

Abstract
The dengue virus (DENV) nonstructural protein 5 (NS5) is composed of two globular domains separated by a 10-residue linker. The N-terminal domain participates in the synthesis of a messenger RNA cap 1 structure (7MeGpppA2'OMe) at the 5' end of the viral genome, and possesses guanylyltransferase, guanine-N7-methyltransferase, and nucleoside-2'O-methyltransferase activities. The C-terminal domain is an RNA-dependent RNA polymerase responsible for viral RNA synthesis. Although crystal structures of the two isolated domains have been obtained, there are no structural data for the full-length NS5. It is also unclear whether the two NS5 domains interact with each other to form a stable structure in which the relative orientation of the two domains is fixed. To investigate the structure and dynamics of DENV type 3 NS5 in solution, small-angle X-ray scattering (SAXS) experiments were carried out on the full-length protein. NS5 was found to be monomeric and well-folded under the conditions tested. The results of these experiments also suggest that NS5 adopts multiple conformations in solution, ranging from compact to more extended forms wherein the two domains do not seem to interact with each other. We interpret the multiple conformations of NS5 observed in solution as resulting from weak interactions between the two NS5 domains and flexibility of the linker in the absence of other components of the replication complex.
Dengue infection is the most prevalent arthropod-borne disease in the world, with 50 to 100 million cases of infection annually and approximately 2.5 billon people at risk of infection1, 2. Due to the dramatic increase in incidence around the world over the past 25 years, dengue is classified by the Centers for Disease Control and Prevention (CDC) as an emerging infectious disease3, 4. Dengue infection results in a wide spectrum of clinical manifestations that range from asymptomatic to life-threatening disease, often associated with unpredictable clinical evolution and outcome2. The World Health Organization (WHO) currently classifies symptomatic infections in three categories: undifferentiated fever, a flu-like dengue fever, and dengue hemorrhagic fever (DHF) characterized by plasma leakage. The most serious complication of DHF is dengue shock syndrome (DSS), which occurs when signs of circulatory failure are detected in addition to other DHF symptoms2, 5. Dengue is caused by four serologically different dengue virus types, DENV-1 to DENV-4, and while infection with one of the four DENV serotypes provides lifelong immunity to that serotype, a secondary infection with another serotype results in a greater risk for developing DHF and DSS. Despite the significant health impact of dengue infections, neither an effective vaccine, which must confer immunity to all four serotypes, nor a specific antiviral therapy is available2, 3.
Dengue viruses belong to the flavivirus genus in the Flaviviridae family, which includes other major human pathogens such as yellow fever, West Nile, Japanese encephalitis or tick-borne encephalitis viruses1, 4. The flavivirus genome is a positive sense single-stranded RNA that acts as a messenger RNA upon infection6. Similar to most cellular mRNAs, the flavivirus genome is capped on the 5' end with a cap 1 structure that consists of a 7-methylguanosine linked to the genome via a 5'-5' triphosphate link with a methyl group added onto the 2'O of the genomic 5'-terminal nucleotide, which is an adenine in all flaviviruses7. The genome thus consists of the 5'-cap 1 structure (7MeGpppA2'OMe), a 5'-untranslated region (5'-UTR), a single open reading frame (ORF), and a 3'-untranslated region (3'UTR), but unlike cellular mRNAs, flavivirus genomes do not contain a poly-A tail on their 3' ends6. After translation of the ORF by the host machinery, the resulting polyprotein is processed by cellular and viral proteases into three structural proteins (C, prM, and E) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5)6. The nonstructural proteins NS1, NS2A, NS3, NS4A, and NS5, along with the viral RNA and host proteins, associate into a viral replicase in a modified membrane structure derived from the endoplasmic reticulum6, 8. Among the nonstructural proteins, the two-domain proteins NS3 and NS5 are the key enzymes in the replication complex, as together they account for all activities needed for genome replication and cap synthesis9, 10. NS3 (~70 kDa) consists of an N-terminal serine protease domain, which requires NS2B as a cofactor, and a C-terminal domain possessing three distinct activities: an RNA helicase, RNA-stimulated nucleoside triphosphatase (NTPase), and 5'-RNA triphosphatase (5'-RTPase)10. NS5, the largest NS protein (~103 kDa), consists of an N-terminal domain possessing three activities necessary for cap synthesis (guanylyltransferase, guanine-N7-methyltransferase, and nucleoside-2'O-methyltransferase) and a C-terminal domain that harbors the primer-independent RNA-dependent RNA polymerase (RdRp) activity10–13. This latter domain is responsible for the replication of the positive-strand RNA genome in an asymmetric and semi-conservative process in which the antigenome is only present in a double-stranded RNA replication intermediate6. During replication, the helicase activity of the NS3 C-terminal domain is thought to be involved in unwinding the replicative form. After replication of the viral genome by the NS5 RdRp domain, the cap structure is added onto the 5' end of the genome by four enzyme activities14. First, the 5' γ-phosphate of the RNA genome is cleaved by the NS3 C-terminal domain to produce a diphosphate-terminated RNA (ppAXn, where Xn represents the RNA). Second, the NS5 guanylyltransferase/methyltransferase (GTase/MTase) domain catalyzes the transfer of a GMP moiety from GTP to the RNA. The third and fourth activities are two S-adenosyl-L-methionine (SAM)-dependent methylations, also catalyzed by the NS5 GTase/MTase domain. These methylations occur sequentially, with guanine-N7-methylation preceding the nucleoside-2'O-methylation14. Genome capping likely occurs during the initial stages of replication, but little is known about how flaviviruses coordinate these two processes.
The interdependence of the NS3 and NS5 activities, suggested by the sequence of reactions during genome replication and capping, was shown by experiments in which NS5 stimulates NS3's NTPase and 5'-RTPase activities15–17, and NS3 stimulates NS5's GTase activity12. Moreover, multiple experiments (e.g., co-immunoprecipitation and pull-down assays in DENV-infected cells) demonstrate that NS3 and NS5 interact with each other16–18. The modularity of NS3 is functionally relevant since the protease domain influences the activities and specificity of the helicase/NTPase/5'-RTPase domain16, 19–22. In contrast, the linkage between the two NS5 domains does not seem to be relevant to the function of these two domains, since the RdRp activity is not altered by the presence of the GTase/MTase domain, and the GTase and MTase activities are not affected by the RdRp domain12, 23. While three structures of the full-length NS3 have been solved in two different conformations20, 22, 24, only the structures of the two isolated NS5 domains have been solved25, 26, and there is no structural information available for the full-length NS5. The NS5 N-terminal GTase/MTase domain has a globular shape with dimensions of 55×45×40 Å, and has a fold similar to the SAM-dependent methyltransferase fold25, 27 (Figure 1). Biochemical and structural analyses identified K61-D146-K182-E218 (KDKE motif) as the active site residues of the nucleoside-2'O-MTase activity, while only D146 is essential for the guanine-N7-MTase activity28. The guanylyltransferase catalytic residue has been proposed to be K2912, 13, though this residue is not conserved among the flavivirus NS5 sequences and the reaction mechanism remains uncertain. The NS5 C-terminal RdRp domain has the characteristic shape of the DNA/RNA polymerase superfamily (structural classification of proteins database)29, commonly compared to a right hand with fingers, palm, and thumb subdomains and overall dimensions of 65×60×40 Å26 (Figure 1). However, RdRps differ from other polymerases of the same superfamily in that they adopt a “closed-hand” conformation, which results from an extension of the fingers subdomain, called the fingertips, connecting the thumb subdomain26, 30, 31. The active site of the RdRp domain is contained in the palm subdomain where the conserved RdRp motif C (GDD motif: G662-D663-D664) is found. In all primer-independent RdRp structures, the active site is encircled by the fingers and thumb subdomains, the fingertips, and a thumb protrusion that occlude the proposed dsRNA product exit site30, 31. Thus, structural and biochemical analyses of Flaviviridae RdRps suggest that large conformational changes are required during the exit of the dsRNA product30–33.
Figure 1. Crystal structures of NS5 GTase/MTase (left) and RdRp (right) domains.
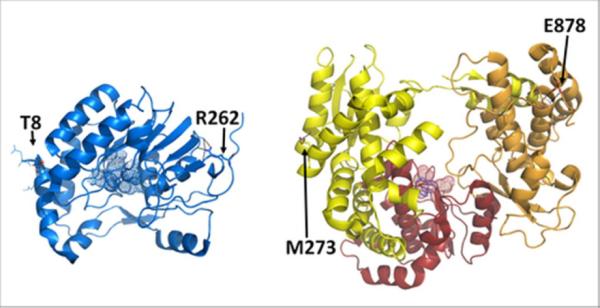
The RdRp subdomains are shown in yellow, red and orange for the fingers, palm and thumb subdomains, respectively. Active site residues are represented by spheres, and residues at the extremities of each domain are represented by sticks and labeled accordingly.
Herein, we report the first structural analysis of the full-length DENV-3 NS5 in solution using small-angle X-ray scattering (SAXS). Our results indicate that NS5 is monomeric in solution, as described previously34. The low resolution shape of NS5 in solution, which resembles an asymmetric and elongated spheroid, has a volume that can accommodate the crystal structures of the DENV NS5 GTase/MTase and RdRp domains very well. However, the relative orientation of the two domains could not be determined accurately. Because the refinement of an atomic model directly against the experimental data is more reliable than docking domain structures into a low resolution SAXS derived shape, we used this strategy to compare our SAXS data to a previously published model of a flavivirus NS535 and to generate alternate models by rigid-body modeling. In addition, we analyze the flexibility of NS5 in solution and demonstrate that it can adopt conformations ranging from compact to more extended forms, with a predominance of compact forms. Overall, our results suggests that in absence of any other protein or nucleic acids, the two domains of NS5 do not interact tightly with each other, and that the non-conserved linker is flexible in solution.
EXPERIMENTAL PROCEDURES
Expression and purification of NS51-878
The DNA fragment encoding DENV-3 NS51-878 (residues 1 to 878) was PCR-amplified from the full-length clone (900 residues) and subcloned into a pET28a vector (EMD biosciences) containing the coding sequence for six histidines and a thrombin cleavage site at the 5' end of the cloning site. Recombinant NS51-878 was produced in BL21-CodonPlus®-RIL Escherichia coli cells (Stratagene). The culture was grown in Luria Broth (LB) medium supplemented with 25 μg/mL chloramphenicol and 50 μg/mL kanamacyn at 37°C until an absorbance (at 600 nm) of 0.6 was reached. Protein expression was induced by addition of 1 mM isopropyl 1-thio-β-D-galactopyranoside at 18°C overnight. Cell pellets were collected by centrifugation (5,000 rpm for 30 min) and resuspended in lysis buffer (50 mM sodium phosphate pH 7 and 1.3 M NaCl) complemented with 1 mg/mL lysozyme, 10 μg/mL ribonuclease A, and one tablet of protease inhibitor cocktail (Roche Applied Science). After 20 min of incubation at 4°C, the cell lysate was sonicated and clarified by centrifugation (17,600×g for 30 min). NS51-878 was first purified by cobalt metal affinity chromatography (TALON® metal affinity resin, Clontech) using an imidazole gradient of 5 to 150 mM in 0.75 M NaCl and 25 mM sodium phosphate buffer, pH 7. This was followed by size-exclusion chromatography (SEC) purification using HiLoad 16/60 Superdex 200 preparative grade (GE Healthcare) equilibrated in SEC buffer (25 mM Tris-HCl pH 7, 400 mM NaCl, 5% glycerol, and 2 mM DTT). The protein homogeneity was then assessed by densitometry on denaturing polyacrylamide gel electrophoresis (SDS-PAGE) and by detection of a single species by analytical ultracentrifugation (AUC; data not shown), in accordance with previous published results for NS51-90034. The protein concentration was determined by UV absorbance at 280 nm with a NanoDrop 1000 spectrophotometer (Thermo Scientific) and a theoretical molar extinction coefficient of 210,708 M−1.cm−1.
Collection and evaluation of SAXS data
SAXS experiments were performed at the ALS beamline 12.3.1 (SIBYLS; Lawrence Berkeley National Laboratory Berkeley, CA, USA). Scattering intensities I(q) for protein and buffer samples were recorded as a function of scattering vector q (q = 4πsinθ/λ, where 2θ is the scattering angle and λ is the X-ray wavelength). The sample-to-detector distance was set to 1.5 m, which resulted in a q range of 0.01 Å−1 to 0.32 Å−1, λ was 1.0 Å and all experiments were performed at 20°C. The data collection strategy described by Hura et al.36, was used in this study. Briefly, SAXS data were collected for three protein concentrations (1.2, 1.8, and 3.5 mg/ml) to evaluate concentration effects on the scattering curves and for two buffer samples (SEC buffer) to minimize errors due to instrumentation. For each sample measurement, SAXS data were collected for three X-ray exposures – one long exposure (5 s.) flanked by two short exposures (0.5 s.) to assess radiation damage. Scattering contributions of the SEC buffer were subtracted from sample scattering data using the program ogreNew (SIBYLS beamline). Data analysis was performed using the program package PRIMUS from the ATSAS suite 2.337. Experimental SAXS data obtained for the different protein concentrations were analyzed for aggregation and folding state using Guinier and Kratky plots, respectively. The forward scattering intensity I(0) (also called the extrapolated scattering intensity at zero angle) and the radius of gyration RG were evaluated using the Guinier approximation: I(q) ≈ I(0) exp(−q2RG2/3), with the limits qRG < 1.3. These parameters were also determined from the pair-distance distribution function P(R), which was calculated from the entire scattering patterns via indirect Fourier inversion of the scattering intensity I(q) using the program GNOM38. The maximum particle diameter Dmax was also estimated from the P(R). The hydrated volume VP of the particle was computed from the Porod invariant, and was used to estimate the molecular mass of a globular protein. Indeed, it was empirically found that the hydrated volume in nm3 should numerically be between 1.5 to 2 times the molecular mass in kDa39. The apparent molecular mass was also determined from I(0) using the following formula: MMp = MMst×[(I(0)p/cp)/(I(0)st/cst)], where MM, I(0) and c are the molecular mass, the forward scattering intensity and the concentration of the protein of interest (p) or the standard protein (st)40. Lysozyme (2, 3 and 6 mg/mL) and bovine serum albumin (1 and 2 mg/mL) were used as standard proteins.
SAXS data analysis
The overall shape of the protein was modeled ab initio by fitting the SAXS data to the calculated SAXS profile of a chain-like ensemble of dummy residues in reciprocal space using the program GASBOR, version 2.2i41.The radius of the volume in which dummy atoms are placed is determined by Dmax/2. Ten independent calculations were performed with no symmetry restrictions. The agreement between the 3D model and the SAXS data was determined using the discrepancy χ2, defined according to Konarev et al.37 and the molecular envelope showing the lower χ2 was chosen as the reference model. The suite DAMAVER was then used to align the molecular envelopes, select the most typical ones and build an averaged model42. The alignment of two molecular envelopes was conducted by minimizing the value of the normalized spatial discrepancy (NSD), which indicates that two shapes are similar when the NSD value is close to one42. The RG and Dmax values of the averaged model were calculated using the program CRYSOL43. The atomic structures of both domains (PDB code 1L9K for the GTase/MTase domain and 2J7U for the RdRp domain) were fit into the averaged molecular envelope using the program SUPCOMB44. SASREF45 was used to calculate a model that best fits to the experimental SAXS data (minimization of the discrepancy χ2) while agreeing with a previously proposed model based on results from genetic interaction experiments35. The tertiary structure of NS51-878 was also modeled using the crystal structures of the individual domains and the program BUNCH45. This program performs modeling of multidomain proteins against SAXS data using a combined rigid body for the atomic structures of the domains, plus an ab initio modeling approach for regions of unknown structure. As with the GASBOR calculations, ten independent models were generated, and the comparison between atomic structures of models and the envelope calculated by GASBOR was conducted using SUPCOMB as described above. To assess the flexibility of this two-domain protein, we employed the EOM (Ensemble Optimization Method) suite46, which can be used to select the best-fitting conformational ensemble when the atomic structures of globular domains are known. First, the program RanCh (Random Chain) creates 10,000 randomized models with different conformations of the linker and protein extremities. The program GAJOE (Genetic Algorithm Judging Optimization of Ensembles) selects a pool of models that best fit the experimental curve. The dimensions of models from the selected pool are then compared to those from the random pool in order to evaluate protein flexibility. For rigid-body calculations, domain extremities were chosen based on the comparison of all available crystal structures for each flavivirus NS5 domain13, 47–50. The GTase/MTase domain included residues between T8 and R262, and the RdRp domain comprised residues between M273 and E878 (last residue of the construct).
RESULTS
NS51-878 is monomeric in solution, as determined by SAXS experiments
The quality of the SAXS measurements was estimated by comparing data from different exposure times and different concentrations. No radiation damage was detected over the 5 s. exposure time, so the data obtained after the long exposure was used for analysis. Several parameters describing shape, size and volume can be extracted directly from scattering curves, but only if the solution is monodisperse. NS5 eluted from SEC as a single peak corresponding to the size of a monomer. After concentration of the protein fractions, the sample homogeneity and purity were estimated by densitometry on SDS-PAGE (>97% purity). The monodispersity as well as the monomeric state of NS51-878 were then confirmed by analytical ultracentrifugation (data not shown). The linearity of the Guinier plot (ln(I(q)) vs. q2) for qRG<1.3 also indicated the quality of the sample51 (Figure 2A). A minor concentration dependence was observed at very small angle for scattering vectors (q<0.035 Å) (Figures 2B and 2C), while at larger angles, the scattering curves superimposed well when the data were scaled for concentration (Figure 2B). This concentration dependence, indicating a slight attractive inter-particle interference, appeared to be linear to a good approximation (R2 = 0.9988) (Figure 2C), and so can be eliminated by extrapolating the experimental data to zero concentration52. The SAXS parameters extrapolated to zero concentration were within the margin of error with those calculated from 1.2 and 1.8 mg/mL samples (Table 1), suggesting that the scattering curves at these concentrations can be considered free from any inter-particle interference.
Figure 2. SAXS data analysis.

A) Guinier plot of the scattered intensities of NS51-878 at 1.2 mg/mL (black dots). The Guinier region (qRG<1.3) is delimited by the gray vertical lines. The red solid line is the linear regression fit in the Guinier region, expanded by a red broken line. The residuals are shown in green. B) Experimental scattering patterns for three concentrations of NS51-878 scaled to concentration. The concentration dependence of the scattering curves at very low scattering angles is shown in the inset. C) Concentration dependence of I(0)/Concentration, where I(0) is the forward scattering intensity derived using the Guinier law. D) Pair-distance distribution function P(R) of NS51-878 at 1.2 and 1.8 mg/mL. Kratky plots of NS51-878 SAXS data at 1.2 and 1.8 mg/mL are shown in the inset.
Table 1.
Parameters derived from SAXS data for three concentrations of NS51–878 and extrapolated to zero concentrationa.
| Concentration (mg/mL) | “0” | 1.2 | 1.8 | 3.5 |
|---|---|---|---|---|
| RG [Guinier37] (Å) | 34.4 ± 1 | 35.1 ± 1 | 35.4 ± 1 | 36.4 ± 1 |
| RG [P(R)38] (Å) | 35.0 ± 1 | 35.6 ± 1 | 36.1 ± 1 | 36.9 ± 1 |
| Dmax [P(R)38] (Å) | - | 125 | 125 | 135 |
| VP [Porod37] (nm3) | 151.4 | 155.6 | 155.9 | 162.2 |
| Molecular mass [I(0)37] (kDa) | 95.5 ± 10 | 97.4 ± 10 | 98.4 ± 10 | 101.1 ± 10 |
| Molecular mass [Porod37] (kDa) | 88.3 ± 13 | 90.8 ± 13 | 90.9 ± 13 | 94.6 ± 13 |
| Oligomerization state | Monomer | Monomer | Monomer | Monomer |
The methods used to determine the parameters are indicated in brackets.
Two parameters addressing the size of the particle in solution, the radius of gyration (RG) and the maximal dimension (Dmax), were determined from the SAXS data. First, the value of RG can be obtained using two independent methods: the Guinier approximation, where only the innermost portion of the scattering curve is used, and the pair-distribution function P(R), which has the advantage of taking into account the whole scattering curve. Both methods provided values in good agreement to each other: 35.1 ± 1 Å and 35.6 ± 1 Å for the 1.2 mg/mL data, and 35.4 ± 1 Å and 36.1 ± 1 Å at 1.8 mg/mL. These values are also in agreement with those obtained by extrapolating to zero concentration − 34.4 ± 1 Å and 35.0 ± 1 Å, respectively (Table 1). Next, the maximal dimension of the particle, Dmax, was estimated from the pair-distance distribution function P(R), which represents the probable distribution of interatomic distances. Dmax was found to be 125 ± 10 Å for both protein solutions of 1.2 mg/mL and 1.8 mg/mL (Figure 2D). The molecular mass of an object in solution can also be determined from two independent measurements: the forward scattering intensity I(0) and the hydrated particle volume (VP). Apparent molecular masses estimated from I(0) were 97.4 ± 10 kDa and 98.4 ± 10 kDa for samples of 1.2 mg/mL and 1.8 mg/mL, respectively. The hydrated particle volumes estimated from Porod plots were 155.6 nm3 at 1.2 mg/mL and 155.9 nm3 at 1.8 mg/mL, corresponding to average molecular masses of 90.8 ± 13 kDa and 90.9 ± 13 kDa, respectively (Table 1). These values, as well as the extrapolated molecular mass at zero concentration (95.5 ± 10 kDa; Table 1), are consistent with the value 103 kDa, calculated from the amino-acid sequence. These results thus indicate that NS51-878 is monodisperse and monomeric in solution at both 1.2 and 1.8 mg/mL. The shape and other modeling calculations presented below were based on the experimental data collected with the 1.2 mg/mL sample, which are very similar to those based on the dataset of the 1.8 mg/mL sample (data not shown).
Ab initio shape calculations of NS51-878
The shape of NS51-878 in solution can be estimated from the shapes of the Kratky plot and the pair-distance distribution function, P(R). The Kratky plot (I(q)×q2 vs. q) (Figure 2D, inset) shows a bell-shaped peak at low angles that indicates a well-folded protein, and the pair-distance distribution function, P(R), shows a characteristic shape of a prolate spheroid (Figure 3)39, 53. A better approximation of the NS51-878 shape was then obtained using the program GASBOR. Ten independent ab initio reconstructions of the NS51-878 envelope produced a similar ellipsoidal envelope (NSD = 1.2 ± 0.1, see “experimental procedures” section), in accordance with the prediction from the shape of the pair-distance distribution function. These models fit the experimental data well, as shown by the fit between the calculated SAXS profile from the reference model and the measured SAXS data (χ2 of 1.3, Figure 3). The averaged model has a relatively flat and asymmetric shape, with a larger bulge at one extremity (Figure 3, inset). The RG and Dmax of the averaged shape are 34.6 Å and 125.6 Å, respectively as calculated from the coordinate file (see “experimental procedures” section). The crystal structures of the individual domains have disc-like shapes, although the GTase/MTase domain (55×45×40 Å; PDB code 1L9K) is significantly smaller than the RdRp domain (65×60×40 Å; PDB code 2J7U). Based on these crystal structures, the averaged model was divided into two portions that seem to correspond to the GTase/MTase domain for the smaller part and to the RdRp domain for the larger part (Figure 3). The crystal structures fit the shape well, but the precise mutual arrangement of the domains could not be determined without ambiguity, even when using a distance restriction (between 10 and 35 Å54) that mimics the 10-residue linker. This is likely due to the overall globular shape of each domain and the low resolution of SAXS-derived shapes.
Figure 3. Ab initio shape determination of NS51-878.

Fit of the SAXS profile derived from the ab initio reference model (red curve, χ2=1.3) to the experimental scattering curve of NS51-878 at 1.2 mg/mL (black). Inset, Two views of the averaged molecular envelope rotated by 90° from each other are shown as surface representations, with fitted crystal structures of GTase/MTase and RdRp domains (same color code as in Figure 1).
Comparison of the SAXS data with a previously published model
In 2007, Malet et al. published a structural model of the full-length NS5 from West Nile virus, another member of the Flavivirus genus35. The model was constructed based on reverse genetic experiments that identified potentially interacting residues in the dengue virus GTase/MTase and RdRp domains. Specifically, the authors used the proposed interaction between residues K46, R47 and E49 from the GTase/MTase domain and residue L512 from the RdRp domains to build their model by protein-protein docking. Two distance restraints were applied during modeling: (a) A restraint of 36 Å between the C-terminal end of the GTase/MTase domain and the N-terminal end of the RdRp domain, and (b) a restraint of 6 Å between residues K46, R47 and E49 from the GTase/MTase domain and residue L512 from the RdRp domain. To determine whether the published model of the full-length NS5 structure is consistent with the SAXS data, we used rigid-body refinement implemented in the program SASREF. Indeed, rather than comparing the structure of the published model directly with the SAXS data, SASREF was used to perform rigid-body modeling to generate a model that satisfies the same distance restraints as the published model while fitting the SAXS data. The structure of the DENV-3 GTase/MTase domain was generated by homology modeling based on the DENV-2 GTase/MTase crystal structure (PDB code: 1L9K). This homology model and the DENV-3 RdRp crystal structure (PDB code: 2J7U) were used in the SASREF calculations. The model that complies with the distance restraints and best fits the data does so very poorly, with χ2 = 6 (Figure 4A). This indicates that the previously proposed model alone could not explain the experimental SAXS data presented herein.
Figure 4. Comparison of the NS51-878 models with SAXS data.

A) Fit of the SAXS profile derived from the “genetic interaction” restrained rigid-body model (red curve, χ2=6.0) to the experimental scattering curve of NS51-878 at 1.2 mg/mL (black). Two views of the atomic model are also shown (same color code as in Figure 1). B) Fit of the scattering profile of the BUNCH model with the best χ2 (red curve, χ2=2.0) to the experimental scattering curve of NS51-878 at 1.2 mg/mL (black curve) is shown. Four models out of ten generated by the program BUNCH are superimposed. These four models were selected to highlight the variety of conformations obtained. The RdRp domains are colored as in Figure 1, and the GTase/MTase domains are colored in different shades of blue. The flexible regions are represented by spheres.
Rigid-body modeling of NS51-878
Ab initio calculations of the NS51-878 molecular envelope and comparison of SAXS data with a published NS5 model were not successful in producing an atomic model of NS51-878 in solution. Because it is more reliable to directly refine the relative positions of the two domains against the experimental data, a combination of rigid body and ab initio modeling was performed, using the program BUNCH45. As described in the experimental procedures section, rigid-body modeling was applied to the domain structures (T8 to R262 for the GTase/MTase domain and M273 to E878 for the RdRp domain), and ab initio modeling was used for the N-terminal extremity (a total of 28 residues including a hexahistidine tag, a thrombin cleavage site, and residues G1 to E7) as well as for the 10-residue linker (H263 to N272). All 10 independent models showed a similar fit to the data, with a χ2 ranging from 2.0 to 2.6 (Figure 4B). While the 10 calculations led to very different models in which the inter-domain interaction surface varied significantly from one to the other, all the models present similarities to the averaged ab initio shape since they can be described as elongated, with the two domains arranged in a side-by-side configuration. In addition, the two NS5 domains interact with each other via a relative small surface area (Figure 4B). In all these models, the fingers subdomain of the RdRp domain was always involved at the intermolecular interface, while the GTase/MTase domain displayed a variety of orientations.
Conformational flexibility of NS51-878 in solution
The modular organization of proteins is often associated with flexibility and multiple conformations in solution55. For NS51-878, the prolate ellipsoidal shape determined by GASBOR, the small and various interface between the two domains in the BUNCH models, and the absence of marked features (i.e. several peaks) in the Kratky plot and the P(R) function, may suggest a weak interaction between the two domains, and thus the presence of flexibility in domain orientation39. We used the program EOM46 to determine if these observations are due to linker flexibility that permits NS51-878 to adopt different conformations in solution. From an ensemble of random NS51-878 models in which the inter-domain linker and the N-terminal extremity were allowed to have different conformations, EOM selected a sub-ensemble of about 20 models that best fit the experimental data (χ2=1.5, Figure 5). Comparing the RG distribution of the selected ensemble against that of the random ensemble gives an indication of the flexibility of NS51-878 in solution. The random RG distribution covers the RG range from about 30 to 50 Å, corresponding to compact and very extended conformations, respectively. In contrast, the RG distribution of the selected pool has a bimodal distribution that could result from a linker region switching between two major conformations in solution. The first peak is relatively sharp and centered around RG = 33−34 Å, and represents about 80% of the selected ensemble of models. This suggests that the most highly populated conformations of NS51-878 are compact in solution. However, the RG distribution contains a second small peak at RG = 44−45 Å, which accounts for about 20% of the sub-ensemble of models (Figure 5).
Figure 5. Multiple conformations of NS51-878.

A) Distributions of radius of gyration values for the random (black) and selected pools (colored area) of NS51-878 structures for each concentration (red: 1.2 mg/mL; blue: 1.8 mg/mL; green: 3.5 mg/mL). B) Fit of the scattering profile derived from the selected ensemble of conformations to the experimental scattering curve of NS51-878 at 1.2 mg/mL (red, χ2=1.5), 1.8 mg/mL (blue, χ2=1.2), and 3.5 mg/mL (green, χ2=1.5). C) Front and side views of two structures in the selected pool (1.2 mg/mL sample). A “compact conformation” (left, RG = 33.9 Ǻ, Dmax = 120.3 Ǻ) and an “extended conformation” (right, RG = 44.6 Ǻ, Dmax = 163.3 Ǻ) are shown with the same color code as in Figure 1. The flexible regions are represented by gray spheres.
To determine whether the presence of an extended conformation an artifact from protein aggregation, EOM analysis was also performed on SAXS data obtained for higher protein concentrations (1.8 and 3.5 mg/mL; Figure 5). In both cases, the selected ensemble fit the SAXS data well with χ2 of 1.2 and 1.5 for the 1.8 and 3.5 mg/mL sample concentrations, respectively (Figure 5B). Similar to the EOM results from the 1.2 mg/mL sample, the RG distribution is bimodal for both concentrations (Figure 5A). Additionally, the frequency of some “compact conformations” (34 Å < RG < 41 Å) increased, while the proportion of “extended” conformations (RG > 41 Å) decreased at larger concentrations. The “extended conformations” represent 18 % and 12 % of the selected pool of structures for the 1.8 and 3.5 mg/mL concentrations, respectively (Figure 5A). Taken together with the EOM results obtained for the 1.2 mg/mL concentration, we found that the percentage of the population with “extended conformations” is linearly dependent on NS5 concentration to a good approximation (coefficient of determination R2 of 0.996). Therefore, the percentage of the population in “extended conformations” was calculated to be 23.3 % when extrapolated to zero concentration. Since the proportion of extended conformations does not increase as the protein concentration increases, we conclude that the observed “extended” population is not an artifact of inter-particle interference or protein aggregation.
DISCUSSION
The dengue virus NS5 protein is the catalytic subunit of the viral replication complex, and so is essential for viral transcription and genome replication. This critical protein is composed of two globular domains tethered by a 10-residue linker. Although their crystal structures have been solved separately25, 26, little is known about the potential interaction between the two domains. To tackle this question in the absence of a crystal structure of the full-length NS5, SAXS experiments were performed, since they are a powerful tool for obtaining structural information regarding the overall shape of objects in solution. Analysis of SAXS and analytical ultracentrifugation data clearly show that NS51-878 is monomeric and monodisperse in solution under our experimental conditions. Multidomain proteins consisting of two or more folded domains joined by linkers represent a high fraction of proteins in the three kingdoms of life, with a prediction of 65% of the proteins in eukaryotes and 40% in bacteria and archaea55. This modular organization is often associated with flexibility and the presence of several protein conformations in solution46. Flexibility between the NS5 domains was established from SAXS data. Therefore, models obtained from ab initio shape calculations and rigid-body modeling represent an “average” of all NS51-878 conformations and do not reflect a unique structure of the protein in solution. About 80% of NS51-878 ensemble structures have a relatively compact structure wherein the two domains likely interact with each other. However, the broad RG (from 30 to 41 Å) of this population hints at the existence of multiple conformations in solution, possibly with diverse contacts between the GTase/MTase and RdRp domains. More extended conformations of NS51-878, in which the two domains do not interact, represent ~20% of NS51-878 ensemble structures in solution. These calculations suggest that the interaction between the GTase/MTase and RdRp domains may be transient and heterogeneous. Thus, even if the model proposed by Malet et al.35 does not fit well with our SAXS data by itself, we cannot exclude the existence of such a conformation in solution and/or in the replication complex. The NS5 linker region (residues 263 to 273) was rarely observed in crystal structures of flavivirus NS5 domains. When it is seen in the structure, this region does not have any secondary structure and shows conformational disorder, as observed in the crystal structures of the Wesselbron virus GTase/MTase domain13. In this latter structure, the linker is a loop connected to the first helix of the RdRp domain (residues 275 to 285), and these two regions show a higher degree of dynamics compared to the rest of the protein13. The high mobility of the C-terminal region of the Wesselbron virus GTase/MTase domain is in accordance with the SAXS data presented here indicating flexibility between the NS5 GTase/MTase and RdRp domains. Furthermore, activity assays indicate that the two domains are functionally independent from each other, since it was shown that the GTase/MTase domain does not affect RdRp activity23 and that the RdRp domain does not affect GTase activity12. In order to fully understand the dynamics of NS5 and the importance of the linker for viral replication, it would be informative to test different sequences, lengths or even the absence of the linker, in vitro and in the context of viral replication. Moreover, trans-complementation experiments between the two domains of NS5 would also be valuable to understand why the GTase/MTase and RdRp domains are linked together. On the other hand, it has been shown that NS5 modulates NS3 NTPase and 5'-RTPase activities, and NS3 modulates NS5 GTase activity12, 15–17. Interactions between NS5 and NS3 were also identified by different methods. For example, the NS3 C-terminal domain was shown to interact with residues from the NS5 fingers domain (residues 320 to 368) using the yeast two-hybrid system56, and a genetic interaction was demonstrated between the NS3 C-terminal domain and the NS5 GTase/MTase domain17, 57. From these data, we propose that NS5 can adopt different conformations in solution, and the flexibility of the linker may be restrained upon interaction with NS3 or other components of the replication complex during the viral life cycle. A structure of the complex between NS5 and NS3 will provide key information regarding essential interaction sites that could become a target for the design of dengue replication inhibitors.
ACKNOWLEDGMENT
The authors wish to acknowledge the SIBYLS beamline 12.3.1 at the Advanced Light Source Lawrence (Berkeley National Laboratory), where SAXS data was collected. We specially thank Michal Hammel for assistance during data collection and Luis Holthauzen for help with AUC experiments and analysis. We are also very grateful to Mark White for helpful discussions and critical reading of the manuscript, and David Konkel for critically editing it.
Funding information: This work was supported by a UTMB Sealy Memorial Endowment Fund research pilot grant and a NIH Research Grant R01 AI-57363 (to K.H.C) and supported in part through a Sealy and Smith foundation grant to the Sealy Center for Structural Biology and Molecular Biophysics.
ABBREVIATIONS
- AUC
analytical ultracentrifugation
- DENV
dengue virus
- DHF
dengue hemorrhagic fever
- Dmax
maximal dimension
- DSS
dengue shock syndrome
- GTase
guanylyltransferase
- MTase
methyltransferase
- NS
nonstructural protein
- NSD
normalized spatial discrepancy
- NTPase
RNA-stimulated nucleoside triphosphatase
- ORF
open reading frame
- P(R)
pair-distribution function
- RdRp
RNA-dependent RNA polymerase
- RG
radius of gyration
- 5'RTPase
5'-RNA triphosphatase
- SEC
size-exclusion chromatography
- SAM
S-adenosyl-L-methionine
- SAXS
small-angle X-ray scattering
- UTR
untranslated region
- VP
hydrated particle volume
REFERENCES
- 1.Gould EA, Solomon T. Pathogenic flaviviruses. Lancet. 2008;371:500–509. doi: 10.1016/S0140-6736(08)60238-X. [DOI] [PubMed] [Google Scholar]
- 2.WHO . Dengue: guidelines for diagnosis, treatment, prevention and control. World Health Organization; 2009. [PubMed] [Google Scholar]
- 3.Mackenzie JS, Gubler DJ, Petersen LR. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat Med. 2004;10:S98–109. doi: 10.1038/nm1144. [DOI] [PubMed] [Google Scholar]
- 4.Vasilakis N, Weaver SC. The history and evolution of human dengue emergence. Adv Virus Res. 2008;72:1–76. doi: 10.1016/S0065-3527(08)00401-6. [DOI] [PubMed] [Google Scholar]
- 5.Rigau-Perez JG, Clark GG, Gubler DJ, Reiter P, Sanders EJ, Vorndam AV. Dengue and dengue haemorrhagic fever. Lancet. 1998;352:971–977. doi: 10.1016/s0140-6736(97)12483-7. [DOI] [PubMed] [Google Scholar]
- 6.Paranjape SM, Harris E. Control of dengue virus translation and replication. Curr Top Microbiol Immunol. 2010;338:15–34. doi: 10.1007/978-3-642-02215-9_2. [DOI] [PubMed] [Google Scholar]
- 7.Khromykh AA, Kondratieva N, Sgro JY, Palmenberg A, Westaway EG. Significance in replication of the terminal nucleotides of the flavivirus genome. J Virol. 2003;77:10623–10629. doi: 10.1128/JVI.77.19.10623-10629.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernandez-Garcia MD, Mazzon M, Jacobs M, Amara A. Pathogenesis of flavivirus infections: using and abusing the host cell. Cell Host Microbe. 2009;5:318–328. doi: 10.1016/j.chom.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Perera R, Kuhn RJ. Structural proteomics of dengue virus. Curr Opin Microbiol. 2008;11:369–377. doi: 10.1016/j.mib.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bollati M, Alvarez K, Assenberg R, Baronti C, Canard B, Cook S, Coutard B, Decroly E, de Lamballerie X, Gould EA, Grard G, Grimes JM, Hilgenfeld R, Jansson AM, Malet H, Mancini EJ, Mastrangelo E, Mattevi A, Milani M, Moureau G, Neyts J, Owens RJ, Ren J, Selisko B, Speroni S, Steuber H, Stuart DI, Unge T, Bolognesi M. Structure and functionality in flavivirus NS-proteins: perspectives for drug design. Antiviral Res. 2009;87:125–148. doi: 10.1016/j.antiviral.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egloff MP, Decroly E, Malet H, Selisko B, Benarroch D, Ferron F, Canard B. Structural and functional analysis of methylation and 5'-RNA sequence requirements of short capped RNAs by the methyltransferase domain of dengue virus NS5. J Mol Biol. 2007;372:723–736. doi: 10.1016/j.jmb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Issur M, Geiss BJ, Bougie I, Picard-Jean F, Despins S, Mayette J, Hobdey SE, Bisaillon M. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA. 2009;15:2340–2350. doi: 10.1261/rna.1609709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bollati M, Milani M, Mastrangelo E, Ricagno S, Tedeschi G, Nonnis S, Decroly E, Selisko B, de Lamballerie X, Coutard B, Canard B, Bolognesi M. Recognition of RNA cap in the Wesselsbron virus NS5 methyltransferase domain: implications for RNA-capping mechanisms in Flavivirus. J Mol Biol. 2009;385:140–152. doi: 10.1016/j.jmb.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 14.Liu L, Dong H, Chen H, Zhang J, Ling H, Li Z, Shi PY, Li H. Flavivirus RNA cap methyltransferase: structure, function, and inhibition. Front Biol. 2010;5:286–303. doi: 10.1007/s11515-010-0660-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui T, Sugrue RJ, Xu Q, Lee AK, Chan YC, Fu J. Recombinant dengue virus type 1 NS3 protein exhibits specific viral RNA binding and NTPase activity regulated by the NS5 protein. Virology. 1998;246:409–417. doi: 10.1006/viro.1998.9213. [DOI] [PubMed] [Google Scholar]
- 16.Yon C, Teramoto T, Mueller N, Phelan J, Ganesh VK, Murthy KH, Padmanabhan R. Modulation of the nucleoside triphosphatase/RNA helicase and 5'-RNA triphosphatase activities of Dengue virus type 2 nonstructural protein 3 (NS3) by interaction with NS5, the RNA-dependent RNA polymerase. J Biol Chem. 2005;280:27412–27419. doi: 10.1074/jbc.M501393200. [DOI] [PubMed] [Google Scholar]
- 17.Davidson AD. Advances in Virus Research. Academic Press; 2009. Chapter 2 New Insights into Flavivirus Nonstructural Protein 5; pp. 41–101. [DOI] [PubMed] [Google Scholar]
- 18.Kapoor M, Zhang L, Ramachandra M, Kusukawa J, Ebner KE, Padmanabhan R. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J Biol Chem. 1995;270:19100–19106. doi: 10.1074/jbc.270.32.19100. [DOI] [PubMed] [Google Scholar]
- 19.Xu T, Sampath A, Chao A, Wen D, Nanao M, Chene P, Vasudevan SG, Lescar J. Structure of the Dengue virus helicase/nucleoside triphosphatase catalytic domain at a resolution of 2.4 A. J Virol. 2005;79:10278–10288. doi: 10.1128/JVI.79.16.10278-10288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo D, Xu T, Hunke C, Gruber G, Vasudevan SG, Lescar J. Crystal structure of the NS3 protease-helicase from dengue virus. J Virol. 2008;82:173–183. doi: 10.1128/JVI.01788-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chernov AV, Shiryaev SA, Aleshin AE, Ratnikov BI, Smith JW, Liddington RC, Strongin AY. The two-component NS2B-NS3 proteinase represses DNA unwinding activity of the West Nile virus NS3 helicase. J Biol Chem. 2008;283:17270–17278. doi: 10.1074/jbc.M801719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo D, Wei N, Doan DN, Paradkar PN, Chong Y, Davidson AD, Kotaka M, Lescar J, Vasudevan SG. Flexibility between the protease and helicase domains of the dengue virus NS3 protein conferred by the linker region and its functional implications. J Biol Chem. 2010;285:18817–18827. doi: 10.1074/jbc.M109.090936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selisko B, Dutartre H, Guillemot JC, Debarnot C, Benarroch D, Khromykh A, Despres P, Egloff MP, Canard B. Comparative mechanistic studies of de novo RNA synthesis by flavivirus RNA-dependent RNA polymerases. Virology. 2006;351:145–158. doi: 10.1016/j.virol.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 24.Assenberg R, Mastrangelo E, Walter TS, Verma A, Milani M, Owens RJ, Stuart DI, Grimes JM, Mancini EJ. Crystal structure of a novel conformational state of the flavivirus NS3 protein: implications for polyprotein processing and viral replication. J Virol. 2009;83:12895–12906. doi: 10.1128/JVI.00942-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Egloff MP, Benarroch D, Selisko B, Romette JL, Canard B. An RNA cap (nucleoside-2'-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21:2757–2768. doi: 10.1093/emboj/21.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yap TL, Xu T, Chen YL, Malet H, Egloff MP, Canard B, Vasudevan SG, Lescar J. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J Virol. 2007;81:4753–4765. doi: 10.1128/JVI.02283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001;29:3784–3795. doi: 10.1093/nar/29.18.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ray D, Shah A, Tilgner M, Guo Y, Zhao Y, Dong H, Deas TS, Zhou Y, Li H, Shi PY. West Nile virus 5'-cap structure is formed by sequential guanine N-7 and ribose 2'-O methylations by nonstructural protein 5. J Virol. 2006;80:8362–8370. doi: 10.1128/JVI.00814-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lo Conte L, Ailey B, Hubbard TJ, Brenner SE, Murzin AG, Chothia C. SCOP: a structural classification of proteins database. Nucleic Acids Res. 2000;28:257–259. doi: 10.1093/nar/28.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrer-Orta C, Arias A, Escarmis C, Verdaguer N. A comparison of viral RNA-dependent RNA polymerases. Curr Opin Struct Biol. 2006;16:27–34. doi: 10.1016/j.sbi.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Choi KH, Rossmann MG. RNA-dependent RNA polymerases from Flaviviridae. Curr Opin Struct Biol. 2009;19:746–751. doi: 10.1016/j.sbi.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 32.Ferron F, Bussetta C, Dutartre H, Canard B. The modeled structure of the RNA dependent RNA polymerase of GBV-C virus suggests a role for motif E in Flaviviridae RNA polymerases. BMC Bioinformatics. 2005;6:255. doi: 10.1186/1471-2105-6-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cameron CE, Moustafa IM, Arnold JJ. Dynamics: the missing link between structure and function of the viral RNA-dependent RNA polymerase? Curr Opin Struct Biol. 2009;19:768–774. doi: 10.1016/j.sbi.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szymanski MR, Jezewska MJ, Bujalowski PJ, Bussetta C, Ye M, Choi KH, Bujalowski W. Full-length Dengue virus RNA-dependent RNA polymerase-RNA/DNA complexes: stoichiometries, intrinsic affinities, cooperativities, base, and conformational specificities. J Biol Chem. 2011;286:33095–33108. doi: 10.1074/jbc.M111.255034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malet H, Egloff MP, Selisko B, Butcher RE, Wright PJ, Roberts M, Gruez A, Sulzenbacher G, Vonrhein C, Bricogne G, Mackenzie JM, Khromykh AA, Davidson AD, Canard B. Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J Biol Chem. 2007;282:10678–10689. doi: 10.1074/jbc.M607273200. [DOI] [PubMed] [Google Scholar]
- 36.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr., Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konarev PV, Volkov VV, Sokolova AV, Koch MH, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003;36:1277–1282. [Google Scholar]
- 38.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- 39.Mertens HD, Svergun DI. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. Journal of Structural Biology. 2010;172:128–141. doi: 10.1016/j.jsb.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 40.Mylonas E, Svergun DI. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. Journal of Applied Crystallography. 2007;40:s245–s249. [Google Scholar]
- 41.Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Svergun D, Barberato C, Koch MHJ. CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. Journal of applied crystallography. 1995;28:768–773. [Google Scholar]
- 44.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. Journal of Applied Crystallography. 2001;34:33–41. [Google Scholar]
- 45.Petoukhov MV, Svergun DI. Global Rigid Body Modeling of Macromolecular Complexes against Small-Angle Scattering Data. Biophysical journal. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernadó P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural Characterization of Flexible Proteins Using Small-Angle X-ray Scattering. Journal of the American Chemical Society. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 47.Assenberg R, Ren J, Verma A, Walter TS, Alderton D, Hurrelbrink RJ, Fuller SD, Bressanelli S, Owens RJ, Stuart DI, Grimes JM. Crystal structure of the Murray Valley encephalitis virus NS5 methyltransferase domain in complex with cap analogues. J Gen Virol. 2007;88:2228–2236. doi: 10.1099/vir.0.82757-0. [DOI] [PubMed] [Google Scholar]
- 48.Geiss BJ, Stahla H, Hannah AM, Gari AM, Keenan SM. Focus on flaviviruses: current and future drug targets. Future Med Chem. 2009;1:327–344. doi: 10.4155/fmc.09.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jansson AM, Jakobsson E, Johansson P, Lantez V, Coutard B, de Lamballerie X, Unge T, Jones TA. Structure of the methyltransferase domain from the Modoc virus, a flavivirus with no known vector. Acta Crystallogr D Biol Crystallogr. 2009;65:796–803. doi: 10.1107/S0907444909017260. [DOI] [PubMed] [Google Scholar]
- 50.Bollati M, Milani M, Mastrangelo E, de Lamballerie X, Canard B, Bolognesi M. Crystal structure of a methyltransferase from a no-known-vector Flavivirus. Biochem Biophys Res Commun. 2009;382:200–204. doi: 10.1016/j.bbrc.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 51.Vachette P, Koch MH, Svergun DI. Looking behind the beamstop: X-ray solution scattering studies of structure and conformational changes of biological macromolecules. Methods Enzymol. 2003;374:584–615. doi: 10.1016/S0076-6879(03)74024-5. [DOI] [PubMed] [Google Scholar]
- 52.Pilz I. In: Small Angle X-ray Scattering, chapter 8: Proteins. Glatter O, Kratky O, editors. Academic Press Inc., London Ltd.; 1982. pp. 239–293. [Google Scholar]
- 53.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 54.George RA, Heringa J. An analysis of protein domain linkers: their classification and role in protein folding. Protein Eng. 2002;15:871–879. doi: 10.1093/protein/15.11.871. [DOI] [PubMed] [Google Scholar]
- 55.Bernadó P. Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur Biophys J. 2010;39:769–780. doi: 10.1007/s00249-009-0549-3. [DOI] [PubMed] [Google Scholar]
- 56.Johansson M, Brooks AJ, Jans DA, Vasudevan SG. A small region of the dengue virus-encoded RNA-dependent RNA polymerase, NS5, confers interaction with both the nuclear transport receptor importin-beta and the viral helicase, NS3. J Gen Virol. 2001;82:735–745. doi: 10.1099/0022-1317-82-4-735. [DOI] [PubMed] [Google Scholar]
- 57.Kroschewski H, Lim SP, Butcher RE, Yap TL, Lescar J, Wright PJ, Vasudevan SG, Davidson AD. Mutagenesis of the dengue virus type 2 NS5 methyltransferase domain. J Biol Chem. 2008;283:19410–19421. doi: 10.1074/jbc.M800613200. [DOI] [PubMed] [Google Scholar]