

Abstract
Colorectal cancer is one of the most common cancers in the world. Dietary fat intake is a major risk factor for colorectal cancer. Some nuclear hormone receptors play an important role in regulating nutrient metabolism and energy homeostasis. Among these receptors, special attention has been focused on the role of peroxisome proliferator-activated receptors (PPARs) in colorectal cancer, because PPARs are involved in regulation of lipid and carbohydrate metabolism. PPARs are ligand-activated intracellular transcription factors. The PPAR subfamily consists of three subtypes encoded by distinct genes named PPARα, PPARβ/δ, and PPARγ. PPARγ is the most extensively studied subtype of PPARs. Even though many investigators have studied the expression and clinical implications of PPARs in colorectal cancer, there are still many controversies about the role of PPARs in colorectal cancer. In this paper, the recent progresses in understanding the role of PPARs in colorectal cancer are summarized.
1. Introduction
Colorectal cancer is one of the most common cancers in the world. Its incidence appears to be increasing, particularly in developed countries [1–3]. Colorectal carcinogenesis results from the loss of the normal regulatory pathways involved in cell proliferation and cell death. Especially, molecular alterations of multiple pathways including Wnt (Wingless type)/adenomatous polyposis coli (APC), cyclooxygenase-2 (COX-2), and Ras are known to play important roles in progression of colorectal cancer. Recent progresses in the development of new chemotherapeutic agents have improved the prognosis of colorectal cancer patients [4]. However, for most patients with advanced colorectal cancer, it is still difficult to achieve a complete remission, especially with surgery or chemotherapy. Therefore, significant effort has been exerted to identify novel drug targets for both the prevention and treatment of colorectal cancer.
The peroxisome proliferator-activated receptors (PPARs) belong to members of the nuclear hormone receptor superfamily including receptors for steroid, retinoid, vitamin D, and thyroid hormones [5]. PPARs have received the attention of investigators interested in studying about the intracellular pathways that control signal transduction and gene transcription since their discovery in 1990. The name of PPARs was derived from its property to proliferate peroxisomes in rodent liver, where PPARα plays the major role. However, none of the PPARs could be contributed to peroxisome proliferation in humans [6]. PPARs are metabolic regulators involved in the regulation of glucose and lipid homeostasis. Ligand-activated PPAR forms heterodimer with the retinoid X receptor (RXR) and binds to a PPAR response element (PPRE) to regulate the transcription of numerous target genes [7, 8]. The target genes are involved in cell differentiation, proliferation, immune/inflammation response, and lipid metabolism. PPAR subfamily consists of three members such as PPARα, PPARβ/δ, and PPARγ. PPAR isoforms consist of activation domain (A/B), DNA-binding domain (C), hinge region (D), and ligand-binding domain (E). Each subtype has different characteristics as summarized in Figure 1. PPARα is expressed in brown adipose tissue, liver, kidney, heart, skeletal muscle, and enterocyte. Ligands for PPARα are fibrates, leukotriene B4, and so on. PPARα is involved in peroxisome proliferation, lipid catabolism, lipid-lowering effect, anti-inflammation, keratinocyte differentiation and proliferation, and skin wound healing. PPARβ/δ is ubiquitously expressed and is involved in reverse cholesterol transport, cell proliferation, apoptosis, and so on. PPARγ is expressed in adipose tissue, colon, immune system, hematopoietic cells, and retina. PPARγ is involved in lipid anabolism, adipocyte differentiation, control of inflammation, macrophage maturation, embryo implantation, and molecular targets of antidiabetic thiazolidinediones (Reviewed in [9]). Of the three PPARs identified to now, PPARγ represents the most promising target in view of the many reports implicating this molecule in cancer cell growth.
Figure 1.
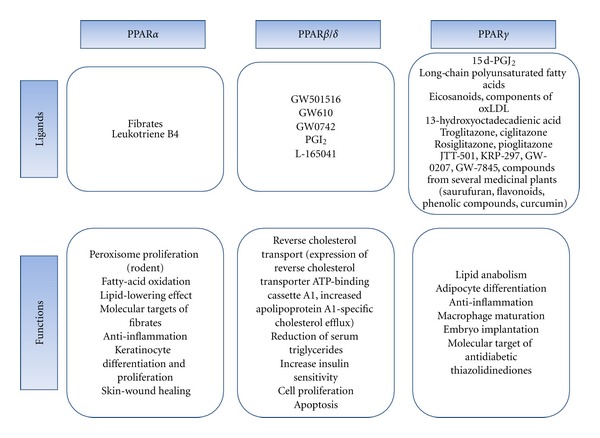
Summary of ligands and functions of each PPAR.
2. The Role of PPARα in Colorectal Cancer
Although the procarcinogenic effects of PPARα in rodent hepatocarcinoma are evident, less is known about the role of PPARα in human colorectal cancer. Previous studies showed that activation of PPARα by exogenous agonists causes inhibition of tumor cell growth in cell lines derived from colorectal cancer [10]. However, there is no evidence showing that PPARα expression is elevated in human cancers. Recent studies have shown that aspirin and other nonsteroidal anti-inflammatory drugs reduce the relative risk of developing colorectal cancers [11, 12]. The products of COX activity are known to be involved in carcinogenesis [13–15]. COX-2 is not expressed in most normal tissues but is induced upon stimulation by inflammatory agents, and also by oncogenes, growth factors, carcinogens, and tumor promoters [16–21]. Overexpression of COX-2 contributes to colorectal carcinogenesis by promoting the invasiveness of malignant cells, inhibiting apoptosis, and supporting angiogenesis [22–24]. Furthermore, human colorectal carcinoma patients with COX-2 positive tumors show a significantly poorer prognosis than those with tumors negative for COX-2 [25]. It was recently demonstrated that bile acids, particularly secondary bile acids such as lithocholic acid and chenodeoxycholic acid, can stimulate cell proliferation [26] and act as tumor promoters in colon carcinogenesis [27, 28]. Previous reports have suggested that endogenous bile acids are ligands for nuclear receptors such as farnesoid X receptor (FXR), pregnane X receptor (PXR), and vitamin D receptor (VDR) [29–32]. A recent study reported that bile acids also induce the expression of the PPARα gene via activation of FXR and leads to expression of COX-2 contributing to colorectal carcinogenesis [33]. These data suggest that PPARα has the tumor-promoting activity.
There is a growing importance of chemotherapy for malignant colon cancers. However, resistance to anticancer drugs is still a major obstacle in the failure of chemotherapy in colorectal cancer patients. Tong et al. demonstrated that decreased expression of PPARα confers resistance to hydroxycamptothecin, an inhibitor of topoisomerase I [34]. Thus, they suggest that increased expression of PPARα is necessary to overcome hydroxycamptothecin resistance even though its reason is not clarified.
3. The Role of PPARγ in Colorectal Cancer
The PPARγ is a ligand-activated transcription factor of the nuclear receptor superfamily [35, 36] and is expressed in a variety of malignant tissues including prostate, breast, and colon [37–41]. Upon activation, PPARγ forms heterodimer with RXR and mediate transcriptional activation by binding to the PPRE [7, 8]. In the inactive state, association of various corepressor molecules with PPARγ (e.g., nuclear receptor corepressor or silencing mediator for retinoid receptor and thyroid hormone receptors) prevents this complex from binding to DNA. For transcriptional transactivation of PPARγ, recruitment of coactivators (e.g., CCAAT/enhancer-binding protein, cyclic adenosine monophosphate response-element-binding protein, steroid receptor coactivator-1, receptor-interacting protein 140, PPARγ coactivator-1, and PPARγ binding protein) is required which replace co-repressors from the heterodimer complex. Transcriptional transrepression occurs through a genome independent mechanism and is mediated via physical association of the heterodimer with other activated transcription factors (STAT, NF-κB, and AP-1) thereby blocking their functions (reviewed in [42]). PPARγ has been known to be related to inflammation, immune response, and pathogenesis of some disorders including obesity, atherosclerosis, cancer, and so on [43]. There are natural ligands for PPARγ, including long-chain polyunsaturated fatty acids, eicosanoids, components of oxidized low density lipoproteins (oxLDL) [44], and oxidized alkyl phospholipids. The prostaglandin J2 derivative, 15d-PGJ2 is the most potent endogenous ligand for the PPARγ receptor. The antidiabetic thiazolidinedione (TZD) class of drugs including troglitazone, rosiglitazone, pioglitazone and ciglitazone are synthetic ligands for PPARγ [44]. Recent studies have focused on the effect of PPARγ ligands as anticancer agents. However, there are still controversies about the antitumor activity of PPARγ agonists. Thus, this paper describes the role of PPARγ in colorectal cancer and its detailed mechanisms clarified until now.
3.1. The Role of PPARγ as a Tumor Suppressor in Colorectal Cancer
Several studies have focused on the putative association between the various polymorphisms and mutations of the PPARγ gene and the occurrence of cancer. It was described that 4 somatic PPARγ gene mutations resulting in reducing its function occurred in 55 sporadic colon cancers [45]. However, Ikezoe et al. [46] analyzed 397 clinical samples and cell lines including colon, breast, and lung cancers for mutations of PPARγ gene and showed the absence of PPARγ gene mutations. These data suggest that PPARγ mutations may occur in cancers but they are rare.
There has been substantial accumulation of experimental data supporting that synthetic PPARγ ligands as well as 15d-PGJ2 induce apoptosis in several types of cancer cells [41, 43]. Although increasing evidence has established that PPARγ agonist induces growth arrest in cancer cells, the molecular mechanism of the growth inhibition by PPARγ agonist is not well understood and complicated. This paper describes some of the molecular mechanisms for anticancer activity of PPARγ (Table 1 and Figure 2) as follows.
Table 1.
Potential molecular mechanisms for anticancer activity of PPARγ.
| Actions and molecular mechanisms | References |
|---|---|
| (1) Inhibition of cell proliferation and induction of apoptosis | |
| (1) Upregulation of PTEN | [47, 48] |
| (2) Downregulation of survivin | [49] |
| (3) Downregulation of XIAP | [50, 51] |
| (4) Suppression of NF-κB | [52] |
| (5) Up-regulation of cyclin-dependent kinase (CDK) inhibitors, down-regulation of CDK, and down-regulation of cyclin D1 |
[51, 53–57] |
| (6) Down-regulation of COX-2 | [51] |
| (7) Up-regulation of Krüppel-like Factor4 (KLF4) | [58, 59] |
| (8) Up-regulation of Bax and down-regulation of Bcl-2 | [51, 60, 61] |
| (9) Inhibition of telomerase activity and hTERT expression | [62] |
| (2) Induction of cellular differentiation | |
| Up-regulation of E-cadherin and Drg-1 gene expression | [63] |
| (3) Inhibition of angiogenesis | |
| (1) Down-regulation of vascular endothelial growth factor (VEGF) | [64] |
| (2) Down-regulation of matrix metalloproteinases (MMP) | [65] |
| (3) Down-regulation of iNOS and COX-2 | [66–72] |
| (4) Down-regulation of proinflammatory mediators | [73] |
Figure 2.
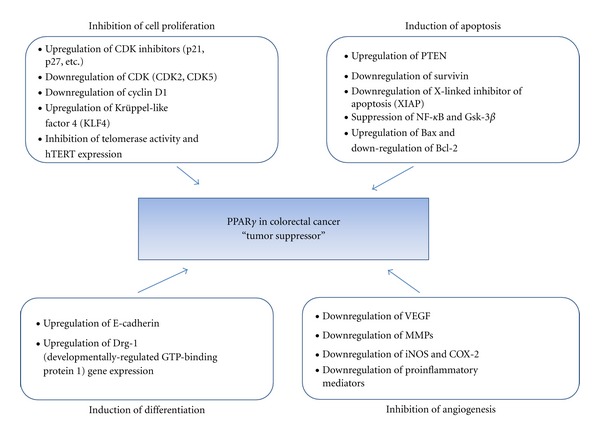
Potential molecular mechanisms for PPARγ as tumor suppressor in colorectal cancer.
3.1.1. Inhibition of Cell Proliferation and Induction of Apoptosis
(1) Upregulation of PTEN —
The Phosphatase and Tensin Homolog (PTEN) tumor suppressor gene modulates several cellular functions, including cell migration, survival, and proliferation by inhibiting phosphatidylinositol 3-kinase (PI-3K)-mediated signaling cascades [74]. Previous studies have demonstrated that rosiglitazone, a synthetic ligand for PPARγ, upregulates PTEN expression in Caco2 colorectal cancer cells [47]. Dai et al. also show that treatment of colon cancer cells with rosiglitazone stimulates expression of tumor suppressor gene PTEN. This effect is probably mediated through the binding of PPARγ on PPRE in the promoter of PTEN [48]. Inhibition of the PI-3K/Akt pathway by increased PTEN expression is believed to underlie this effect of the PPARγ ligand.
(2) Downregulation of Survivin —
Survivin is one of the inhibitors of apoptosis protein (IAP) family since it is overexpressed in almost every human tumor that has been studied, but is barely detectable in most normal adult tissues [75]. Overexpression of survivin is associated with poor clinical outcome with reduced tumor cell apoptosis in patients with colorectal cancer [76, 77]. PPARγ agonist GW7845 induced cell death through downregulation of survivin in colorectal cancer cells [49].
(3) Downregulation of X-Linked Inhibitor of Apoptosis (XIAP) —
XIAP can inhibit apoptosis by binding and thereby inactivating caspases including caspase-9 and the effector caspases (-3 and -7) [78]. Qiao et al. showed that 15d-PGJ2 and troglitazone mediate XIAP downregulation in colon cancer cells by facilitating ubiquitination and proteasomal degradation [50]. In addition, Lee et al. demonstrated that pioglitazone induces apoptosis through downregulation of XIAP via unknown mechanism in colorectal cancer cell lines [51].
(4) Suppression of NF-κB and GSK-3β —
The transcription factor NF-κB is involved in the regulation of various genes, including metalloproteinases (MMPs), inflammatory response genes, and a number of antiapoptotic genes including cIAP1, cIAP2, and glycogen synthase kinase-3 (GSK-3) [79]. Its activation is also associated with cell proliferation, cell cycle progression, promotion of tumor growth, angiogenesis, and metastasis through the expression of genes participating in malignant conversion and tumor promotion [80–82]. Ban et al. showed that PPARγ agonist, troglitazone inhibits colon cancer cell growth via inactivation of NF-κB by suppressing GSK-3β activity [52].
(5) Upregulation of Cyclin-Dependent Kinase (CDK) Inhibitors, Downregulation of CDK and Downregulation of Cyclin D1 —
Interestingly, CDK5 protein expression and kinase activity were significantly inhibited by ciglitazone, which was associated with ciglitazone-induced antiproliferation in colon cancer HT-29 cells [53]. Cyclin D1 is involved in G1/S progression and increased proliferation. PPARγ activation in intestinal epithelial cells results in the inhibition of cell cycle and S-phase entry though a decrease in cyclin D1 expression [54, 55]. PPARγ ligand treatment not only decreases the protein level of cyclin D1, but also increases the CDK inhibitors p21CIP and p27KIP1 through both increased transcriptional activity and inhibition of proteasome degradation in colorectal cancer cells [56, 57]. Ciglitazone also inhibited G1/S cell cycle progression through upregulation of p27 and inhibition of Cdk2 activity in HT-29 cells [56]. Fajas et al. [83] suggested that PPARγ activation in the presence of RB results in G1 arrest, whereas in the absence of RB, cells accumulate in G2/M, endoduplicate, and undergo apoptosis. Lee et al. [51] also showed that pioglitazone treatment leads to G2/M block through downregulation of cyclin B1 and cdc2 and upregulation of p21 in RB-deficient human colorectal cancer SNU-C4 and SNU-C2A cells. Thus, these studies suggest that the antiproliferative or proapoptotic effects of PPARγ agonist are associated with its ability to regulate the expression of various genes which are involved in controlling the cell cycle and cell survival/death.
(6) Downregulation of COX-2 —
Most of the current studies showed that COX-2 contributes to tumorigenesis through various mechanisms and overexpression of COX-2 can stimulate tumor growth, invasion, and metastasis [84, 85]. A previous study showed that pioglitazone induces apoptosis through the downregulation of COX-2, activation of caspase-3, downregulation of Bcl-2 and upregulation of Bax in RB-deficient human colorectal cancer cells [51].
(7) Upregulation of Krüppel-Like Factor 4 (KLF4) —
KLF4 is a member of the Krüppel-like zinc finger transcription factor family. It is extensively expressed in the epithelial cells of the gastrointestinal tract [86–88]. Over-expression of KLF4 in colon cancer cells caused inhibition of DNA synthesis and cell growth [89, 90]. Zhi and Tseng demonstrated that 15d-PGJ2 inhibits proliferation of HT-29 human colon cancer cells and induces upregulation of KLF4 mRNA and protein through the activation of MEK/ERK and STAT-dependent pathway [58]. They provided a novel mechanism for the antitumorigenic actions of 15d-PGJ2. In addition, rosiglitazone treatment of colorectal cancer cells caused to G1 arrest because increased expression of KLF4 by rosiglitazone leads to increased expression of p21 and decreased expression of cyclin D1 [59]. These data suggest that KLF4 is a nodal player in a network of PPARγ-regulated genes.
(8) Upregulation of Bax and Downregulation of Bcl-2 —
In colon cancer cells, treatment of the PPARγ ligands (pioglitazone, troglitazone) induces apoptosis through upregulation of the proapoptotic protein Bax and downregulation of the antiapoptpotic protein Bcl-2 [51, 60, 61]. Alternative expression of Bax and Bcl-2 causes apoptosis by the release of cytochrome c and subsequent activation of several effector caspases.
(9) Inhibition of Telomerase Activity and hTERT Expression through Modulation of the Myc/Mad/Max Network —
The telomerase stabilizes telomere length by adding TTAGGG repeats to telomeres [91, 92]. Telomerase activity has been detected in almost all human tumors [93, 94] but not in adjacent normal cells [95, 96]. Human telomerase is composed of human telomerase RNA, telomerase-associated protein 1 and human telomerase reverse transcriptase (hTERT) [91, 97]. The forced expression of hTERT in normal human cells has been reported to increase their lifespan [98], while the expression of dominant-negative hTERT in human cancer cells has been known to inhibit telomerase and cause telomere shortening [99, 100]. A recent study shows that 15d-PGJ2 and rosiglitazone inhibit Caco-2 colon cancer cell proliferation through the inhibition of telomerase activity and hTERT expression. In addition, it was demonstrated that the inhibition of hTERT expression in Caco-2 cells depends on the downregulation of c-Myc and the upregulation of Mad 1 by PPARγ ligands [62].
3.1.2. Induction of Cellular Differentiation
PPARγ has been demonstrated to induce differentiation in solid tumors both in vitro and in vivo [101]. In colon cancer cells, activation of PPARγ by troglitazone treatment inhibits growth and metastasis through differentiation-promoting effects, such as the marked increase in p21 Waf-1, developmentally regulated GTP-binding protein 1 (DRG-1), and E-cadherin in human colon cancer cells [63]. These effects involve modulation of the E-cadherin/β-catenin system and upregulation of Drg-1 gene expression.
3.1.3. Inhibition of Angiogenesis
Angiogenesis, a formation of new capillaries from the preexisting vessels, is a complex process involved in the degradation of the basement membrane by cellular proteases, the penetration and migration of endothelial cells into the extracellular matrix, endothelial cell proliferation, tube formation, and vessel stabilization [102]. Inhibition of angiogenesis may contribute to the mechanism by which PPARγ agonists halt the cancer process. Several studies demonstrated that PPARγ agonist inhibits angiogenesis through the following mechanisms.
(1) Downregulation of Vascular Endothelial Growth Factor (VEGF) —
VEGF is involved in angiogenesis [103, 104]. VEGF expression is increased in several cancers including colorectal and other tumors [105, 106]. It was shown that rosiglitazone inhibited angiogenesis via the downregulation of VEGF and VEGF mRNA in pancreatic cancer xenografts [64].
(2) Downregulation of Matrix Metalloproteinases (MMPs) —
The process of cancer cell invasion is dependent on the degradation of the extracellular matrix (ECM) by MMPs. MMPs are a family of proteases cleaving several macromolecules of the ECM [107]. 15d-PGJ2 has been reported to have inhibitory effects on the proliferation and invasiveness of colon cancer cell lines which are associated with G1 cell cycle arrest and downregulation of MMP-7 synthesis [65].
(3) Downregulation of iNOS and COX-2 —
It has been shown that both COX-2 and inducible nitric oxide synthase (iNOS) are overexpressed in various human cancers [108]. It was reported that iNOS is associated with altered expression of important modulators of angiogenesis [108]. 15d-PGJ2 downregulates iNOS [66–68] and COX-2 [69–71]. The expression of COX-2 and iNOS is regulated by NF-κB. The recent several studies have demonstrated that 15d-PGJ2 can act as a negative regulator of proinflammatory signaling through blocking the NF-κB activation pathway at multiple levels via covalent modification of NF-κB or its regulators [72]. Thus, antiangiogenic effects of 15d-PGJ2 might be associated with disruption of NF-κB and subsequent blockade of iNOS and COX-2 expression.
(4) Downregulation of Proinflammatory Mediators —
The potential mechanism of angiogenesis inhibition by 15d-PGJ2 may involve downregulation of pro-inflammatory mediators. Both physiological and pathological angiogenesis can be stimulated by pro-inflammatory cytokines, such as IL-1 and TNF-α. Certain cytokines (e.g., IL-6 and CSF-1) can influence the phenotype and the function of tumor-associated macrophages and indirectly stimulate tumor invasiveness and angiogenesis [109]. Tumor-associated macrophages play an important role in tumor progression due to production of several angiogenic factors, such as VEGF, IL-8, inflammatory cytokines (IL-1 and IL-10) and proteases (MMP-2 and MMP-9) [109]. Thus, 15d-PGJ2 inhibits angiogenesis through suppression of such pro-inflammatory cytokines [73]. Induction of several pro-inflammatory cytokines, such as TNF-α, IL-1, and IL-8, is regulated at the transcription level by NF-κB. It is still unclear whether 15d-PGJ2 exerts an anti-angiogenic effect through inhibition of NF-κB-dependent induction of pro-inflammatory mediators or through downregulation of cancer cell-derived pro-inflammatory cytokine release which is NF-κB-independent. Hence, further investigations are necessary to clarify the signaling pathways that delineate the anti-angiogenic effects of 15d-PGJ2.
3.2. The Role of PPARγ as a Tumor Promoter in Colorectal Cancer
In contrast to above described, PPARγ has been known to have procarcinogenic activity such as stimulation of tumor cell growth and induction of angiogenesis. This review describes some mechanisms for it as summarized in Table 2 and Figure 3.
Table 2.
Potential molecular mechanisms for procarcinogenic activity of PPARγ.
Figure 3.
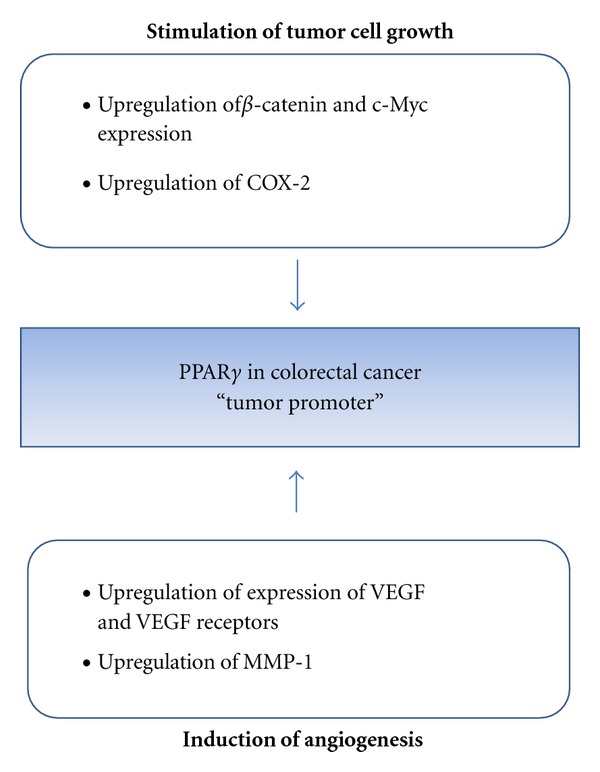
Potential molecular mechanisms for PPARγ as tumor promoter in colorectal cancer.
3.2.1. Stimulation of Tumor Cell Growth
Although the majority of publications indicate that PPARγ agonists have potent antiproliferative properties in several types of cancer cells, there are some reports demonstrating the cell growth promoting effects of 15d-PGJ2 and other PPARγ ligands. It was shown that activation of PPARγ by troglitazone increased the frequency and the size of colon tumors in C57BL/6J-APCMin/+ mice [116, 117]. In addition, a recent study shows that low concentration of 15d-PGJ2 and pioglitazone can promote the growth of APC-mutated HT-29 colon cancer cells in vitro and in vivo [110].
(1) Upregulation of β-Catenin and c-Myc Expression —
The Wnt/β-catenin pathway plays a critical role in the development of colon cancer [118]. Choi et al. showed that low concentrations of 15d-PGJ2 and pioglitazone promote the HT-29 colon cancer cells in vitro and in vivo through increase in β-catenin and c-Myc expression [110].
(2) Upregulation of COX-2 —
As previously mentioned, 15d-PGJ2 is one of the major final products of COX-2. Since abnormal overexpression of COX-2 was observed in several cancer cells, COX-2 has been shown to contribute to carcinogenesis by promoting cell proliferation and angiogenesis as well as by protecting cells from apoptosis [119]. The regulation of COX-2 synthesis occurs mainly at the transcriptional level, although mRNA stabilization is also involved. A recent study has shown that 15d-PGJ2 enhances COX-2 expression through ROS-Akt-driven AP-1 activation in human breast cancer cells [111].
3.2.2. Induction of Angiogenesis
It has been reported that PPARγ agonist can induce angiogenesis in various cell lines. Several studies provided some molecular mechanisms for induction of angiogenesis by PPARγ agonist. Here, this paper summarizes the potential molecular mechanisms for enhanced metastasis and invasion by PPARγ agonist clarified until now.
(1) Upregulation of Expression of VEGF and VEGF Receptors —
It was shown that the mRNA expression of VEGF was augmented by 15d-PGJ2 and troglitazone in vascular smooth muscle cell, human monocytes/macrophages, human colorectal cancer cells and human coronary artery endothelial cells [112, 113]. More recently, 15d-PGJ2 and troglitazone have been reported to increase the expression of VEGF and its receptors (Flt-1 and KDR) in myofibroblasts [114].
(2) Upregulation of MMP-1 —
Kim et al. reported that 15d-PGJ2 enhances the angiogenesis by upregulation of MMP-1 [115]. MMP-1 is a major proteinase degrading native fibrillar collagens. MMP-1 is produced by a variety of cell types, including endothelium. It is implicated in several pathological processes such as tumor invasion and restenosis [120]. In addition, Kim et al. suggested that iron may contribute to increased metastasis and invasiveness by 15d-PGJ2 in human breast cancer cells [115]. Thus, these studies suggest the regulation of MMP-1 expression by 15d-PGJ2 may be more complex than expected.
4. The Role of PPARβ/δ in Colorectal Cancer
PPARβ/δ is also expressed in the colon and can be activated by fatty acids. In recent studies, it was shown that PPARβ/δ plays a central role in the differentiation of Paneth cells and innate immunity [121]. The role of PPARβ/δ in colorectal cancer is more controversial than that of PPARγ. Recent studies have shown that PPARβ/δ is involved in the pathogenesis of colorectal cancer [122]. Inactivation of APC upregulates PPARβ/δ expression in colorectal cancer cells [122]. It has also been reported that PPARβ/δ levels increase in colorectal tumor after treatment with the potent carcinogen azoxymethane (AOM) [123]. The increased expression of PPARβ/δ could potentially be activated by endogenous ligands such as COX-derived prostacyclin [123]. It was proposed that PPARβ/δ activation would initiate the expression of target genes, which still remain to be identified, and enhance cell growth. In support of this model, PPARβ/δ-null HCT116 cells have reduced tumorigenecity in a xenograft model [124]. PPARβ/δ expression levels in colorectal cancers are higher than in normal mucosa, supporting the hypothesis that APC suppresses activity of β-catenin/Tcf-4 transcription of target genes, including PPARβ/δ, c-myc, and cyclin D1 [122, 123]. PPARβ/δ expression and activity are also induced by oncogenic K-ras in rat intestinal epithelial cells [125]. These studies support a procarcinogenic role of PPARβ/δ in colorectal cancer.
A few mechanisms have been proposed to explain the procarcinogenic effect of PPARβ/δ. Di-Poï et al. suggested that PPARβ/δ activation increases the expression of 3-phosphoinositide-dependent-protein kinae 1 (PDPK1) and integrin-linked kinase (ILK), and decreases the expression of PTEN, causing increased phosphorylation of AKT, leading to antiapoptotic signaling and enhanced cell survival [126]. Another related mechanism is derived from the observation that ligand activation of PPARβ/δ increases the expression of VEGF through a PPARβ/δ-dependent mechanism, causing increased phosphorylation of AKT, which promotes cell survival by blocking apoptosis [127]. In addition, Kwak et al. [128] demonstrated that PPARβ/δ-binding aptamers suppressed transcription from natural promoters of VEGF-A and COX-2 and inhibited tumorigenic potential of colon-cancer cells. These data suggest that PPARβ/δ play an important role in transcription of tumor-promoting genes such as VEGF-A and COX-2.
However, other studies conflict with those reports. Targeted deletion of APC alleles reduces PPARβ/δ expression in mouse intestine [129]. PPARβ/δ expression in human colorectal cancers or intestinal polyps of APCMin/+ mice are either unchanged or downregulated as compared with normal controls (reviewed in [130, 131]).
The conflicting results about the effect of PPARβ/δ on intestinal tumorigenesis in APCMin/+- and AOM-treated mice may be related to differences in the specific targeting strategy employed to delete PPARβ/δ [127]. Deletion of PPARβ/δ exon 4 and/or 5, which encodes an essential portion of the DNA-binding domain, is thought to disrupt PPARβ/δ function as a nuclear transcriptional factor and to inhibit colonic carcinogenesis [127, 132]. Increased expression of VEGF in colon tumors was suppressed by loss of PPARβ/δ expression [133]. These findings indicate that PPARβ/δ has an important role in promoting colonic tumorigenesis. The deletion of exon 8 [134, 135], the last PPARβ/δ exon, is postulated to generate a hypomorphic PPARβ/δ protein that remains at least partly functional.
In a mouse mammary tumor model, treatment with the PPARβ/δ agonist GW501516 accelerated tumor formation, while a PPARγ agonist GW7845 delayed tumor growth [136]. This observation suggests that there are distinct mechanistic differences between PPARγ and PPARβ/δ in regulating tumor progression. A recent study showed that PPARβ/δ confers resistance to PPARγ-induced apoptosis by increasing the expression of survivin [49].
Recently, Yang et al. [137] showed that the specific knockdown of PPARβ/δ in colon-cancer cell lines results in more malignant morphologies, larger colonies and less CEA production, and enhances cell-fibronectin adhesion, without effects on cell invasion and migration. These findings indicate that PPARβ/δ may facilitate differentiation and inhibit the cell-fibronectin adhesion of colon cancer, having a protective role in the carcinogenesis and progression of colon cancer. Further immunohistochemistry data reveal that the expression of PPARβ/δ is closely associated with the differentiation and tumor-node-metastasis stage of rectal cancer. It was also shown that PGI2 and L-165041, a synthetic PPARδ ligand, activate PPARδ and upregulate PPARδ-mediated 14-3-3ε expression. 14-3-3ε binds and sequesters Bad in cytosol. PGI2-induced 14-3-3ε upregulation is accompanied by augmented Bad sequestration and protects HT-29 cells from Bad-triggered mitochondrial leakage of proapoptotic factors and the consequent apoptosis [138].
5. Conclusion and Future Directions
Even though the extensive studies to clarify the role of PPARs in colorectal cancer using several PPAR agonists and gene knockout experiments were performed, there are still many controversies about them. PPAR ligands induce many physiological changes, including increased oxidation of fatty acids, which contributes to decreasing serum lipids and reducing body weight; and inhibition of inflammatory signaling. There are good reasons to suggest that PPAR agonists should be potential candidates for treating and preventing colorectal cancer, because obesity and chronic inflammation are major risk factors for colorectal cancer. It is interesting to note that there is an overlap in target genes regulated by each PPAR, but the physiological effects induced by selective PPAR agonists are unique owing to the complexity of the PPAR-dependent and the PPAR-independent effects that each agonist induces. To completely understand the role of PPARs in colorectal cancer, it is necessary to dissect the complex regulation of PPAR expression and to examine interactions of each PPAR with other nuclear receptors and signalling molecules involved in cell proliferation and cell death in the near future.
Acknowledgment
This study was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korea Ministry Education, Science and Technology (MEST) (R13-2002-044-05002-0).
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer Journal for Clinicians. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer Journal for Clinicians. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer Journal for Clinicians. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 4.Waters J, Cunningham D. The changing face of chemotherapy in colorectal cancer. British Journal of Cancer. 2001;84(1):1–7. doi: 10.1054/bjoc.2000.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor super-family: the second decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 7.Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochimica et Biophysica Acta. 1996;1302(2):93–109. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- 8.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. Journal of Medicinal Chemistry. 2000;43(4):527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 9.Park JI. Cellular and Genetic Practices for Translational Medicine. Research Signpost; 2011. The role of 15d-PGJ2, a natural ligand for peroxisome proliferator-activated receptor γ (PPARγ), in cancer; pp. 169–195. [Google Scholar]
- 10.Grau R, Punzón C, Fresno M, Iñiguez MA. Peroxisome-proliferator-activated receptor α agonists inhibit cyclo-oxygenase 2 and vascular endothelial growth factor transcriptional activation in human colorectal carcinoma cells via inhibition of activator protein-1. Biochemical Journal. 2006;395(1):81–88. doi: 10.1042/BJ20050964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenberg L, Palmer JR, Zauber AG, Warshauer ME, Stolley PD, Shapiro S. A hypothesis: nonsteroidal anti-inflammatory drugs reduce the incidence of large bowel cancer. Journal of the National Cancer Institute. 1991;83(5):355–358. doi: 10.1093/jnci/83.5.355. [DOI] [PubMed] [Google Scholar]
- 12.Reddy BS, Rao CV, Seibert K. Evaluation of cyclooxygenase-2 inhibitor for potential chemopreventive properties in colon carcinogenesis. Cancer Research. 1996;56(20):4566–4569. [PubMed] [Google Scholar]
- 13.Thun MJ, Namboodiri MM, Heath CW., Jr. Aspirin use and reduced risk of fatal colon cancer. The New England Journal of Medicine. 1991;325(23):1593–1596. doi: 10.1056/NEJM199112053252301. [DOI] [PubMed] [Google Scholar]
- 14.Logan RFA, Little J, Hawtin PG, Hardcastle JD. Effect of aspirin and non-steroidal anti-inflammatory drugs on colorectal adenomas: case-control study of subjects participating in the Nottingham faecal occult blood screening programme. British Medical Journal. 1993;307(6899):285–289. doi: 10.1136/bmj.307.6899.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schreinemachers DM, Everson RB. Aspirin use and lung, colon, and breast cancer incidence in a prospective study. Epidemiology. 1994;5(2):138–146. doi: 10.1097/00001648-199403000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. The Journal of Biological Chemistry. 1991;266(20):12866–12872. [PubMed] [Google Scholar]
- 17.Jones DA, Carlton DP, McIntyre TM, Zimmerman GA, Prescott SM. Molecular cloning of human prostaglandin endoperoxide synthase type II and demonstration of expression in response to cytokines. The Journal of Biological Chemistry. 1993;268(12):9049–9054. [PubMed] [Google Scholar]
- 18.DuBois RN, Awad J, Morrow J, Roberts LJ, II, Bishop PR. Regulation of eicosanoid production and mitogenesis in rat intestinal epithelial cells by transforming growth factor-α and phorbol ester. Journal of Clinical Investigation. 1994;93(2):493–498. doi: 10.1172/JCI116998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue H, Yokoyama C, Hara S, Tone Y, Tanabe T. Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. The Journal of Biological Chemistry. 1995;270(42):24965–24971. doi: 10.1074/jbc.270.42.24965. [DOI] [PubMed] [Google Scholar]
- 20.Subbaramaiah K, Telang N, Ramonetti JT, et al. Transcription of cyclooxygenase-2 is enhanced in transformed mammary epithelial cells. Cancer Research. 1996;56(19):4424–4429. [PubMed] [Google Scholar]
- 21.Mestre JR, Subbaramaiah K, Sacks PG, et al. Retinoids suppress epidermal growth factor-induced transcription of cyclooxygenase-2 in human oral squamous carcinoma cells. Cancer Research. 1997;57(14):2890–2895. [PubMed] [Google Scholar]
- 22.Tsujii M, Kawano S, Dubois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(7):3336–3340. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsujii M, Dubois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83(3):493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 24.Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, Dubois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93(5):705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 25.Masunaga R, Kohno H, Dhar DK, et al. Cyclooxygenase-2 expression correlates with tumor neovascularization and prognosis in human colorectal carcinoma patients. Clinical Cancer Research. 2000;6(10):4064–4068. [PubMed] [Google Scholar]
- 26.Craven PA, Pfanstiel J, DeRubertis FR. Role of activation of protein kinase C in the stimulation of colonic epithelial proliferation and reactive oxygen formation by bile acids. Journal of Clinical Investigation. 1987;79(2):532–541. doi: 10.1172/JCI112844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narisawa T, Magadia NE, Weisburger JH, Wynder EL. Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-methyl-N′-nitro-N-nitrosoguanidine in rats. Journal of the National Cancer Institute. 1974;53(4):1093–1097. doi: 10.1093/jnci/53.4.1093. [DOI] [PubMed] [Google Scholar]
- 28.Allinger UG, Johansson GK, Gustafsson JA, Rafter JJ. Shift from a mixed to a lactovegetarian diet: influence on acidic lipids in fecal water—a potential risk factor for colon cancer. American Journal of Clinical Nutrition. 1989;50(5):992–996. doi: 10.1093/ajcn/50.5.992. [DOI] [PubMed] [Google Scholar]
- 29.Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bite acids. Science. 1999;284(5418):1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 30.Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284(5418):1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 31.Staudinger JL, Goodwin B, Jones SA, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(6):3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makishima M, Lu TT, Xie W, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296(5571):1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 33.Oshio H, Abe T, Onogawa T, et al. Peroxisome proliferator-activated receptor α activates cyclooxygenase-2 gene transcription through bile acid transport in human colorectal cancer cell lines. Journal of Gastroenterology. 2008;43(7):538–549. doi: 10.1007/s00535-008-2188-3. [DOI] [PubMed] [Google Scholar]
- 34.Tong JL, Zhang CP, Nie F, et al. MicroRNA 506 regulates expression of PPARα in hydroxycamptothecin-resistant human colon cancer cells. FEBS Letters. 2011;585(22):3560–3568. doi: 10.1016/j.febslet.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 35.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240(4854):889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schoonjans K, Martin G, Staels B, Auwerx J. Peroxisome proliferator-activated receptors, orphans with ligands and functions. Current Opinion in Lipidology. 1997;8(3):159–166. doi: 10.1097/00041433-199706000-00006. [DOI] [PubMed] [Google Scholar]
- 37.Mansure JJ, Nassim R, Kassouf W. Peroxisome proliferator-activated receptor γ in bladder cancer: a promising therapeutic target. Cancer Biology and Therapy. 2009;8(7):1–9. doi: 10.4161/cbt.8.7.7853. [DOI] [PubMed] [Google Scholar]
- 38.Dubois RN, Gupta R, Brockman J, Reddy BS, Krakow SL, Lazar MA. The nuclear eicosanoid receptor, PPARγ, is aberrantly expressed in colonic cancers. Carcinogenesis. 1998;19(1):49–53. doi: 10.1093/carcin/19.1.49. [DOI] [PubMed] [Google Scholar]
- 39.Sarraf P, Mueller E, Jones D, et al. Differentiation and reversal of malignant changes in colon cancer through PPARγ . Nature Medicine. 1998;4(9):1046–1052. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- 40.Mueller E, Sarraf P, Tontonoz P, et al. Terminal differentiation of human breast cancer through PPARγ . Molecular Cell. 1998;1(3):465–470. doi: 10.1016/s1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- 41.Kubota T, Koshizuka K, Williamson EA, et al. Ligand for peroxisome proliferator-activated receptor γ (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer Research. 1998;58(15):3344–3352. [PubMed] [Google Scholar]
- 42.Krishnan A, Nair SA, Pillai MR. Biology of PPARγ in cancer: a critical review on existing lacunae. Current Molecular Medicine. 2007;7(6):532–540. doi: 10.2174/156652407781695765. [DOI] [PubMed] [Google Scholar]
- 43.de la Lastra CA, Sánchez-Fidalgo S, Villegas I, Motilva V. New pharmacological perspectives and therapeutic potential of PPAR-γ agonists. Current Pharmaceutical Design. 2004;10(28):3505–3524. doi: 10.2174/1381612043382909. [DOI] [PubMed] [Google Scholar]
- 44.Wang T, Xu J, Yu X, Yang R, Han ZC. Peroxisome proliferator-activated receptor γ in malignant diseases. Critical Reviews in Oncology/Hematology. 2006;58(1):1–14. doi: 10.1016/j.critrevonc.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 45.Sarraf P, Mueller E, Smith WM, et al. Loss-of-function mutations in PPARγ associated with human colon cancer. Molecular Cell. 1999;3(6):799–804. doi: 10.1016/s1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 46.Ikezoe T, Miller CW, Kawano S, et al. Mutational analysis of the peroxisome proliferator-activated receptor γ in human malignancies. Cancer Research. 2001;61(13):5307–5310. [PubMed] [Google Scholar]
- 47.Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARγ agonists are mediated via upregulation of PTEN. Current Biology. 2001;11(10):764–768. doi: 10.1016/s0960-9822(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 48.Dai Y, Qiao L, Chan KW, et al. Loss of XIAP sensitizes rosiglitazone-induced growth inhibition of colon cancer in vivo. International Journal of Cancer. 2008;122(12):2858–2863. doi: 10.1002/ijc.23443. [DOI] [PubMed] [Google Scholar]
- 49.Wang D, Ning W, Xie D, Guo L, Dubois RN. Peroxisome proliferator-activated receptor δ confers resistance to peroxisome proliferator-activated receptor γ-induced apoptosis in colorectal cancer cells. Oncogene. 2012;31(8):1013–1023. doi: 10.1038/onc.2011.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiao L, Dai Y, Gu Q, et al. Downregulation of X-linked inhibitor of apoptosis synergistically enhanced peroxisome proliferator-activated receptor γ ligand-induced growth inhibition in colon cancer. Molecular Cancer Therapeutics. 2008;7(7):2203–2211. doi: 10.1158/1535-7163.MCT-08-0326. [DOI] [PubMed] [Google Scholar]
- 51.Lee CJ, Han JS, Seo CY, et al. Pioglitazone, a synthetic ligand for PPARγ, induces apoptosis in RB-deficient human colorectal cancer cells. Apoptosis. 2006;11(3):401–411. doi: 10.1007/s10495-006-4003-z. [DOI] [PubMed] [Google Scholar]
- 52.Ban JO, Kwak DH, Oh JH, et al. Suppression of NF-κB and GSK-3β is involved in colon cancer cell growth inhibition by the PPAR agonist troglitazone. Chemico-Biological Interactions. 2010;188(1):75–85. doi: 10.1016/j.cbi.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 53.Kim E, Chen F, Wang CC, Harrison LE. CDK5 is a novel regulatory protein in PPARγ ligand-induced antiproliferation. International Journal of Oncology. 2006;28(1):191–194. [PubMed] [Google Scholar]
- 54.Kitamura S, Miyazaki Y, Hiraoka S, et al. PPARγ agonists inhibit cell growth and suppress the expression of cyclin D1 and EGF-like growth factors in ras-transformed rat intestinal epithelial cells. International Journal of Cancer. 2001;94(3):335–342. doi: 10.1002/ijc.1470. [DOI] [PubMed] [Google Scholar]
- 55.Chen A, Xu J. Activation of PPARγ by curcumin inhibits Moser cell growth and mediates suppression of gene expression of cyclin D1 and EGFR. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2005;288(3):G447–G456. doi: 10.1152/ajpgi.00209.2004. [DOI] [PubMed] [Google Scholar]
- 56.Chen F, Harrison LE. Ciglitazone-induced cellular anti-proliferation increases p27 kip1 protein levels through both increased transcriptional activity and inhibition of proteasome degradation. Cellular Signalling. 2005;17(7):809–816. doi: 10.1016/j.cellsig.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 57.Chen F, Kim E, Wang CC, Harrison LE. Ciglitazone-induced p27 gene transcriptional activity is mediated through Sp1 and is negatively regulated by the MAPK signaling pathway. Cellular Signalling. 2005;17(12):1572–1577. doi: 10.1016/j.cellsig.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 58.Zhi YC, Tseng CC. 15-Deoxy-Δ12,14 prostaglandin J2 upregulates krüppel-like factor 4 expression independently of peroxisome proliferator-activated receptor γ by activating the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signal transduction pathway in HT-29 colon cancer cells. Molecular Pharmacology. 2005;68(5):1203–1213. doi: 10.1124/mol.105.014944. [DOI] [PubMed] [Google Scholar]
- 59.Rageul J, Mottier S, Jarry A, et al. KLF4-dependent, PPARγ-induced expression of GPA33 in colon cancer cell lines. International Journal of Cancer. 2009;125(12):2802–2809. doi: 10.1002/ijc.24683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen GG, Xu H, Lee JFY, et al. 15-Hydroxy-eicosatetraenoic acid arrests growth of colorectal cancer cells via a peroxisome proliferator-activated receptor gamma-dependent pathway. International Journal of Cancer. 2003;107(5):837–843. doi: 10.1002/ijc.11447. [DOI] [PubMed] [Google Scholar]
- 61.Chen GG, Lee JFY, Wang SH, Chan UPF, Ip PC, Lau WY. Apoptosis induced by activation of peroxisome-proliferator activated receptor-gamma is associated with Bcl-2 and NF-κB in human colon cancer. Life Sciences. 2002;70(22):2631–2646. doi: 10.1016/s0024-3205(02)01510-2. [DOI] [PubMed] [Google Scholar]
- 62.Toaldo C, Pizzimenti S, Cerbone A, et al. PPARγ ligands inhibit telomerase activity and hTERT expression through modulation of the Myc/Mad/Max network in colon cancer cells. Journal of Cellular and Molecular Medicine. 2010;14(6):1347–1357. doi: 10.1111/j.1582-4934.2009.00966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshizumi T, Ohta T, Ninomiya I, et al. Thiazolidinedione, a peroxisome proliferator-activated receptor-gamma ligand, inhibits growth and metastasis of HT-29 human colon cancer cells through differentiation-promoting effects. International Journal of Oncology. 2004;25(3):631–639. [PubMed] [Google Scholar]
- 64.Dong YD, Wang XP, Wu K. Suppression of pancreatic carcinoma growth by activating peroxisome proliferator-activated receptor γ involves angiogenesis inhibition. World Journal of Gastroenterology. 2009;15(4):441–448. doi: 10.3748/wjg.15.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shen D, Deng C, Zhang M. Peroxisome proliferator-activated receptor γ agonists inhibit the proliferation and invasion of human colon cancer cells. Postgraduate Medical Journal. 2007;83(980):414–419. doi: 10.1136/pmj.2006.052761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giri S, Rattan R, Singh AK, Singh I. The 15-deoxy-Δ12,14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via downregulating multiple steps in phosphatidylinositol 3-kinase-Akt-NF-κB-p300 pathway independent of peroxisome proliferator-activated receptor γ . Journal of Immunology. 2004;173(8):5196–5208. doi: 10.4049/jimmunol.173.8.5196. [DOI] [PubMed] [Google Scholar]
- 67.Petrova TV, Akama KT, van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: downregulation of inducible nitric-oxide synthase by 15-deoxy-Δ12,14-prostaglandin J2 . Proceedings of the National Academy of Sciences of the United States of America. 1999;96(8):4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chatterjee PK, Patel NSA, Cuzzocrea S, et al. The cyclopentenone prostaglandin 15-deoxy-Δ12,14- prostaglandin J2 ameliorates ischemic acute renal failure. Cardiovascular Research. 2004;61(3):630–643. doi: 10.1016/j.cardiores.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 69.Inoue H, Tanabe T, Umesono K. Feedback control of cyclooxygenase-2 expression through PPARγ . The Journal of Biological Chemistry. 2000;275(36):28028–28032. doi: 10.1074/jbc.M001387200. [DOI] [PubMed] [Google Scholar]
- 70.Fahmi H, Pelletier JP, Mineau F, Martel-Pelletier J. 15d-PGJ2 is acting as a “dual agent” on the regulation of COX-2 expression in human osteoarthritic chondrocytes. Osteoarthritis and Cartilage. 2002;10(11):845–848. doi: 10.1053/joca.2002.0835. [DOI] [PubMed] [Google Scholar]
- 71.Subbaramaiah K, Lin DT, Hart JC, Dannenberg AJ. Peroxisome proliferator-activated receptor γ ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence for involvement of activator protein-1 and CREB-binding protein/p300. The Journal of Biological Chemistry. 2001;276(15):12440–12448. doi: 10.1074/jbc.M007237200. [DOI] [PubMed] [Google Scholar]
- 72.Kim EH, Surh YJ. 15-Deoxy-Δ12,14-prostaglandin J2 as a potential endogenous regulator of redox-sensitive transcription factors. Biochemical Pharmacology. 2006;72(11):1516–1528. doi: 10.1016/j.bcp.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 73.Azuma Y, Shinohara M, Wang PL, Ohura K. 15-Deoxy-Δ12,14-prostaglandin J2 inhibits IL-10 and IL-12 production by macrophages. Biochemical and Biophysical Research Communications. 2001;283(2):344–346. doi: 10.1006/bbrc.2001.4783. [DOI] [PubMed] [Google Scholar]
- 74.Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. Journal of Cell Science. 2001;114(13):2375–2382. doi: 10.1242/jcs.114.13.2375. [DOI] [PubMed] [Google Scholar]
- 75.Altieri DC. Validating survivin as a cancer therapeutic target. Nature Reviews Cancer. 2003;3(1):46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 76.Kawasaki H, Altieri DC, Lu CD, Toyoda M, Tenjo T, Tanigawa N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Research. 1998;58(22):5071–5074. [PubMed] [Google Scholar]
- 77.Sarela AI, Scott N, Ramsdale J, Markham AF, Guillou PJ. Immunohistochemical detection of the anti-apoptosis protein, survivin, predicts survival after curative resection of stage II colorectal carcinomas. Annals of Surgical Oncology. 2001;8(4):305–310. doi: 10.1007/s10434-001-0305-0. [DOI] [PubMed] [Google Scholar]
- 78.Kaufmann T, Strasser A, Jost PJ. Fas death receptor signalling: roles of Bid and XIAP. Cell Death and Differentiation. 2012;19(1):42–50. doi: 10.1038/cdd.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ghosh S, May MJ, Kopp EB. NF-κB and rel proteins: evolutionarily conserved mediators of immune responses. Annual Review of Immunology. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 80.Greten FR, Eckmann L, Greten TF, et al. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 81.Pikarsky E, Porat RM, Stein I, et al. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 82.Garg A, Aggarwal BB. Nuclear transcription factor-κB as a target for cancer drug development. Leukemia. 2002;16(6):1053–1068. doi: 10.1038/sj.leu.2402482. [DOI] [PubMed] [Google Scholar]
- 83.Fajas L, Egler V, Reiter R, Miard S, Lefebvre AM, Auwerx J. PPARγ controls cell proliferation and apoptosis in an RB-dependent manner. Oncogene. 2003;22(27):4186–4193. doi: 10.1038/sj.onc.1206530. [DOI] [PubMed] [Google Scholar]
- 84.Lu S, Yu G, Zhu Y, Archer MC. Cyclooxygenase-2 overexpression in MCF-10F human breast epithelial cells inhibits proliferation, apoptosis and differentiation, and causes partial transformation. International Journal of Cancer. 2005;116(6):847–852. doi: 10.1002/ijc.21142. [DOI] [PubMed] [Google Scholar]
- 85.Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Seminars in Oncology. 2004;31(7):2–11. doi: 10.1053/j.seminoncol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 86.Dang DT, Pevsner J, Yang VW. The biology of the mammalian Krüppel-like family of transcription factors. International Journal of Biochemistry and Cell Biology. 2000;32(11-12):1103–1121. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shie JL, Chen ZY, O’Brien MJ, Pestell RG, Lee ME, Tseng CC. Role of gut-enriched Krüppel-like factor in colonic cell growth and differentiation. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2000;279(4):G806–G814. doi: 10.1152/ajpgi.2000.279.4.G806. [DOI] [PubMed] [Google Scholar]
- 88.Bieker JJ. Krüppel-like factors: three fingers in many pies. The Journal of Biological Chemistry. 2001;276(37):34355–34358. doi: 10.1074/jbc.R100043200. [DOI] [PubMed] [Google Scholar]
- 89.Chen ZY, Shie JL, Tseng CC. Upregulation of gut-enriched krüppel-like factor by interferon-γ in human colon carcinoma cells. FEBS Letters. 2000;477(1-2):67–72. doi: 10.1016/s0014-5793(00)01764-6. [DOI] [PubMed] [Google Scholar]
- 90.Dang DT, Chen X, Feng J, Torbenson M, Dang LH, Yang VW. Overexpression of Krüppel-like factor 4 in the human colon cancer cell line RKO leads to reduced tumorigenecity. Oncogene. 2003;22(22):3424–3430. doi: 10.1038/sj.onc.1206413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dhaene K, van Marck E, Parwaresch R. Telomeres, telomerase and cancer: an up-date. Virchows Archiv. 2000;437(1):1–16. doi: 10.1007/s004280000189. [DOI] [PubMed] [Google Scholar]
- 92.Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nature Reviews Drug Discovery. 2006;5(7):577–584. doi: 10.1038/nrd2081. [DOI] [PubMed] [Google Scholar]
- 93.Hiyama E, Yokoyama T, Tatsumoto N, et al. Telomerase activity in gastric cancer. Cancer Research. 1995;55(15):3258–3262. [PubMed] [Google Scholar]
- 94.Chadeneau C, Hay K, Hirte HW, Gallinger S, Bacchetti S. Telomerase activity associated with acquisition of malignancy in human colorectal cancer. Cancer Research. 1995;55(12):2533–2536. [PubMed] [Google Scholar]
- 95.Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279(5349):349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 96.Vaziri H, Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Current Biology. 1998;8(5):279–282. doi: 10.1016/s0960-9822(98)70109-5. [DOI] [PubMed] [Google Scholar]
- 97.Meeker AK, de Marzo AM. Recent advances in telomere biology: implications for human cancer. Current Opinion in Oncology. 2004;16(1):32–38. doi: 10.1097/00001622-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 98.Morales CP, Holt SE, Ouellette M, et al. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nature Genetics. 1999;21(1):115–118. doi: 10.1038/5063. [DOI] [PubMed] [Google Scholar]
- 99.Hahn WC, Stewart SA, Brooks MW, et al. Inhibition of telomerase limits the growth of human cancer cells. Nature Medicine. 1999;5(10):1164–1170. doi: 10.1038/13495. [DOI] [PubMed] [Google Scholar]
- 100.Delhommeau F, Thierry A, Feneux D, et al. Telomere dysfunction and telomerase reactivation in human leukemia cell lines after telomerase inhibition by the expression of a dominant-negative hTERT mutant. Oncogene. 2002;21(54):8262–8271. doi: 10.1038/sj.onc.1206054. [DOI] [PubMed] [Google Scholar]
- 101.Kawamata H, Tachibana M, Fujimori T, Imai Y. Differentiation-inducing therapy for solid tumors. Current Pharmaceutical Design. 2006;12(3):379–385. doi: 10.2174/138161206775201947. [DOI] [PubMed] [Google Scholar]
- 102.Risau W. Mechanisms of angiogenesis. Nature. 1997;386(6626):671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 103.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature Medicine. 2003;9(6):669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 104.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(3):4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 105.Lee JC, Chow NH, Wang ST, Huang SM. Prognostic value of vascular endothelial growth factor expression in colorectal cancer patients. European Journal of Cancer. 2000;36(6):748–753. doi: 10.1016/s0959-8049(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 106.Poon RTP, Fan ST, Wong J. Clinical implications of circulating angiogenic factors in cancer patients. Journal of Clinical Oncology. 2001;19(4):1207–1225. doi: 10.1200/JCO.2001.19.4.1207. [DOI] [PubMed] [Google Scholar]
- 107.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annual Review of Cell and Developmental Biology. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: how hot is the link? Biochemical Pharmacology. 2006;72(11):1605–1621. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 109.Minuzzo S, Moserle L, Indraccolo S, Amadori A. Angiogenesis meets immunology: cytokine gene therapy of cancer. Molecular Aspects of Medicine. 2007;28(1):59–86. doi: 10.1016/j.mam.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 110.Choi IK, Kim YH, Kim JS, Seo JH. PPAR-γ ligand promotes the growth of APC-mutated HT-29 human colon cancer cells in vitro and in vivo. Investigational New Drugs. 2008;26(3):283–288. doi: 10.1007/s10637-007-9108-x. [DOI] [PubMed] [Google Scholar]
- 111.Kim EH, Na HK, Kim DH, et al. 15-Deoxy-Δ12,14-prostaglandin J2 induces COX-2 expression through Akt-driven AP-1 activation in human breast cancer cells: a potential role of ROS. Carcinogenesis. 2008;29(4):688–695. doi: 10.1093/carcin/bgm299. [DOI] [PubMed] [Google Scholar]
- 112.Yamakawa K, Hosoi M, Koyama H, et al. Peroxisome proliferator-activated receptor-γ agonists increase vascular endothelial growth factor expression in human vascular smooth muscle cells. Biochemical and Biophysical Research Communications. 2000;271(3):571–574. doi: 10.1006/bbrc.2000.2665. [DOI] [PubMed] [Google Scholar]
- 113.Inoue M, Itoh H, Tanaka T, et al. Oxidized LDL regulates vascular endothelial growth factor expression in human macrophages and endothelial cells through activation of peroxisome proliferator-activated receptor-γ . Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(4):560–566. doi: 10.1161/01.atv.21.4.560. [DOI] [PubMed] [Google Scholar]
- 114.Chintalgattu V, Harris GS, Akula SM, Katwa LC. PPAR-γ agonists induce the expression of VEGF and its receptors in cultured cardiac myofibroblasts. Cardiovascular Research. 2007;74(1):140–150. doi: 10.1016/j.cardiores.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 115.Kim DH, Kim JH, Kim EH, et al. 15-Deoxy-Δ12,14-prostaglandin J2 upregulates the expression of heme oxygenase-1 and subsequently matrix metalloproteinase-1 in human breast cancer cells: possible roles of iron and ROS. Carcinogenesis. 2009;30(4):645–654. doi: 10.1093/carcin/bgp012. [DOI] [PubMed] [Google Scholar]
- 116.Lefebvre AM, Chen I, Desreumaux P, et al. Activation of the peroxisome proliferator-activated receptor γ promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nature Medicine. 1998;4(9):1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- 117.Saez E, Tontonoz P, Nelson MC, et al. Activators of the nuclear receptor PPARγ enhance colon polyp formation. Nature Medicine. 1998;4(9):1058–1061. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- 118.Karim RZ, Tse GMK, Putti TC, Scolyer RA, Lee CS. The significance of the Wnt pathway in the pathology of human cancers. Pathology. 2004;36(2):120–128. doi: 10.1080/00313020410001671957. [DOI] [PubMed] [Google Scholar]
- 119.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55(1):115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Józkowicz A, Huk I, Nigisch A, Cisowski J, Weigel G, Dulak J. Prostaglandin-J2 upregulates expression of matrix metalloproteinase-1 independently of activation of peroxisome proliferator-activated receptor-γ . Acta Biochimica Polonica. 2003;50(3):677–689. [PubMed] [Google Scholar]
- 121.Varnat F, Heggeler BB, Grisel P, et al. PPARβ/δ regulates paneth cell differentiation via controlling the hedgehog signaling pathway. Gastroenterology. 2006;131(2):538–553. doi: 10.1053/j.gastro.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 122.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARδ is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99(3):335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gupta RA, Tan J, Krause WF, et al. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor δ in colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(24):13275–13280. doi: 10.1073/pnas.97.24.13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Park BH, Vogelstein B, Kinzler KW. Genetic disruption of PPARδ decreases the tumorigenicity of human colon cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2598–2603. doi: 10.1073/pnas.051630998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shao J, Sheng H, Dubois RN. Peroxisome proliferator-activated receptors modulate K-Ras-mediated transformation of intestinal epithelial cells. Cancer Research. 2002;62(11):3282–3288. [PubMed] [Google Scholar]
- 126.Di-Poï N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARβ in keratinocytes via transcriptional control of the Akt1 signaling pathway. Molecular Cell. 2002;10(4):721–733. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- 127.Wang D, Wang H, Guo Y, et al. Crosstalk between peroxisome proliferator-activated receptor δ and VEGF stimulates cancer progression. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(50):19069–19074. doi: 10.1073/pnas.0607948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kwak H, Hwang I, Kim JH, Mee YK, Yang JS, Jeong S. Modulation of transcription by the peroxisome proliferator-activated receptor δ-binding RNA aptamer in colon cancer cells. Molecular Cancer Therapeutics. 2009;8(9):2664–2673. doi: 10.1158/1535-7163.MCT-09-0214. [DOI] [PubMed] [Google Scholar]
- 129.Reed KR, Sansom OJ, Hayes AJ, et al. PPARδ status and Apc-mediated tumourigenesis in the mouse intestine. Oncogene. 2004;23(55):8992–8996. doi: 10.1038/sj.onc.1208143. [DOI] [PubMed] [Google Scholar]
- 130.Peters JM, Gonzalez FJ. Sorting out the functional role(s) of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) in cell proliferation and cancer. Biochimica et Biophysica Acta. 2009;1796(2):230–241. doi: 10.1016/j.bbcan.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Peters JM, Hollingshead HE, Gonzalez FJ. Role of peroxisome-proliferator-activated receptor β/δ (PPARβ/δ) in gastrointestinal tract function and disease. Clinical Science. 2008;115(3-4):107–127. doi: 10.1042/CS20080022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nadra K, Anghel SI, Joye E, et al. Differentiation of trophoblast giant cells and their metabolic functions are dependent on peroxisome proliferator-activated receptor β/δ . Molecular and Cellular Biology. 2006;26(8):3266–3281. doi: 10.1128/MCB.26.8.3266-3281.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zuo X, Peng Z, Moussalli MJ, et al. Targeted genetic disruption of peroxisome proliferator-activated receptor-δ and colonic tumorigenesis. Journal of the National Cancer Institute. 2009;101(10):762–767. doi: 10.1093/jnci/djp078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Harman FS, Nicol CJ, Marin HE, Ward JM, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-δ attenuates colon carcinogenesis. Nature Medicine. 2004;10(5):481–483. doi: 10.1038/nm1026. [DOI] [PubMed] [Google Scholar]
- 135.Peters JM, Lee SST, Li W, et al. Growths, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β(δ) Molecular and Cellular Biology. 2000;20(14):5119–5128. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kopelovich L, Yin Y, Russell RG, et al. Peroxisome proliferator-activator receptor δ and γ agonists differentially alter tumor differentiation and progression during mammary carcinogenesis. Cancer Research. 2005;65(9):3950–3957. doi: 10.1158/0008-5472.CAN-04-3990. [DOI] [PubMed] [Google Scholar]
- 137.Yang L, Olsson B, Pfeifer D, et al. Knockdown of peroxisome proliferator-activated receptor-β induces less differentiation and enhances cell-fibronectin adhesion of colon cancer cells. Oncogene. 2010;29(4):516–526. doi: 10.1038/onc.2009.370. [DOI] [PubMed] [Google Scholar]
- 138.Wu KK, Liou JY. Cyclooxygenase inhibitors induce colon cancer cell apoptosis via PPARδ→14-3-3ε pathway. Methods in Molecular Biology. 2009;512:295–307. doi: 10.1007/978-1-60327-530-9_16. [DOI] [PubMed] [Google Scholar]