

Abstract
Because of the ubiquity of the secondary carbinol subunit, the development of new methods for its enantioselective synthesis remains an important ongoing challenge. In this report, we describe the first non-enzymatic method for the dynamic kinetic resolution (DKR) of secondary alcohols (specifically, aryl alkyl carbinols) through enantioselective acylation, and we substantially expand the scope of this approach, vis-à-vis enzymatic reactions. Simply combining an effective process for the kinetic resolution of alcohols with an active catalyst for the racemization of alcohols did not lead to DKR, due to the incompatibility of the ruthenium-based racemization catalyst with the acylating agent (Ac2O) used in the kinetic resolution. A mechanistic investigation revealed that the ruthenium catalyst is deactivated through the formation of a stable ruthenium-acetate complex; this deleterious pathway was circumvented through the appropriate choice of acylating agent (an acyl carbonate). Mechanistic studies of this new process point to reversible N-acylation of the nucleophilic catalyst, acyl transfer from the catalyst to the alcohol as the rate-determining step, and carbonate anion serving as the Brønsted base in that acyl-transfer step.
INTRODUCTION
Dynamic kinetic resolution (DKR) is a powerful strategy in asymmetric synthesis, allowing the stereoconvergent transformation of both enantiomers of a racemic substrate into a single enantiomer of a target molecule.1 This approach overcomes a limitation of simple kinetic resolutions, which provide a maximum direct yield of 50% for a particular enantiomer. For an efficient DKR, the reaction conditions for kinetic resolution and for racemization of the substrate must be compatible, and racemization must occur rapidly relative to conversion of the slow-reacting enantiomer to product.
Because secondary carbinols are a common subunit in bioactive compounds,2 a great deal of effort has been dedicated to the development of efficient methods for their synthesis in enantioenriched form.3 Enzymatic resolution, including DKR, of racemic alcohols is one of the more widely used approaches, especially in industry.4 With respect to enantioselective acylation,5 although impressive progress has been described for enzymatic DKR’s,6 as well as for non-enzymatic (and non-dynamic) kinetic resolutions,7 we are not aware of any reports of useful non-enzymatic DKR’s.
In previous studies, we established that a planar-chiral DMAP derivative serves as an effective catalyst for the kinetic resolution of a variety of racemic aryl alkyl carbinols,8 propargylic alcohols,9 and allylic alcohols10 via acylation with Ac2O, providing selectivity factors as high as ~100 (eq 1). Our early attempts to develop a dynamic variant were stymied either by the incompatibility of the acylation and the racemization processes, or else by the need for elevated temperature in order to achieve racemization, which led to a substantial erosion in enantioselectivity.
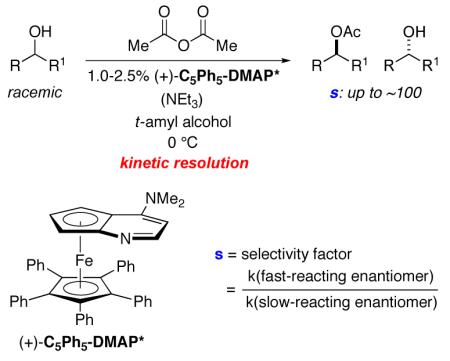 |
(1) |
Recent discoveries of catalysts that rapidly racemize secondary alcohols at room temperature11 provided fresh impetus for us to revisit the unsolved challenge of developing an effective method for the non-enzymatic DKR of alcohols via enantioselective acylation. In this report, we describe the achievement of this objective through the use of a planar-chiral DMAP derivative (acylation catalyst) in combination with a ruthenium complex (racemization catalyst) in the presence of an acyl carbonate (eq 2), and we present mechanistic studies, some of which were critical to the successful development of this process.
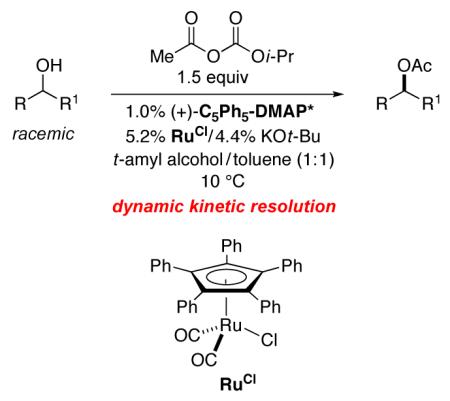 |
(2) |
RESULTS AND DISCUSSION
A variety of transition-metal complexes have been reported to catalyze the racemization of secondary alcohols.11 We were particularly drawn to the recent systematic studies of Bäckvall, who has established that (C5Ph5)Ru(CO)2(Ot-Bu) (RuOt-Bu), generated in situ from (C5Ph5)Ru(CO)2Cl (RuCl) and KOt-Bu, is an active racemization catalyst at room temperature, and that it can be applied to enzymatic DKR’s of secondary alcohols.12
Bäckvall generally employs toluene as the solvent when utilizing RuOt-Bu as a racemization catalyst, whereas t-amyl alcohol is the solvent of choice for kinetic resolutions catalyzed by (C5Ph5)-DMAP* (eq 1). One could envision that use of an alcohol as the solvent might inhibit racemization of a secondary alcohol by RuOt-Bu; we have, however, determined that racemization of 1-phenylethanol proceeds smoothly in t-amyl alcohol even at 0 °C (eq 3; we employ a small excess of RuCl in order to decrease the likelihood that adventitious KOt-Bu will have a deleterious impact on our DKR’s, e.g., by facilitating a Brønsted base-mediated non-enantioselective acylation). Furthermore, planar-chiral DMAP derivative (C5Ph5)-DMAP* is compatible with the racemization process (eq 3), and no interaction between RuOt-Bu (or the ruthenium alkoxide derived from 1-phenylethanol) and (C5Ph5)-DMAP* can be detected in the 1H NMR spectrum of a mixture of the two compounds in t-amyl alcohol/toluene.
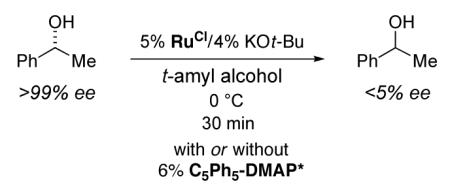 |
(3) |
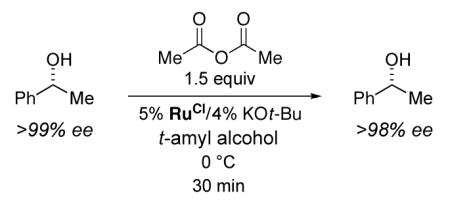
Unfortunately, when we applied the conditions that we had developed earlier for the kinetic resolution of secondary alcohols (eq 1) to the DKR of 1-phenylethanol, it became clear that the ruthenium racemization catalyst is not compatible with the acylation process (eq 4). Thus, at partial conversion, the unreacted alcohol was substantially enantiomerically enriched (98% ee after 24 hours; >99% ee after 48 hours).
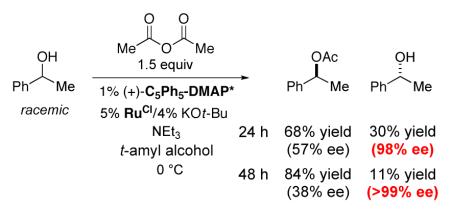 |
(4) |
An additional control experiment revealed that RuOt-Bu is deactivated by Ac2O (eq 5 vs. eq 3). We hypothesized that the ruthenium alkoxide might be acylated by Ac2O to generate t-butyl acetate and a ruthenium-acetate complex (RuOAc), and indeed that has proved to be the case (eq 6). RuOAc is sufficiently stable that it can be purified by column chromatography and characterized by X-ray crystallography, which reveals an η1-acetate ligand as part of an 18-electron ruthenium complex.
 |
(5) |
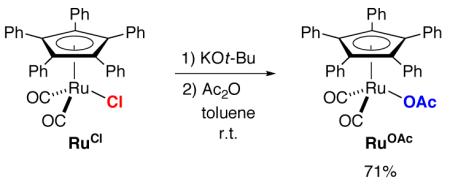 |
(6) |
As we had conjectured, RuOAc is not an active racemization catalyst for 1-phenylethanol in t-amyl alcohol at 0 °C (eq 7; cf. eq 3). Furthermore, we have confirmed that RuOAc forms rapidly under the conditions that we had originally explored for the DKR of 1-phenylethanol (eq 4).13
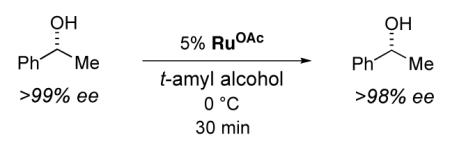 |
(7) |
Thus, we needed to identify an acylating agent that would not deactivate the ruthenium-alkoxide racemization catalyst through the formation of a stable ruthenium carboxylate. Acyl carbonates14 were among the array of candidates, since they are less electrophilic than anhydrides and, if acylation of the ruthenium alkoxide were to occur, the resulting ruthenium carbonate might decarboxylate and thereby regenerate a ruthenium alkoxide (eq 8).15,16
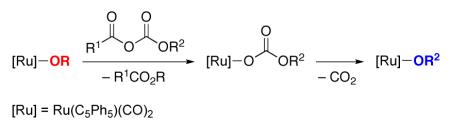 |
(8) |
Indeed, we have determined that, with acetyl isopropyl carbonate17 as the acylating agent in a mixture of the preferred solvents for (C5Ph5)-DMAP*-catalyzed acylation (t-amyl alcohol) and RuOt-Bu-catalyzed racemization (toluene), the DKR of 1-phenylethanol proceeds with good ee and in excellent yield (eq 9). We observe lower enantioselectivity and yield when other acyl carbonates are employed as the acylating agent.
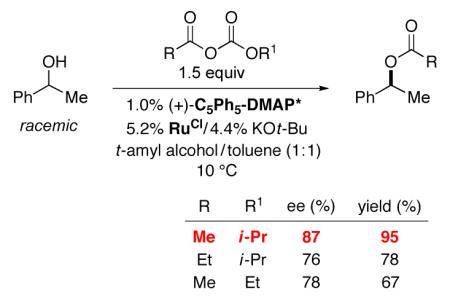 |
(9) |
This method achieves the DKR of an array of aryl alkyl carbinols with ~90% ee and in high yield (Table 1, entries 1–9).18 The scope of the process is relatively broad: for example, the alkyl substituent of the carbinol can range in size from Me to isopropyl. In contrast, corresponding enzymatic DKR’s of aryl alkyl carbinols are only effective when the alkyl group is not branched (in the α, β, or even the γ position).6
Table 1. Non-Enzymatic Dynamic Kinetic Resolution of Secondary Alcohols (for the reaction conditions, see eq 2)a.
| entry | alcohol | ee (%) | yield (%)b |
|---|---|---|---|
| 1 |
|
87 | 95 (85) |
| 2 | 90 | 96 (95) | |
| 3 | 82 | 90 (86) | |
| 4 | 91 | 98 (95) | |
| 5 |
|
85 | 89 (88) |
| 6 | 88 | 93 (92) | |
| 7 |
|
91 | 97 (96) |
| 8 |

|
93 | 97 (92) |
| 9 |
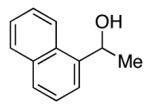
|
90 | 99 (94) |
| 10 |
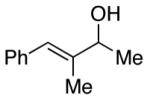
|
88 | 97 (90) |
All data are the average of two experiments. The predominant product is the S enantiomer.
The yield was determined by GC analysis with the aid of a calibrated internal standard. The yield of purified product is provided in parentheses.
Effective DKR is observed when the aromatic ring of the aryl alkyl carbinol is electron-poor or electron-rich (Table 1, entries 5 and 6), para-, meta-, or ortho-substituted (entries 5–8), or an extended π system (entry 9). Furthermore, the non-enzymatic DKR of an allylic alcohol can be achieved with useful enantioselectivity and yield (entry 10).
This combination of planar-chiral DMAP derivative (C5Ph5)-DMAP* and a ruthenium racemization catalyst can also be applied to the stereoconvergent acylation of a diol. Thus, when three isomers of a diol are subjected to our DKR conditions, the C2-symmetric bis(acetate) can be isolated in excellent ee (eq 10).
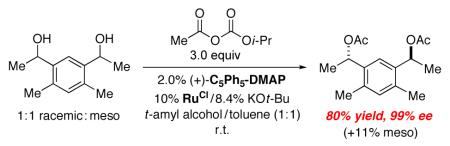 |
(10) |
The rate law for the DKR of 1-phenylethanol is first order in 1-phenylethanol, first order in the acylation catalyst ((C5Ph5)-DMAP*), “fractional” (first order at low concentration, approaching zeroth order at higher concentration) order in acetyl isopropyl carbonate, and zeroth order in the racemization catalyst. According to an NMR spectroscopic study, the resting state of the planar-chiral DMAP derivative during a DKR is a mixture of the free catalyst and the N-acylated catalyst,19 which accounts for the “fractional” dependence of rate on the concentration of acetyl isopropyl carbonate. Acyl transfer from the catalyst to the alcohol is likely the rate-determining step of the DKR (Figure 1).
Figure 1.
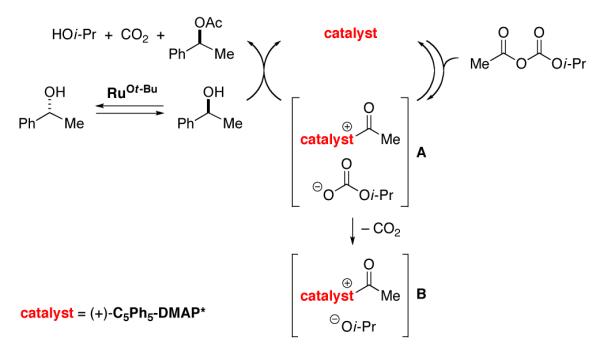
Outline of a mechanism for the non-enzymatic dynamic kinetic resolution of a secondary alcohol catalyzed by (+)-(C5Ph5)-DMAP* and RuOt-Bu.
When (C5Ph5)-DMAP* is treated with two equivalents of acetyl isopropyl carbonate at −20 °C, the acylpyridinium salt is generated almost quantitatively, according to 1H NMR spectroscopy (eq 11).20 The salt is stable for at least 6 hours at this temperature.
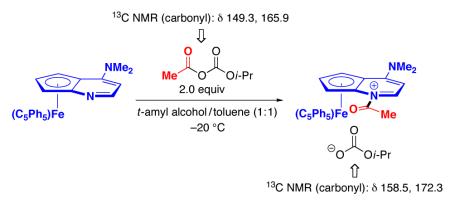 |
(11) |
We hypothesize that ion pair A, rather than B, is the species that reacts with the secondary alcohol in the rate-determining acyl-transfer step of the reaction (Figure 1). Consistent with this suggestion, the decarboxylation of acetyl isopropyl carbonate in the presence of (C5Ph5)-DMAP* proceeds significantly more slowly (eq 12; 5% after three hours) than the acylation of 1-phenylethanol under DKR conditions (37% conversion after three hours).
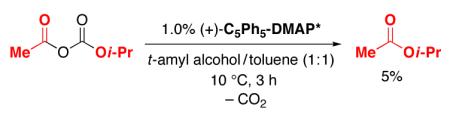 |
(12) |
CONCLUSIONS
Dynamic kinetic resolution is a powerful approach to the enantioselective synthesis of secondary alcohols, an important family of bioactive compounds. We have developed the first non-enzymatic method for the DKR of alcohols through enantioselective acylation, and we have established that this process is effective for a variety of aryl alkyl carbinols (including a diol); this study complements corresponding enzymatic DKR’s, which are not useful when the alkyl substituent is branched. We were not able to address this challenge simply by combining an effective method for the kinetic resolution of alcohols with an active catalyst for the racemization of alcohols—the two processes proved to be incompatible, specifically, the ruthenium-based racemization catalyst was poisoned by the acylating agent (Ac2O) employed in the kinetic resolution. Nevertheless, we determined the pathway for deactivation (formation of a stable, inactive ruthenium-acetate adduct) and developed a method that circumvents this problem. Mechanistic studies (reactivity, kinetics, and spectroscopic) of this new process for the DKR of aryl alkyl carbinols indicate that N-acylation of the nucleophilic catalyst is reversible, acyl transfer from the catalyst to the alcohol is the rate-determining step, and a carbonate anion serves as the Brønsted base in that acyl-transfer step. The development of additional applications of planar-chiral DMAP derivatives in asymmetric catalysis is underway.
Supplementary Material
ACKNOWLEDGMENTS
Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences: R01-GM057034 for G.C.F. and F32 GM087889 for J.M.M.) and by the Global COE in Chemistry, Nagoya University (fellowship to A.U.). We thank Dr. Jeffrey H. Simpson (Department of Chemistry Instrumentation Facility) for assistance with NMR spectroscopy, Dr. Peter Müller for assistance with X-ray crystallography (and NSF grant CHE-0946721 for the purchase of an X-ray diffractomer), and Dr. Gerald B. Rowland for preliminary studies.
Footnotes
ASSOCIATED CONTENT
Experimental procedures and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Pellissier H. Chirality from Dynamic Kinetic Resolution. Royal Society of Chemistry; Cambridge: 2011. [Google Scholar]; (b) Pellissier H. Tetrahedron. 2011;67:3769–3802. [Google Scholar]
- (2).For an example, see: Grundy S, editor. Atorvastatin in the Management of Cardiovascular Risk: From Pharmacology to Clinical Evidence. Kluwer; Auckland, New Zealand: 2007.
- (3).For example, see: Molander GA, editor. Science of Synthesis, Stereoselective Synthesis. Thieme; New York: 2011. p. 2.
- (4).For leading references, see: Fischer T, Pietruszka J. Top. Curr. Chem. 2010;297:1–43. doi: 10.1007/128_2010_62. Blaser H-U, Federsel H-J, editors. Asymmetric Catalysis on Industrial Scale. Wiley-VCH; New York: 2010. Pollard D, Kosjek B. In: Organic Synthesis with Enzymes in Non-Aqueous Media. Carrea G, Riva S, editors. Wiley-VCH; New York: 2008. pp. 169–188.
- (5).For some leading references, see: Oriyama T. In: Science of Synthesis, Stereoselective Synthesis. De Vries JG, Molander GA, Evans PA, editors. Vol. 3. Thieme; New York: 2011. pp. 829–849. Müller CE, Schreiner PR. Angew. Chem. Int. Ed. 2011;50:6012–6042. doi: 10.1002/anie.201006128. Spivey AC, Arseniyadis S. Top. Curr. Chem. 2010;291:233–280. doi: 10.1007/978-3-642-02815-1_25.
- (6).For reviews and leading references, see: Martín-Matute B, Bäckvall JE, Gotor B, Alfonso I, Garcia-Urdiales E. Asymmetric Organic Synthesis with Enzymes. Wiley-VCH; New York: 2008. pp. 89–113. Lee JH, Han K, Kim M-J, Park J. Eur. J. Org. Chem. 2010:999–1015.
- (7).For a review, see: Pellissier H. Adv. Synth. Catal. 2011;353:1613–1666.
- (8).Ruble JC, Latham HA, Fu GC. J. Am. Chem. Soc. 1997;119:1492–1493. Ruble JC, Tweddell J, Fu GC. J. Org. Chem. 1998;63:2794–2795. Chen Y-H, McDonald FE. J. Am. Chem. Soc. 2006;128:4568–4569. doi: 10.1021/ja061082f. For an application of this method, see:
- (9).Tao B, Ruble JC, Hoic DA, Fu GC. J. Am. Chem. Soc. 1999;121:5091–5092. [Google Scholar]
- (10).Bellemin-Laponnaz S, Tweddell J, Ruble JC, Breitling FM, Fu GC. Chem. Commun. 2000:1009–1010. For a recent application of this method, see: Francais A, Leyva A, Etxebarria-Jardi G, Ley SV. Org. Lett. 2010;12:340–343. doi: 10.1021/ol902676t.
- (11).For a review of racemization catalysts for the DKR of alcohols and amines, see: Ahn Y, Ko S-B, Kim M-J, Park J. Coord. Chem. Rev. 2008;252:647–658.
- (12).Martín-Matute B, Edin M, Bogár K, Kaynak FB, Bäckvall J-E. J. Am. Chem. Soc. 2005;127:8817–8825. doi: 10.1021/ja051576x. [DOI] [PubMed] [Google Scholar]
- (13).Notes: (a) RuOt-Bu reacts with AcOH within 10 minutes at room temperature (in t-amyl alcohol/toluene (1:1)) to generate RuOAc and t-BuOH. (b) In the presence of propionic acid and 1-phenylethanol, RuOAc does not react to form a ruthenium-propionate adduct after 20 hours at room temperature (in t-amyl alcohol/toluene (1:1)).
- (14).For an example of the use of an acyl carbonate in an enzymatic kinetic resolution of an alcohol, see: Guibe-Jampel E, Chalecki Z, Bassir M, Gelo-Pujic M. Tetrahedron. 1996;52:4397–4402.
- (15).We subsequently determined that RuOt-Bu does not react with acetyl isopropyl carbonate after 20 hours at room temperature, in the absence or in the presence of 1-phenylethanol (in t-amyl alcohol/toluene (1:1)).
- (16).Kim has reported that DMAP catalyzes the decarboxylation of acyl carbonates to form esters: Kim S, Lee JI, Kim YC. J. Org. Chem. 1985;50:560–565.
- (17).Notes: (a) Acetyl isopropyl carbonate can be synthesized in one step from acetic acid and isopropyl chloroformate. After five days at room temperature under nitrogen, there is no detectable decomposition. (b) We are not aware of previous reports of the use of this acyl carbonate as an acylating agent for alcohols. (c) Whereas acylations of alcohols by Ac2O generally include a stoichiometric Brønsted base as an additive (to capture AcOH), when acetyl isopropyl carbonate is used as the acylating agent, isopropanol is produced, and no added base is required.
- (18).Notes: (a) On a gram scale, the DKR illustrated in entry 8 of Table 1 proceeded in 92% ee and >99% yield (1.25 g of product; catalyst recovery for C5Ph5-DMAP*: 74%). (b) During the course of the DKR of 1-phenylethanol (Table 1, entry 1), the ee of the product was constant, and the ee of the unreacted starting material was less than 15%. (c) Under the conditions described in eq 2, a simple kinetic resolution (i.e., without RuCl and KOt-Bu) of 1-phenylethanol proceeded with a selectivity factor of 14, a value consistent with the 87% ee that we observe for the DKR of 1-phenylethanol (Table 1, entry 1). (d) Under our standard conditions, DKR’s of phenyl t-butyl carbinol and of phenyl chloromethyl carbinol were not effective. (e) Both enantiomers of C5Ph5-DMAP* are available from Strem Chemicals.
- (19).Notes: (a) For spectroscopic characterization and X-ray structural analysis of N-acetylated C5Ph5-DMAP*, see ref. 9. (b) This DKR was conducted with 3.6% (C5Ph5)-DMAP*, in order to facilitate analysis by 1H NMR spectroscopy.
- (20).For the reaction of acetyl isopropyl carbonate with (C5Ph5)-DMAP*, lower temperature favors N-acylation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.