2× PBS, 2 mM MgCl2, 0.5 mM DTT, 0.1 mM EDTA, 10 μg/mL torula yeast RNA (type VI [Sigma-Aldrich]), 50 μg/mL BSA, and 6% glycerol. Filter binding was performed as previously described (Wong and Lohman 1993; Hall and Kranz 1999) to determine the percent of RNA bound at each concentration, and results were combined and plotted using GraphPad Prism 5 software. Apparent Kd and Bmax were calculated by nonlinear regression assuming single-site specific binding and using the equation Y = Bmax × X/(Kd + X), fitting by the method of least squares.
Generation of cell lines and growth curves
Gene fragments cloned into the p2T7-177 RNAi vector (Wickstead et al. 2002) for generating single and double MRB8170 and MRB4160 knockdown cell lines were PCR-amplified from the T. brucei genomic DNA using the primers given in Supplemental Table 1, which also indicates the restriction sites used for ligation into the construct as well as the corresponding region of the fragment in ORF (Fig. 2). This table also lists the primers used to amplify the whole ORFs of MRB8170 and MRB4160 1 and 2 that were cloned into the pLew79-MHTAP vector (Jensen et al. 2007). Both types of constructs were linearized with the NotI restriction enzyme and electroporated as previously described (Hashimi et al. 2008) into the parental procyclic strain 29-13, and clones obtained by limiting dilution were selected for phleomycin (2.5 μg/mL) resistance. The 29-13-derived cell lines were grown in SDM-79 medium supplemented with hygromycin (50 μg/mL) and G418 (15 μg/mL). RNAi was induced by the addition of 1 μg/mL tetracycline. The growth dynamics of the tetracycline-induced cells compared with their noninduced counterparts was performed as described elsewhere (Hashimi et al. 2009).
Western blot analysis and antibodies
Proteins were separated on 10% or 12% SDS-polyacrylamide gels and transferred onto PVDF membranes. Forty micrograms of proteins from hypotonically isolated mt vesicles were loaded on gels for assessing of single and double knockdowns with the affinity-purified MRB8170 (Tb927.8.8170) antibody by Western analysis, which is described by Ammerman and coworkers (2012). The other polyclonal antibodies used is this study were generated against MRB3010, in particular, its oligopeptide CEALQMKKLHQERGGNPM (Bethyl Laboratories), GAP1 (Hashimi et al. 2009; Ammerman et al. 2011), TbRGG2 (Fisk et al. 2008), KREPA6 (Tarun et al. 2008), trCoIV (Maslov et al. 2002), and the Myc protein (Sigma-Aldrich). In addition, monoclonal antibodies immunodecorating KREPA1 and KREL1 (Panigrahi et al. 2001) were also used. All antibodies against RECC proteins (KREPA1 and 6; KREL1) were kindly provided by Ken Stuart (Seattle Biomed).
Glycerol gradient fractionation of mitochondrial lysates
The resolution of MRB1 and RECC macromolecular particles by ultracentrifugation on 10%–30% glycerol gradients was performed as described previously (Ammerman et al. 2011). Hypotonically isolated mt vesicles (Hashimi et al. 2008) from 1010 cells were lysed in a lysis buffer (1% Triton-X, 10 mM Tris at pH 7.2, 10 mM MgCl2, 100 mM KCl, 1 mM DTT, 1 μg/mL pepstatin, 2 μg/mL leupeptin, 1 mM pefabloc) supplemented with either 40 units of RNaseOUT or an RNase cocktail (0.1 unit/μL RNase A, 0.125 unit/μL T1, 0.001 unit/μL V1, 0.03 unit/μL micrococcal nuclease) to eliminate both single-stranded (ss) and double-stranded (ds) RNAs.
Tandem affinity purification and protein analysis
Tandem affinity purification (TAP) of MRB8170 and MRB4160 was performed on ∼5 × 109 tetracycline-induced cells using the protocol described in Hashimi et al. (2008). In the case of RNase treatment during the TAP purification, cleared lysates were split equally in IPP150 buffer (10 mM Tris at pH 8.0, 150 mM NaCl, 0.1% Igepal) that was supplemented either with 40 units of RNaseOUT (Invitrogen) or an RNase cocktail (0.1 unit/μL RNase A, 0.1 unit/μL RNase I, 0.001 unit/μL RNase V1), before loading onto the initial IgG-Sepharose columns. TEV eluates were collected for Western blot analyses, while they were completely processed via a second calmodulin-Sepharose column for submission to mass spectroscopy (LC-MS/MS).
The eluates were scored for the presence of the TAP-tagged MRB8170/4160 protein by Western blot probed with an α-Myc antibody. The positive samples were pooled, acetone-precipitated, and resuspended in an appropriate digestion overnight at 37°C with 20 μg/mL proteomic grade trypsin (Sigma-Aldrich), and the digested peptides were purified using a C18 ZipTip (Millipore) according to the manufacturer's protocol. Subsequent LC-MS/MS was performed on a NanoAcuity ultra performance liquid chromatography device coupled on-line to an electrospray ionization (ESI) Q-Tof Premier mass spectrometer (Waters). One microliter of sample was diluted in 3% acetonitrile/0.1% formic acid (v/v) and subjected to reverse-phase HPLC with a flow rate of 0.4 μL/min through a BEH300 C18 analytical column (Waters). A linear solvent gradient was applied—initially 3% (v/v) solvent B (0.1% (v/v) formic acid in acetonitrile diluted with solvent A (0.1% [v/v] formic acid in water) was gradually increased to 40% solvent B over 30 min. The peptides were then eluted by increasing the concentration of solvent B to 85% over a 2-min period, where it was maintained for another 5 min and then lowered to 3% for the next run. All eluted peptides were applied directly into the ESI source. Raw data were acquired in data-independent MS^e Identity mode. Precursor ion spectra were acquired with a 5 V collision energy and fragment ion spectra with a 20–35 V collision energy ramp in alternating 1-sec scans. The data-dependent analysis mode was subsequently used for secondary analysis. Peptide spectra were acquired with a 5 V collision energy, and peptides with charge states of +2–4 were selected for MS/MS analysis. Fragment spectra were collected with a 20–40 V collision energy ramp. In both modes, peptide and fragment spectra were acquires with a 2- and 5-ppm tolerance, respectively. Raw data were then used to interrogate the T. brucei Uniprot and NCBI protein databases, as well as the one described in Panigrahi et al. (2008), using the PLGS v2.3 program (Waters). Acetyl N-terminal deamidation of N and Q, carbamidomethyl C, and M oxidation were set as variable modifications. Identification of three consecutive y- or b-ions was set as a prerequisite for a positive peptide match.
Quantitative real-time PCR
RNA was isolated and processed for subsequent reverse transcription into cDNA as described previously by Hashimi et al. (2008, 2009) from single and double MRB8170 and MRB4160 knockdown cell lines for 5 and 4 d, respectively, in the presence or absence of tetracycline. The resulting random hexamer-primed cDNA was used as a template for quantitative real-time (q) PCR as described in the aforementioned references using previously designed primers that anneal to the maxicircle mRNA sequences (Carnes et al. 2005). Primers that amplify MRB8170 or MRB4160 encoding cDNAs were designed specifically for this study and are given in Supplemental Table 1. All qPCRs were performed in triplicate and also with a negative control template from a mock reverse transcription reaction to ensure that genomic DNA was adequately degraded before cDNA production. The raw qPCR was used in Pfaffl analysis (Pfaffl 2001) to obtain the relative abundance of the examined transcripts in the RNAi cell lines as compared with those in the untreated controls grown and processed in parallel.
Guanylyltransferase and gRNA Northern analyses
RNA for these assays were isolated and treated as for the above-described qPCR procedure. For the guanylyltransferase-mediated 5′-capping of the gRNAs, 15 μg of total RNA was incubated with the enzyme and 5 μCi of [α-32P]GTP (800 Ci/mmol) and then extracted as described elsewhere (Ammerman et al. 2011). The radiolabeled products of both of these assays were separated on a denaturing 8% acrylamide/7 M urea gel in 1× TBE. For gRNA Northerns, 10 μg of total RNA was run on the aforementioned gel and transferred onto a Zeta-probe membrane (Bio-Rad), which was probed with specific 5′-32P-labeled oligonucleotides hybridizing specific gRNAs as previously performed (Madina et al. 2011).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
Much gratitude is given to Andrew Jackson (Sanger Institute) for enlightening discussions about the nature of the MRB8170/4160 paralogs in the context of the chromosome 4 and 8 duplication event that he described in T. brucei. The efforts of Yan-Zi Wen, Lucie Novotná, and Zdeňka Čičová (Institute of Parasitology) at early stages of this project are appreciated. We thank Ken Stuart (Seattle Biomed) for his kind gift of antibodies and Peter Koník (University of South Bohemia) for help with mass spectrometry. This work was supported by the Grant Agency of the Czech Republic 204/09/1667, the RNPnet FP7 program (289007), and the Praemium Academiae award to J.L.; NIH grants RO1 AI061580 and RO1 AI077520 to L.K.R.; the NIH Postdoctoral Fellowship F32 AI07718501 to J.C.F.; and the Institutional Research Concept AV0Z50200510 to R.S. J.L. is a Fellow of the Canadian Institute for Advanced Research.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.033852.112.
REFERENCES
- Acestor N, Panigrahi AK, Carnes J, Zíková A, Stuart KD 2009. The MRB1 complex functions in kinetoplastid RNA processing. RNA 15: 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonzo JD, Thiemann O, Simpson L 1997. The mechanism of U insertion/deletion RNA editing in kinetoplastid mitochondria. Nucleic Acids Res 25: 3751–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerman ML, Fisk JC, Read LK 2008. gRNA/pre-mRNA annealing and RNA chaperone activities of RBP16. RNA 14: 1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerman ML, Presnyak V, Fisk JC, Foda BM, Read LK 2010. TbRGG2 facilitates kinetoplastid RNA editing initiation and progression past intrinsic pause sites. RNA 16: 2239–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerman ML, Hashimi H, Novotná L, Čičová Z, McEvoy SM, Lukeš J, Read LK 2011. MRB3010 is a core component of the MRB1 complex that facilitates an early step of the kinetoplastid RNA editing process. RNA 17: 865–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerman ML, Downey KM, Hashimi H, Fisk JC, Tomasello DL, Faktorová D, Kafková L, King T, Lukeš J, Read LK 2012. Architecture of the trypanosome RNA editing accessory complex, MRB1. Nucleic Acids Res 40: 5637–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Aphasizhev R 2010. RET1-catalyzed uridylylation shapes the mitochondrial transcriptome in Trypanosoma brucei. Mol Cell Biol 30: 1555–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizhev R, Aphasizheva I, Simpson L 2003. A tale of two TUTases. Proc Natl Acad Sci 100: 10617–10622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Maslov D, Wang X, Huang L, Aphasizhev R 2011. Pentatricopeptide repeat proteins stimulate mRNA adenylation/uridylation to activate mitochondrial translation in trypanosomes. Mol Cell 42: 106–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts GJ, Benne R 1996. Mechanism and evolution of RNA editing in kinetoplastida. Biochim Biophys Acta 1307: 39–54 [DOI] [PubMed] [Google Scholar]
- Bhat GJ, Souza AE, Feagin JE, Stuart K 1992. Transcript-specific developmental regulation of polyadenylation in Trypanosoma brucei mitochondria. Mol Biochem Parasitol 52: 231–240 [DOI] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Ernst NL, Steinberg A, Stuart K 2005. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc Natl Acad Sci 102: 16614–16619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Peltan A, Fleck M, Stuart K 2008. RNA editing in Trypanosoma brucei requires three different editosomes. Mol Cell Biol 28: 122–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Soares CZ, Wickham C, Stuart K 2011. Endonuclease associations with three distinct editosomes in Trypanosoma brucei. J Biol Chem 286: 19320–19330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant JD, Hall L, Swift RV, Landon M, Schnaufer A, Amaro RE 2010. Novel naphthalene-based inhibitors of Trypanosoma brucei RNA editing ligase 1. PLoS Negl Trop Dis 4: e803 doi: 10.1371/journal.pntd.0000803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, et al. 2005. Comparative genomics of trypanosomatid parasitic protozoa. Science 309: 404–409 [DOI] [PubMed] [Google Scholar]
- Etheridge RD, Aphasizheva I, Gershon PD, Aphasizhev R 2008. 3′ adenylation determines mRNA abundance and monitors completion of RNA editing in T. brucei mitochondria. EMBO J 27: 1596–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk JC, Ammerman ML, Presnyak V, Read LK 2008. TbRGG2, an essential RNA editing accessory factor in two Trypanosoma brucei life cycle stages. J Biol Chem 283: 23016–23025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk JC, Presnyak V, Ammerman ML, Read LK 2009. Distinct and overlapping functions of MRP1/2 and RBP16 in mitochondrial RNA metabolism. Mol Cell Biol 29: 5214–5225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden DE, Hajduk SL 2005. The 3′-untranslated region of cytochrome oxidase II mRNA functions in RNA editing of African trypanosomes exclusively as a cis guide RNA. RNA 11: 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KB, Kranz JK 1999. Nitrocellulose filter binding for determination of dissociation constants. Methods Mol Biol 118: 105–114 [DOI] [PubMed] [Google Scholar]
- Hashimi H, Zíková A, Panigrahi AK, Stuart KD, Lukeš J 2008. TbRGG1, an essential protein involved in kinetoplastid RNA metabolism that is associated with a novel multiprotein complex. RNA 14: 970–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimi H, Čičová Z, Novotná L, Wen YZ, Lukeš J 2009. Kinetoplastid guide RNA biogenesis is dependent on subunits of the mitochondrial RNA binding complex 1 and mitochondrial RNA polymerase. RNA 15: 588–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez A, Madina BR, Ro K, Wohlschlegel JA, Willard B, Kinter MT, Cruz-Reyes J 2010. REH2 RNA helicase in kinetoplastid mitochondria: Ribonucleoprotein complexes and essential motifs for unwinding and guide RNA (gRNA) binding. J Biol Chem 285: 1220–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AP 2007. Tandem gene arrays in Trypanosoma brucei: Comparative phylogenomic analysis of duplicate sequence variation. BMC Evol Biol 7: 54 doi: 10.1186/1471-2148-7-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BC, Kifer CT, Brekken DL, Randall AC, Wang Q, Drees BL, Parsons M 2007. Characterization of protein kinase CK2 from Trypanosoma brucei. Mol Biochem Parasitol 151: 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao CY, Read LK 2005. Opposing effects of polyadenylation on the stability of edited and unedited mitochondrial RNAs in Trypanosoma brucei. Mol Cell Biol 25: 1634–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslowsky DJ, Riley GR, Feagin JE, Stuart K 1992. Guide RNAs for transcripts with developmentally regulated RNA editing are present in both life cycle stages of Trypanosoma brucei. Mol Cell Biol 12: 2043–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslowsky DJ, Kutas SM, Stuart K 1996. Distinct differences in the requirements for ribonucleoprotein complex formation on differentially regulated pre-edited mRNAs in Trypanosoma brucei. Mol Biochem Parasitol 80: 1–14 [DOI] [PubMed] [Google Scholar]
- Li F, Herrera J, Zhou S, Maslov DA, Simpson L 2011. Trypanosome REH1 is an RNA helicase involved with the 3′–5′ polarity of multiple gRNA-guided uridine insertion/deletion RNA editing. Proc Natl Acad Sci 108: 3542–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukeš J, Hashimi H, Zíková A 2005. Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr Genet 48: 277–299 [DOI] [PubMed] [Google Scholar]
- Madina BR, Kuppan G, Vashisht AA, Liang YH, Downey KM, Wohlschlegel JA, Ji X, Sze SH, Sacchettini JC, Read LK, et al. 2011. Guide RNA biogenesis involves a novel RNase III family endoribonuclease in Trypanosoma brucei. RNA 17: 1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov DA, Simpson L 1992. The polarity of editing within a multiple gRNA-mediated domain is due to formation of anchors for upstream gRNAs by downstream editing. Cell 70: 459–467 [DOI] [PubMed] [Google Scholar]
- Maslov DA, Zíková A, Kyselová I, Lukeš J 2002. A putative novel nuclear-encoded subunit of the cytochrome c oxidase complex in trypanosomatids. Mol Biochem Parasitol 125: 113–125 [DOI] [PubMed] [Google Scholar]
- Militello KT, Read LK 1999. Coordination of kRNA editing and polyadenylation in Trypanosoma brucei mitochondria: Complete editing is not required for long poly(A) tract addition. Nucleic Acids Res 27: 1377–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshiri H, Acoca S, Kala S, Najafabadi HS, Hogues H, Purisima E, Salavati R 2011. Naphthalene-based RNA editing inhibitor blocks RNA editing activities and editosome assembly in Trypanosoma brucei. J Biol Chem 286: 14178–14189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson D, Gunasekera K, Mani J, Osteras M, Farinelli L, Baerlocher L, Roditi I, Ochsenreiter T 2010. Spliced leader trapping reveals widespread alternative splicing patterns in the highly dynamic transcriptome of Trypanosoma brucei. PLoS Pathog 6: e1001037 doi: 10.1371/journal.ppat.1001037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Schnaufer A, Carmean N, Igo RP Jr, Gygi SP, Ernst NL, Palazzo SS, Weston DS, Aebersold R, Salavati R, et al. 2001. Four related proteins of the Trypanosoma brucei RNA editing complex. Mol Cell Biol 21: 6833–6840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Ernst NL, Domingo GJ, Fleck M, Salavati R, Stuart KD 2006. Compositionally and functionally distinct editosomes in Trypanosoma brucei. RNA 12: 1038–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Zíková A, Dalley RA, Acestor N, Ogata Y, Anupama A, Myler PJ, Stuart KD 2008. Mitochondrial complexes in Trypanosoma brucei: A novel complex and a unique oxidoreductase complex. Mol Cell Proteomics 7: 534–545 [DOI] [PubMed] [Google Scholar]
- Pelletier M, Read LK 2003. RBP16 is a multifunctional gene regulatory protein involved in editing and stabilization of specific mitochondrial mRNAs in Trypanosoma brucei. RNA 9: 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45 doi: 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, Lopez P, Brinkmann H, Budin K, Germot A, Laurent J, Moreira D, Muller M, Le Guyader H 2000. Early-branching or fast-evolving eukaryotes? An answer based on slowly evolving positions. Proc Biol Sci 267: 1213–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifur L, Koslowsky DJ 2008. Trypanosoma brucei ATPase subunit 6 mRNA bound to gA6-14 forms a conserved three-helical structure. RNA 14: 2195–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CM, Read LK 2005. UTP-dependent turnover of Trypanosoma brucei mitochondrial mRNA requires UTP polymerization and involves the RET1 TUTase. RNA 11: 763–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid B, Riley GR, Stuart K, Goringer HU 1995. The secondary structure of guide RNA molecules from Trypanosoma brucei. Nucleic Acids Res 23: 3093–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher MA, Karamooz E, Zíková A, Trantírek L, Lukeš J 2006. Crystal structures of T. brucei MRP1/MRP2 guide-RNA binding complex reveal RNA matchmaking mechanism. Cell 126: 701–711 [DOI] [PubMed] [Google Scholar]
- Shlomai J 2004. The structure and replication of kinetoplast DNA. Curr Mol Med 4: 623–647 [DOI] [PubMed] [Google Scholar]
- Siegel TN, Hekstra DR, Wang X, Dewell S, Cross GA 2010. Genome-wide analysis of mRNA abundance in two life-cycle stages of Trypanosoma brucei and identification of splicing and polyadenylation sites. Nucleic Acids Res 38: 4946–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson L, Aphasizhev R, Gao G, Kang X 2004. Mitochondrial proteins and complexes in Leishmania and Trypanosoma involved in U-insertion/deletion RNA editing. RNA 10: 159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson L, Aphasizhev R, Lukeš J, Cruz-Reyes J 2010. Guide to the nomenclature of kinetoplastid RNA editing: A proposal. Protist 161: 2–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speijer D 2006. Is kinetoplastid pan-editing the result of an evolutionary balancing act? IUBMB Life 58: 91–96 [DOI] [PubMed] [Google Scholar]
- Sprehe M, Fisk JC, McEvoy SM, Read LK, Schumacher MA 2010. Structure of the Trypanosoma brucei p22 protein, a cytochrome oxidase subunit II-specific RNA-editing accessory factor. J Biol Chem 285: 18899–18908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK 2005. Complex management: RNA editing in trypanosomes. Trends Biochem Sci 30: 97–105 [DOI] [PubMed] [Google Scholar]
- Tarun SZ Jr, Schnaufer A, Ernst NL, Proff R, Deng J, Hol W, Stuart K 2008. KREPA6 is an RNA-binding protein essential for editosome integrity and survival of Trypanosoma brucei. RNA 14: 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhamme L, Perez-Morga D, Marchal C, Speijer D, Lambert L, Geuskens M, Alexandre S, Ismaili N, Goringer U, Benne R, et al. 1998. Trypanosoma brucei TBRGG1, a mitochondrial oligo(U)-binding protein that co-localizes with an in vitro RNA editing activity. J Biol Chem 273: 21825–21833 [DOI] [PubMed] [Google Scholar]
- Vondrušková E, van den Burg J, Zíková A, Ernst NL, Stuart K, Benne R, Lukeš J 2005. RNA interference analyses suggest a transcript-specific regulatory role for mitochondrial RNA-binding proteins MRP1 and MRP2 in RNA editing and other RNA processing in Trypanosoma brucei. J Biol Chem 280: 2429–2438 [DOI] [PubMed] [Google Scholar]
- Weng J, Aphasizheva I, Etheridge RD, Huang L, Wang X, Falick AM, Aphasizhev R 2008. Guide RNA-binding complex from mitochondria of trypanosomatids. Mol Cell 32: 198–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstead B, Ersfeld K, Gull K 2002. Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol Biochem Parasitol 125: 211–216 [DOI] [PubMed] [Google Scholar]
- Wong I, Lohman TM 1993. A double-filter method for nitrocellulose-filter binding: application to protein–nucleic acid interactions. Proc Natl Acad Sci 90: 5428–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zíková A, Kopečná J, Schumacher MA, Stuart K, Trantírek L, Lukeš J 2008. Structure and function of the native and recombinant mitochondrial MRP1/MRP2 complex from Trypanosoma brucei. Int J Parasitol 38: 901–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer SL, McEvoy SM, Li J, Qu J, Read LK 2011. A novel member of the RNase D exoribonuclease family functions in mitochondrial guide RNA metabolism in Trypanosoma brucei. J Biol Chem 286: 10329–10340 [DOI] [PMC free article] [PubMed] [Google Scholar]