

The idea that transglutaminase (TG), a posttranslational protein crosslinking enzyme (for reviews, see refs. 1–6), might be involved in the pathology of neurodegenerative diseases, notably of Alzheimer disease (AD), was first suggested by the experiments of Selkoe et al. (7, 8). These authors drew particular attention to the reactions of TG with brain neurofilaments. Miller and Anderton (9) extended the observations by showing that, in addition to the neurofilament triplet proteins, the microtubule-associated proteins were also good substrates for TG. The dynamic quality of the neural intermediate filament network (10) is essential for maintaining the plasticity of the cytoskeleton and cell architecture, in general. Linking together noncovalently assembled segments of the network with intermolecular Nɛ(γ-glutamyl)lysine side chain bridges could impact adversely on the functions and viability of the neuron. Efforts are underway for documenting the TG-mediated covalent polymerization of the microtubule-associated τ proteins (11, 12) which are the main (if not the only) constituents of the intracellular neurofibrillary tangles (or paired helical filaments; PHF) often seen in AD (13) and other diseases (14). With recombinant human τ40 protein as substrate for human TG, a number of potential crosslinking sites were identified by employing dansylcadaverine for the enzyme-directed substitutions of Gln (or acceptor) residues (15) and dansyl-ɛ-aminocaproyl Gln-Gln-Ile-Val for those of the Lys (or donor) residues (16). Listed roughly in the order of reactivities, the following side chains in τ40 were derivatized: Gln-424, -88, -6, -244, -351, -124, -276, and -288; Lys-383, -385, -174, -180, -225, -263, and -24 (S. N. P. Murthy, J. A. Kuret, J. Wilson, T. J. Lukas, and L.L., unpublished). It remains to be determined which of these residues is involved in the homologous polymerization of τ to τ and which is involved in the crosslinking of τ to other neuronal constituents. The in vitro reactions of TG with neurofilament proteins (8) or with τ proteins (11) did not produce PHF. Perhaps some prior modification (e.g., phosphorylation) of the substrates is required for the correct ordering of polymeric assemblies.
However, the cytoskeletal alterations, including the formation of PHF, may only be a secondary response to cerebral insults which, in AD, is mainly attributed to the 40- to 42-residue amyloid β protein (Aβ or βA4; ref. 17), proteolytically processed from a larger transmembrane precursor and secreted to the outside. Aβ has a marked neurotoxic effect that is probably critical for inducing or amplifying the process of brain cell degeneration; the protein is also a main constituent of the amyloid deposits in AD. Aβ is a good substrate for TG (18, 19), and the same has been shown for another peptide component (20) present in the amyloid plaque (called non-Aβ component or NAC, derived from a larger precursor, NACP or synuclein). It has been suggested that the crosslinking of the βA4 amyloid peptide by TG may increase its neurotoxicity*, similarly to the effect of the enzyme on interleukin 2, which converts the cytokine into a factor cytotoxic to oligodendrocytes (21–23).
Green proposed (24) that TG could also be involved in the development of Huntington disease (HD) and introduced the novel idea that the N-terminal Gln repeats of the abnormal gene product (huntingtin) might render the mutant protein a particularly favorable target for TG. Similarly expanded Gln repeats are found in proteins associated with other neurological diseases, e.g., spinocerebellar ataxia, SCA1 (25); Machado–Joseph disease, SCA3 (26); dentato-rubral-pallido-luysian atrophy (DRPLA) (27, 28); and spinal and bulbar muscular atrophy (SBMA) (29). Thus, enhanced TG reactivity of substrates, not present in the normal brain, could have pathogenic implications beyond HD. Kahlem et al. (30) examined a number of model peptides and showed that reactivity to TG increased with the lengths of Gln repeats. In fact, most peptides were inactive at a single glutamine residue, and addition of repeats contributed large increases to the reactivity of each glutamine residue, as long as the peptides remained soluble.
Commercially available guinea pig liver TG (31) is often used in experiments with human proteins as substrates, but the human brain enzyme seems to bear closer similarity to the homospecific red blood cell TG (8). Though both enzymes are cytosolic in nature and are down-regulated by GTP (32–34), requiring only Ca2+ ions to become activated, there are significant differences between the human and guinea pig enzymes with regard to their associations and the selection of Gln targets even in the same protein substrate. In the extracellular environment, a site for extensive changes in AD and HD, coagulation Factor XIII (35) might also become an important player. The A subunits, carrying the masked catalytic sites in the zymogen, have been detected in some microglia and macrophages in neurodegenerative diseases, such as AD (36). Potential extracellular targets include the myelin basic protein (8), although the TG-catalyzed reaction with the protein has not been implicated in the pathology of any neurodegenerative disease thus far. The porcine substrate reacted with activated Factor XIII at Gln-74, -122, -146, and -148 (P. M. Turner and L.L., unpublished).
Which of the TGs might contribute to the progression of a given disease? With the availability of transgenic mouse models (37, 38), which TG gene should be considered for a knockout experiment? Eliminating Factor XIII would represent an obstacle, because the autosomal recessive inheritance of the deficiency in humans is known to be associated with severe bleeding (39, 40). Various TGs (41–44), including a synapse-specific TG (45), were reported to be present in the brain; tetanus toxin is thought to activate the latter. Presumably, one of the brain enzymes would have to catalyze the intracellular, covalent polymerization of τ proteins and the crosslinking of the neurofilament proteins of the cytoskeleton and of synapsin I, a membrane-associated protein involved in neurotransmitter release. It should also be mentioned that Gh, a receptor signaling protein that activates phospholipase C, is a member of the TG family (46, 47). It is difficult to say a priori whether crosslinking of huntingtin occurred intra- or extracellularly. Kahlem et al. showed that aggregation of one of the model peptides (R5Q18R5) was produced and glycine ethylester was incorporated into another (DRPLA) by brain transglutaminase alone (30).
Based on studies with the human red blood cell enzyme, we estimate that about 5 × 10−5 M Ca2+ would be needed for the half-maximal activation of the cytosolic TG (S. N. P. Murthy and L.L., unpublished). At what point during protracted cell death could such an excess of Ca2+ leak into the cell from the outside or be released from internal stores? Would this increase in Ca2+ be a sudden event, akin to the Ca2+ toxicity simulated by experiments for activating the endogenous latent TG (as well as proteases) in red blood cells (48, 49), lens (50, 51), and keratinocytes (52, 53)? Or could TG become activated to a lesser extent by repeated, small transient rises of Ca2+ concentration, allowing for the gradual accumulation of crosslinked products over a longer time period? It may be relevant to this issue that tetanic stimulation was found to increase the crosslink content in rat brain slices (54). When do the Ca2+ pumps fail in AD and HD, and what causes them to fail? Alas, we cannot answer these questions, and we can only play the role of archaeologists, digging among the structural ruins!
An immediate inclination might be to measure the Nɛ(γ-glutamyl)lysine crosslinks, the footprints of prior TG action, in cell fractions or deposits, e.g., amyloid or PHF in AD. Since the stability (i.e., the free energy of hydrolysis) of the side chain isopeptide bridge is not significantly different from that of a peptide in the backbone, there are no chemical procedures for differential cleavage. Serial fragmentation of the protein with proteolytic enzymes is employed for breaking down the backbone to amino acids without hydrolyzing the γ:ɛ isodipeptide. However, biological deposits (such as PHF) often resist complete digestion by proteolytic enzymes; hence, crosslink frequencies can be looked upon only as minimal estimates. The results presented in Fig. 1A indicate that TG functions in some steps of normal brain cell differentiation, laying down a skeleton (which could not be dissolved in 2% sodium dodecylsulfate:0.1 M 2-mercaptoethanol) with a relatively high frequency of cross bridges [about 1 mol of Nɛ(γ-glutamyl)lysine per 50,000 unit weight of protein]. It has also been observed that TG activity increased during mouse brain maturation 2.5-fold from day 3 to adulthood (57). Against the high background in normal brain, measuring crosslink frequencies in neurodegenerative diseases may not be a simple task. In any case, flanking sequences around the crosslinks will have to be determined from partial digests of cell fractions, so that assignments to specific protein constituents could be made (e.g., to τ protein in AD or to the poly-Gln extension of huntingtin in HD). It will have to be proved, as control, that the same crosslinked sequences are absent from normal brain.
Figure 1.
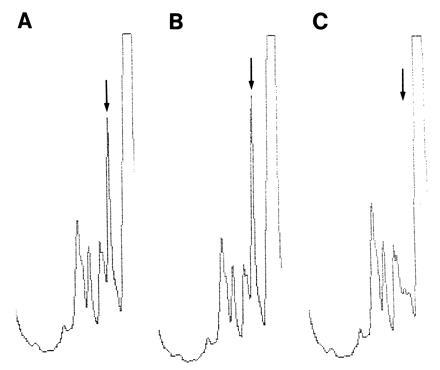
Nɛ(γ-glutamyl)lysine crosslinks are present in normal human brain (J. Wilson, D. Selkoe, and L.L., unpublished). Minced cerebral cortex was incubated (2 hr, room temperature, 50 mM Tris·HCl, pH 7) with sodium dodecylsulfate (2%) and 2-mercaptoethanol (0.1 M), heated (100°C, 10 min), passed through a Nitex sieve (110 μm), and centrifuged (1500 × g; see ref. 7). Proteolytic digestion of the pellet and crosslink analysis were carried out essentially as described (55) but with inclusion of alkaline protease. Only the elution profile around the Nɛ(γ-glutamyl)lysine peak (marked by arrow) is shown. (A) The cortical digest. (B) The cortical digest mixed with the authentic Nɛ(γ-glutamyl)lysine dipeptide (25 pmol). (C) The same mixture as in B, after treatment with γ-glutamylaminocyclotransferase (56), which seen to degrade the isodipeptide. From A, the calculated frequency of Nɛ(γ-glutamyl)lysine in the cortical sample was approximately 1 mol per 500 amino acid residues.
Crosslinking, either through a polyamine linker or through the formation of the Nɛ(γ-glutamyl)lysine bridge (Fig. 2) may not be the only means for changing the solubilities and interactions of proteins by reaction with TG. Concerning the catalytic mechanism, these enzymes are very similar to papain and function also in a hydrolytic mode. The hydrolysis of even a single Gln to a Glu residue in a protein, though representing a change of just a few kilocalories in the thermodynamic behavior of the side chain, can have an enormous effect on the equilibria governing the conformation, the oligomeric associations-dissociations, and the interactions of the macromolecule with other proteins (59). However, searching for TG-mediated Gln-to-Glu conversions in biological specimens would present a formidable project.
Figure 2.
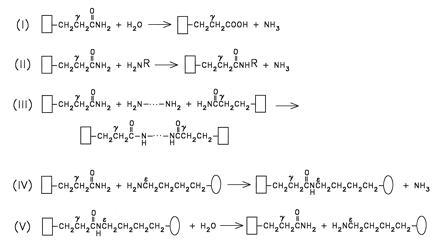
Reactions catalyzed by transglutaminases. The backbones of protein or peptide substrates, carrying TG-reactive Gln (i.e., acceptor) and Lys (i.e., donor) side chains, are illustrated by open rectangles and ovals, respectively. Reactions RI and RV reflect on the hydrolytic nature of this group of enzymes (for the isopeptide hydrolyzing activities indicated in V, see ref. 58). Transamidating possibilities are shown in reactions II–IV. Geometry of crosslinked products, as formed in reaction IV, depends on the number and spatial distribution of TG-reactive Gln and Lys residues in the acceptor and donor protein substrates (3). Synthetic, small molecular weight primary amines (H2NR, reaction II, e.g., glycine ethylester or dansylcadaverine) are frequently used to inhibit protein-to-protein crosslinking events by blocking the TG-reactive Gln residues.
The specificities of TGs are determined mainly by the higher order structure of substrates in their natural environments. No consensus sequences were identified for the Gln (acceptor) and Lys (donor) functionalities with which a TG would react. Thus, taking the interesting findings of Kahlem et al. (30) as a point of departure, the following questions will need to be addressed. (i) Is the reactivity of the Gln repeats in huntingtin different from that of the model peptides? (ii) Does the TG reaction change by formation of a complex between huntingtin and HAP1? This huntingtin-associated protein (60) plays a critical role in the brain-specific pathology of HD and binds to the Gln extensions of the mutant protein. Would the association of huntingtin with HAP1 reduce the accessibility of the Gln repeats to TG for amine substitution, while perhaps promoting its heterologous crosslinking to HAP1? Is HAP1 a unique partner in brain for the TG-catalyzed reaction with huntingtin?
In the present state of knowledge (see also ref. 61), numerous questions may be asked, but most of them seem to be tractable by further experimentation. Also, the possibility exists that an “end run” could be mounted for seeking additional evidence that TG may be involved in the progression of the pathology of some of the neurodegenerative diseases. Nontoxic active center-directed inhibitors of TG are becoming available (62), and Zn2+ is also known to be a potent inhibitor of these enzymes (3). If sufficient concentrations of these inhibitors could be delivered to the brains of the AD and SCA1 transgenic mice (37, 38), we could gain a deeper insight.
Acknowledgments
Research pertaining to this article was supported by Grants HL-02212 and EY-03942 from the National Institutes of Health, U.S. Public Health Service.
Footnotes
Ikura, K., Shimagawa, R., Masuda, S., Takahata, K. & Sasaki, R. (1994) 4th International Conference on Transglutaminase and Protein Crosslinking Reactions, Aug. 28–31, 1994, Debrecen, Hungary.
References
- 1.Lorand L, Stenberg P. In: Handbook of Biochemistry and Molecular Biology. Fasman G D, editor. II. Cleveland: CRC; 1976. pp. 669–689. [Google Scholar]
- 2.Folk J E. Ann Rev Biochem. 1980;49:517–531. doi: 10.1146/annurev.bi.49.070180.002505. [DOI] [PubMed] [Google Scholar]
- 3.Lorand L, Conrad S M. Mol Cell Biochem. 1984;58:9–35. doi: 10.1007/BF00240602. [DOI] [PubMed] [Google Scholar]
- 4.Ichinose A, Bottenus R E, Davie E W. J Biol Chem. 1990;265:13411–13414. [PubMed] [Google Scholar]
- 5.Greenberg C S, Birckbichler P J, Rice R H. FASEB J. 1991;5:3071–3077. doi: 10.1096/fasebj.5.15.1683845. [DOI] [PubMed] [Google Scholar]
- 6.Aeschlimann D, Paulsson M. Thromb Haemostasis. 1994;71:402–415. [PubMed] [Google Scholar]
- 7.Selkoe D J, Ihara Y, Salazar F J. Science. 1982;215:1243–1245. doi: 10.1126/science.6120571. [DOI] [PubMed] [Google Scholar]
- 8.Selkoe D J, Abraham C, Ihara Y. Proc Natl Acad Sci USA. 1982;79:6070–6074. doi: 10.1073/pnas.79.19.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller C C J, Anderton B H. J Neurochem. 1986;46:1912–1922. doi: 10.1111/j.1471-4159.1986.tb08513.x. [DOI] [PubMed] [Google Scholar]
- 10.Straube-West K, Loomis P A, Opal P, Goldman R D. J Cell Sci. 1996;109:2319–2329. doi: 10.1242/jcs.109.9.2319. [DOI] [PubMed] [Google Scholar]
- 11.Dudek S M, Johnson G V W. J Neurochem. 1993;61:1159–1162. doi: 10.1111/j.1471-4159.1993.tb03636.x. [DOI] [PubMed] [Google Scholar]
- 12.Miller M L, Johnson G V W. J Neurochem. 1995;65:1760–1770. doi: 10.1046/j.1471-4159.1995.65041760.x. [DOI] [PubMed] [Google Scholar]
- 13.Goedert M, Trojanowski J Q, Lee V M-Y. In: The Molecular and Genetic Basis of Neurological Disease. 2nd Ed. Rosenberg R N, Prusiner S B, DiMaura S, Barchi R L, editors. Boston: Butterworth–Heinemann; 1996. [Google Scholar]
- 14.Wisniewski K, Jervis G A, Moretz R C, Wisniewski H M. Ann Neurol. 1979;5:288–294. doi: 10.1002/ana.410050311. [DOI] [PubMed] [Google Scholar]
- 15.Murthy S N P, Wilson J, Zhang Y, Lorand L. J Biol Chem. 1994;269:22907–22911. [PubMed] [Google Scholar]
- 16.Lorand L, Velasco P T, Murthy S N P, Wilson J, Parameswaran K N. Proc Natl Acad Sci USA. 1992;89:11161–11163. doi: 10.1073/pnas.89.23.11161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selkoe D J. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- 18.Ikura K, Takahata K, Sasaki R. FEBS Lett. 1993;326:109–111. doi: 10.1016/0014-5793(93)81772-r. [DOI] [PubMed] [Google Scholar]
- 19.Rasmussen L K, Sorensen E S, Peterson T E, Gliemann J, Jensen P H. FEBS Lett. 1994;338:161–166. doi: 10.1016/0014-5793(94)80356-0. [DOI] [PubMed] [Google Scholar]
- 20.Jensen P H, Sorensen E S, Petersen T E, Gliemann J, Rasmussen L K. Biochem J. 1995;310:91–94. doi: 10.1042/bj3100091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eitan S, Schwartz M. Science. 1993;261:106–108. doi: 10.1126/science.8100369. [DOI] [PubMed] [Google Scholar]
- 22.Eitan S, Solomon A, Lavie V, Yoles E, Hirschberg D L, Belkin M, Schwartz M. Science. 1994;264:1764–1768. doi: 10.1126/science.7911602. [DOI] [PubMed] [Google Scholar]
- 23.Eizenberg O, Faber-Elman A, Gottlieb E, Oren M, Rotter V, Schwartz M. EMBO J. 1995;14:1136–1144. doi: 10.1002/j.1460-2075.1995.tb07097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green H. Cell. 1993;74:955–956. doi: 10.1016/0092-8674(93)90718-6. [DOI] [PubMed] [Google Scholar]
- 25.Orr H T, Chung M Y, Banfi S, Kwiatkowski T J, Jr, Servadio A, Beaudet A L, McCall A E, Duvick L A, Ranum L P, Zoghbi H Y. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- 26.Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Kimura J, Narumiya S, Kakizuka A. Nat Genet. 1994;8:221–228. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- 27.Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, Takahashi H, Kondo R, Ishikawa A, Hayashi T, Saito M, Tomoda A, Miike T, Naito H, Ikuta F, Tsuji S. Nat Genet. 1994;6:9–13. doi: 10.1038/ng0194-9. [DOI] [PubMed] [Google Scholar]
- 28.Nagafuchi S, Yanagisawa H, Sato K, Shirayama T, Ohsaki E, et al. Nat Genet. 1994;6:14–18. doi: 10.1038/ng0194-14. [DOI] [PubMed] [Google Scholar]
- 29.La Spada A R, Wilson E M, Lubahn D B, Harding A E, Fischbeck K H. Nature (London) 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 30.Kahlem P, Terré C, Green H, Djian P. Proc Natl Acad Sci USA. 1996;93:14580–14585. doi: 10.1073/pnas.93.25.14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark D D, Mycek M J, Neidle A, Waelsch H. Arch Biochem Biophys. 1959;79:338–354. doi: 10.1016/0003-9861(59)90613-7. [DOI] [PubMed] [Google Scholar]
- 32.Achyuthan K E, Greenberg C S. J Biol Chem. 1987;262:1901–1906. [PubMed] [Google Scholar]
- 33.Lee K N, Birckbichler P J, Patterson M K., Jr Biochem Biophys Res Commun. 1989;162:1370–1375. doi: 10.1016/0006-291x(89)90825-5. [DOI] [PubMed] [Google Scholar]
- 34.Bergamini C M, Signorini M, Caselli L, Melandri P. Biochem Mol Biol Int. 1993;30:727–732. [PubMed] [Google Scholar]
- 35.Lorand L, Jeong J-M, Radek J T, Wilson J. Methods Enzymol. 1993;222:22–35. doi: 10.1016/0076-6879(93)22005-z. [DOI] [PubMed] [Google Scholar]
- 36.Akiyama H, Kondo H, Ikeda K, Arai T, Kato M, McGleer P L. Neurosci Lett. 1995;202:29–32. doi: 10.1016/0304-3940(95)12188-9. [DOI] [PubMed] [Google Scholar]
- 37.Burright E N, Clark H B, Servadio A, Matilla T, Feddersen R M, Yunis W S, Duvick L A, Zoghbi H Y, Orr H T. Cell. 1995;82:937–948. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- 38.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Nature (London) 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 39.Lorand L, Urayama T, Atencio A C, Hsia D Y-Y. Am J Hum Genet. 1970;22:89–95. [PMC free article] [PubMed] [Google Scholar]
- 40.Lorand L, Losowsky M S, Miloszewski K J M. In: Progress in Hemostasis and Thrombosis. Spaet T H, editor. Vol. 5. New York: Grune & Stratton; 1980. pp. 245–290. [PubMed] [Google Scholar]
- 41.Wajda I J, Lee J M, Neidle A. J Neurochem. 1969;16:655–663. doi: 10.1111/j.1471-4159.1969.tb06865.x. [DOI] [PubMed] [Google Scholar]
- 42.Gilad G M, Varon L E. J Neurochem. 1985;45:1522–1526. doi: 10.1111/j.1471-4159.1985.tb07222.x. [DOI] [PubMed] [Google Scholar]
- 43.Perry M J M, Mahoney S-A, Haynes L W. Neuroscience. 1995;65:1063–1076. doi: 10.1016/0306-4522(94)00556-k. [DOI] [PubMed] [Google Scholar]
- 44.Ohashi H, Itoh Y, Birckbichler P J, Takeuchi Y. J Biochem. 1995;118:1271–1278. doi: 10.1093/oxfordjournals.jbchem.a125018. [DOI] [PubMed] [Google Scholar]
- 45.Facchiano F, Benfenati F, Valtorta F, Luini A. J Biol Chem. 1993;268:4588–4591. [PubMed] [Google Scholar]
- 46.Nakaoka H, Perez D M, Baek K J, Das T, Husain A, Misono K, Im M-J, Graham R M. Science. 1994;264:1593–1596. doi: 10.1126/science.7911253. [DOI] [PubMed] [Google Scholar]
- 47.Feng J-F, Rhee S G, Im M-J. J Biol Chem. 1996;271:16451–16454. doi: 10.1074/jbc.271.28.16451. [DOI] [PubMed] [Google Scholar]
- 48.Siefring G E, Jr, Apostol A B, Velasco P T, Lorand L. Biochemistry. 1978;17:2598–2604. doi: 10.1021/bi00606a022. [DOI] [PubMed] [Google Scholar]
- 49.Lorand L, Bjerrum O J, Hawkins M, Lowe-Krentz L, Siefring G E., Jr J Biol Chem. 1983;258:5300–5305. [PubMed] [Google Scholar]
- 50.Lorand L, Hsu L K H, Siefring G E, Jr, Rafferty N S. Proc Natl Acad Sci USA. 1981;78:1356–1360. doi: 10.1073/pnas.78.3.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Velasco P T, Murthy P, Goll D E, Lorand L. Biochim Biophys Acta. 1990;1040:8472–8475. doi: 10.1016/0167-4838(90)90074-p. [DOI] [PubMed] [Google Scholar]
- 52.Rice R H, Green H. J Cell Biol. 1978;76:705–711. doi: 10.1083/jcb.76.3.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Green, H. (1979) in Harvey Lect. 74, 101–139. [PubMed]
- 54.Friedrich P, Fesus L, Tarcsa E, Czeh G. Neuroscience. 1991;43:331–334. doi: 10.1016/0306-4522(91)90297-2. [DOI] [PubMed] [Google Scholar]
- 55.Murthy S N P, Wilson J, Guy S L, Lorand L. Proc Natl Acad Sci USA. 1991;88:10601–10604. doi: 10.1073/pnas.88.23.10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fink M L, Chung S I, Folk J E. Proc Natl Acad Sci USA. 1980;77:4564–4568. doi: 10.1073/pnas.77.8.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maccioni R B, Seeds N W. Mol Cell Biochem. 1986;69:161–168. doi: 10.1007/BF00224763. [DOI] [PubMed] [Google Scholar]
- 58.Cheng X-F, Chen E, Velasco P T, Parameswaran K N, Lorand L. FASEB J. 1996;10:A1120. (abstr.). [Google Scholar]
- 59.Klotz I M, Darnall D W, Langerman N R. In: The Proteins. 3rd Ed. Neurath H, Hill R L, editors. I. New York: Academic; 1975. pp. 293–411. [Google Scholar]
- 60.Li X-J, Sharp A H, Li S-H, Dawson T M, Snyder S H, Ross C A. Proc Natl Acad Sci USA. 1996;93:4839–4844. doi: 10.1073/pnas.93.10.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cariello L, de Cristofaro T, Zanetti L, Cuomo T, Di Maio L, Campanella G, Rinaldi S, Zanetti P, Di Lauro R, Varrone S. Hum Genet. 1996;98:633–635. doi: 10.1007/s004390050273. [DOI] [PubMed] [Google Scholar]
- 62.Freund K F, Doshi K P, Gaul S L, Claremon D A, Remy D C, Baldwin J J, Pitzenberger S M, Stern A M. Biochemistry. 1994;33:10109–10119. doi: 10.1021/bi00199a039. [DOI] [PubMed] [Google Scholar]