

Abstract
The aim of this study was to use comparative modeling to predict the three-dimensional structure of the CHAPK protein (cysteine, histidine-dependent amidohydrolase/peptidase domain of the LysK endolysin, derived from bacteriophage K). Iterative PSI-BLAST searches against the Protein Data Bank (PDB) and nonredundant (nr) databases were used to populate a multiple alignment for analysis using the T-Coffee Expresso server. A consensus Maximum Parsimony phylogenetic tree with a bootstrap analysis setting of 1,000 replicates was constructed using MEGA4. Structural templates relevant to our target (CHAPK) were identified, processed in Expresso and used to generate a 3D model in the alignment mode of SWISS-MODEL. These templates were also processed in the I-TASSER web server. A Staphylococcus saprophyticus CHAP domain protein, 2K3A, was identified as the structural template in both servers. The I-TASSER server generated the CHAPK model with the best bond geometries when analyzed using PROCHECK and the most logical organization of the structure. The predicted 3D model indicates that CHAPK has a papain-like fold. Circular dichroism spectropolarimetry also indicated that CHAPK has an αβ fold, which is consistent with the model presented. The putative active site maintained a highly conserved Cys54-His117-Glu134 charge relay and an oxyanion hole residue Asn136. The residue triplet, Cys-His-Glu, is known to be a viable proteolytic triad in which we predict the Cys residue is used in a nucleophilic attack on peptide bonds at a specific site in the pentaglycine cross bridge of staphylococcal cell wall peptidoglycan. Use of comparative modeling has allowed approximation of the 3D structure of CHAPK giving information on the structure and an insight into the binding and active site of the catalytic domain. This may facilitate its development as an alternative antibacterial agent.
Keywords: bacteriophage, CHAP, endolysin, in silico, peptidase, staphylococcus
Introduction
Staphylococcus aureus is a common pathogen that plays a major role in various human and animal diseases ranging from skin and soft tissue infections to more serious cases of pneumonia, endocarditis, meningitis and osteomyelitis.1 Treatment of these infections has become increasingly difficult due to the worldwide prevalence of multidrug-resistant strains including methicillin resistant S. aureus (MRSA) which is a frequent cause of serious nosocomial infections.2 As a consequence it is critical to develop new and effective antibacterials with the potential of eliminating such infections irrespective of antibiotic sensitivity.
Over the past decade numerous studies have focused on developing recombinant bacteriophage (phage)-encoded cell wall hydrolases, termed endolysins (lysins), as novel antibacterial agents as recently reviewed by Loessner (2005),3 Fischetti (2008)4 and Fenton et al. (2010).5 When applied exogenously as purified recombinant proteins to Gram-positive bacteria, lysins bring about rapid cell lysis and bacterial death.3,6,7 It is this ability to rapidly lyse pathogenic Gram-positive cells upon direct contact with peptidoglycan, also termed “lysis from without,” that has laid the foundation for their exploitation as novel therapeutics.6
The majority of lysins display a modular structure, usually comprising of at least one N-terminal catalytic domain which attacks bacterial cell wall peptidoglycan, combined with a C-terminal cell wall binding domain which directs the lytic domain to its site of action.8,9 In the case of staphylococcal lysins the presence of three domains, comprising an N-terminal cysteine, histidine-dependent amidohydrolase/peptidase (CHAP) domain and an amidase-2 domain linked to a C-terminal SH3b cell wall binding domain, is a common feature. This organization has been observed in LysK,10 LysWMY,11 Φ11 lysin,12 MV-L,13 LysH5,14 LysGH15,15 and SAL-1.16
Studies have shown that of the two lytic domains contained within staphylococcal lysins, the CHAP domain confers the principle enzymatic activity of the protein, whereas the amidase domain contributes minimal detectable activity.17-20 Furthermore, a recent study in our laboratory showed that the activity of the single-domain truncated CHAP (later designated CHAPK), was 2-fold higher than the native three-domain LysK protein.20 Studies have also demonstrated that the CHAP domain of staphylococcal lysins acts as a D-Ala-Gly endopeptidase, specifically cleaving the peptide bond between D-alanine and the first glycine in the pentaglycine cross-bridge of staphylococcal cell wall peptidoglycan17,18 (Fig. 1).

Figure 1. CHAPK cleavage site of staphylococcal cell wall peptidoglycan.
CHAP proteins contain three highly conserved amino acid residues, two of which are an invariant cysteine (Cys) and histidine (His) along with a third, polar residue such as asparagine (Asn), aspartic acid (Asp) or glutamic acid (Glu).21 These residues are principally involved in catalytic activity, forming part of the active site of the enzyme as well as being equivalent to the catalytic triad of papain-like thiol proteases.21,22 Site-directed mutagenesis studies on these conserved residues within the CHAP domain of the LysWMY staphylococcal lysin resulted in reduced activity, suggesting that a Cys-His-Asn catalytic triad is crucial for enzymatic function.11
The CHAP domain is a member of the NlpC/P60 family of peptidases and can be found in proteins from bacteria, archaea and eukaryotes of the Trypanosomidae family.22 However, very little structural information is available in relation to the CHAP domains of phage lysins. To date the crystal structure of five lysins has been elucidated, Cpl-1,23 PlyL,24 PlyPSA,25 PlyB,26 and Ply500,27 none of which are derived from staphylococcal phage and none of which contain a CHAP domain.
In the current study a comparative modeling approach is employed to predict the three dimensional (3D) structure of the 162-amino-acid CHAP domain protein, CHAPK, derived from the staphylococcal phage K lysin; LysK.10 The potential of CHAPK as an alternative antibacterial agent which displays rapid lytic activity against pathogenic staphylococci including MRSA, both in vitro and in vivo, has previously been demonstrated by our group.20,28
Comparative modeling allows accurate prediction of the 3D structure of a target protein (CHAPK) based on its similarity to a related template protein whose structure has been experimentally resolved by either X-ray crystallography or Nuclear Magnetic Resonance (NMR) spectroscopy.29 The basis of this approach is that evolutionary related proteins share similar 3D structures if they have a statistically significant sequence similarity (generally > 25% identity).30 The model prediction process consists of four consecutive steps starting with template selection, followed by target-template alignment, model building and finally model evaluation.31
Resolution of the 3D structure of CHAPK by comparative modeling can provide a valuable insight into the structural basis of interaction with its cell wall peptidoglycan substrate and mechanism of catalytic activity. This knowledge may subsequently be used in the design of novel lysins with altered and improved catalytic domains, or the creation of designer lysins with unique capabilities (e.g., enhanced turnover rate, altered specificity) furthering the development of these unique enzymes as alternative antibacterial agents in the fight against multi-drug resistant bacterial infections.
Results and Discussion
Template selection
The first step in the comparative modeling of the target protein, CHAPK, was to identify potential templates whose structure has been resolved by either X-ray crystallography or NMR. The two main criteria for selecting suitable templates for the target were sequence length (choosing proteins of similar length to CHAPK) and percentage identity between the target and template sequence of > 25% over the entire length of the sequence. Generally, proteins with > 25% sequence identity can be labeled homologous and share at least 80% of their structure.30,32 With the aim of collecting the most suitable template sequences to form a strong multiple alignment in the second step of comparative modeling, a PSI-BLAST (Position-Specific Iterated BLAST) search was performed against the Protein Data Bank (PDB) over a number of iterations. This bank is a repository of the 3D structural data of proteins and nucleic acids which has been experimentally obtained by X-ray crystallography or NMR. The most suitable template obtained from the PDB search was the enzyme 2K3A; a 155-amino-acid secreted CHAP protein from Staphylococcus saprophyticus33 with 28% sequence identity to CHAPK. This template returned the lowest (E)-value (0.01) and largest % coverage (58%). The BLAST search converged after three iterations and no known structure received a significantly high score, over a range of scoring matrix and gap penalty settings. This suggested that not many CHAP domain structures with similarity to phage-derived CHAP domains have been experimentally resolved to date.
In order to populate the multiple alignment in step two, iterative PSI-BLAST searches were also performed in the non-redundant (nr) database. Although the inclusion of homologous sequences from the nr database does not contribute structural information, it aids in constructing a better quality multiple alignment, which is generally regarded as more reliable than a pair-wise structural alignment alone.31
Target-template alignment and model building
In the second step of comparative modeling, the combination of selected template structures from the PDB and sequences from the nr-database, collected in step one, were uploaded to the T-Coffee Expresso server together with the amino acid sequence of the target (CHAPK). The Expresso suite is a multiple sequence alignment server which accurately aligns sequences that include structural information.34 The resulting multiple alignment of the CHAPK target and template proteins was used to construct a phylogenetic tree and identify templates which were most closely related to the target. A consensus maximum parsimony phylogenetic tree with bootstrap analysis inferred from 1,000 replicates was constructed using the MEGA4 software suite35 and taken to represent the evolutionary history of the protein sequences analyzed (Fig. 2A). Upon examination of the tree, proteins in close proximity to the target, including the protein 2K3A (with experimentally derived 3D structure) were selected and processed again using the Expresso alignment algorithm (Fig. 2B).

Figure 2. (A) Phylogenetic tree showing evolutionary relationship of 40 CHAPK homologs (15 from PDB BLAST and 25 from nr BLAST databases). The maximum parsimony tree was obtained using the Close-Neighbor-Interchange algorithm level 3 in MEGA4 with a bootstrap consensus setting of 1,000 replicates. CHAPK and the 2K3A CHAP protein are underlined. (B) Expresso alignment of CHAPK target sequence and 2K3A structural template sequence. Highly-conserved cysteine (C), histidine (H) and glutamic acid (E) active site residues present in both proteins have been identified and are shown in black boxes.
For the third step in the process (model building), the above alignment was submitted to the Alignment mode of the web-based homology modeling server, SWISS-MODEL.36 This program facilitates the construction of 3D models of a target protein using experimentally determined structures of related family members.37 Models were constructed using the 2K3A protein and each structurally resolved protein from the clade containing CHAPK in the phylogenetic tree (Fig. 2A). Inspection of predicted 3D CHAPK models in relation to structural compatibility between the target and template, sharing of conserved amino acids and the location of these residues in the overall structure, resulted in the identification of the 2K3A protein as the most promising candidate for accurate modeling of CHAPK.
In model building, accurate alignment of the target sequence with selected template is a crucial step and poor alignments are commonly responsible for inaccurate models.37 In order to examine the quality of the target-template alignment and the models created in SWISS-MODEL Alignment mode, CHAPK was submitted to an additional web-based server, I-TASSER.38 The results from I-TASSER independently identified the 2K3A protein as the most suitable template for constructing an accurate 3D model of CHAPK. Upon inspection and comparison of the predicted models from both I-TASSER (Fig. 3A) and SWISS-MODEL (Fig. 3B) servers, it was obvious that I-TASSER had generated a better quality alignment as its predicted model had a fold and catalytic residue position that was consistent with enzymatic function33 (Fig. 3A).

Figure 3. Predicted 3D structure of CHAPK by I-TASSER (A) and SwissModel (B) servers. Highly conserved cysteine residue (Cys54) is highlighted in orange and conserved histidine residue (His117) is highlighted in blue for both predicted structures. The distance between Cys54 and His117 catalytic residues was determined to be 3.7 Ångströms (Å) for the I-TASSER model (A) whereas this distance was as great as 20 Å in the Swiss-Model structure (B). Two exposed tyrosine aromatic groups, Tyr112 and Tyr140 are also shown. These residues, located either side of the active site in model (A) may function as rails to anchor the enzymes substrate in position. Figures visualized using PyMOL.
Model Evaluation
In the fourth step, the quality of the I-TASSER generated CHAPK model was evaluated using PROCHECK39 and visualized using PyMOL.40 The predicted model had two short α-helices packed against a β-sheet saddle, which is a typical feature of NplC/P60 papain-related peptidases21 (Fig. 3A). Model refinement was performed using CNS_SOLVE,41 and the resultant structure had good geometries with 54.3% of residues falling in the most favored region of the Ramachandran plot, and only 2.9% outliers. A comparison between the newly constructed CHAPK model and the 2K3A CHAP protein template was also assessed. Superposition of CHAPK on the template structure used LSQMAN.42 The calculated root mean square distance (r.m.s.d) value of 1.9 Ångstroms (Å) on 143 C-α atoms indicated the expected degree of agreement between the two structures. Inspection of the superimposed structures in PyMOL showed good agreement between CHAPK and the S. saprophyticus CHAP domain throughout the structures (Fig. 4).
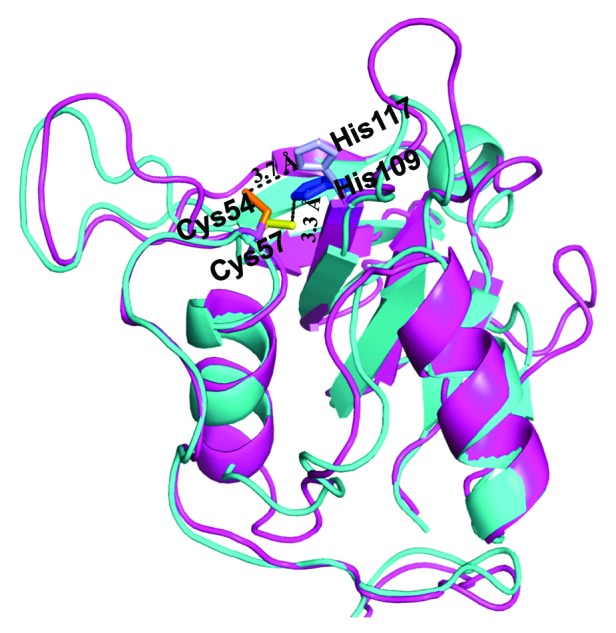
Figure 4. Structural comparison by superposition of the experimentally resolved 2K3A protein (cyan) and predicted CHAPK structure (magenta). Highly conserved active site residues are identified and labeled for both proteins. CHAPK Cys54 (orange) and His117 (light blue), 2K3A Cys57 (yellow), His 109 (blue). Figure created in PyMOL and analyzed in LSQMAN42 returning a value of 1.9 Å over 143 C-α atoms.
Further analysis of secondary structure in the CHAPK model was performed using the software package Digital Shape Sampling and Processing (DSSP).43,44 The predicted structure differed from the parental template model 2K3A in that the region between residues 86 and 102 contains a β-strand in 2K3A (Fig. 5B). However, the CHAPK model has this region somewhat more distant from the sheet (Fig. 5A). As a result these residues are not being categorized as a β-strand in the CHAPK model by DSSP43,44 (Fig. 5A).
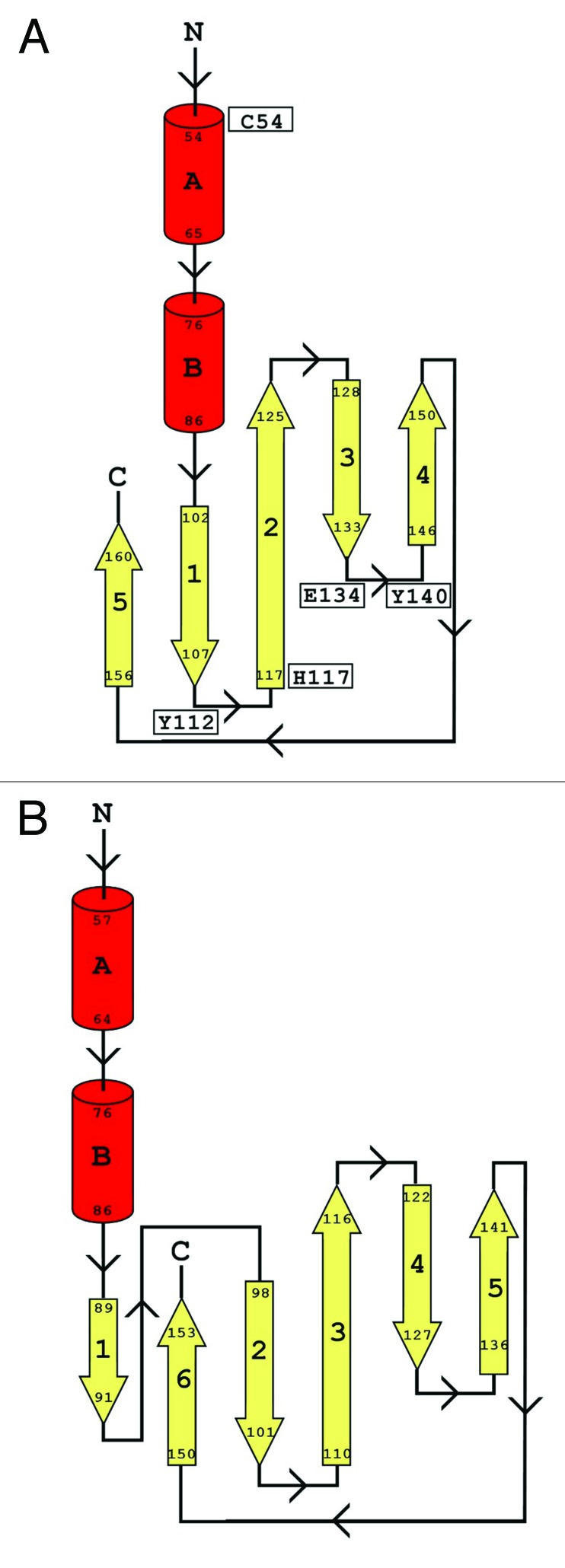
Figure 5. Topology maps for CHAPK and 2K3A proteins. Secondary structure was determined using DSSP. The topology maps show α-helices in red and the strands of the β-sheets in yellow. (A) shows the topology map for CHAPK while (B) shows the topology map for the Staphylococcus saprophyticus CHAP domain (PDB identifier 2K3A). Key residues in CHAPK are indicated in diagram (B).
Circular dichroism (CD) spectropolarimetry was performed to investigate the presence of an αβ fold, predicted in the CHAPK model. The CD spectrum obtained for CHAPK showed characteristic minima in the range 205–220 nm, typical of a protein containing α-helical and β-sheet structure (Fig. 6). Deconvolution of the spectrum using CDPro45,46 and a test set comprising 43 soluble and 13 membrane proteins, indicated that CHAPK contains an αβ fold, which supports the model. The exact α-helix and β-sheet content determined from the spectrum varied depending on which algorithm was used in the CDPro45,46 suite of programs. These data are shown in Table 1. In the case of SELCON and CONTINLL algorithms, the content of α-helix is close to that found in the model (13.9%) while the CDSSTR algorithm is under-estimating the α-helical content significantly. For all components of CDPro45,46 the estimated β-sheet structure is at variance with the amount of β-sheet seen in the model (33.9%, 35.2% and 39% for SELCON, CONTINLL and CDSSTR respectively, compared with an estimated 18.7% in the model). This could be explained by additional secondary structure in the protein that is not well modeled. The region from residues 86–102 (Fig. 5A) in the predicted CHAPK model is an area where there is potentially an additional β-strand. This would bring the CHAPK model closer in structure to the 2K3A structure. In addition, the N-terminal region of the 2K3A protein is an area of extended coil in the NMR structure, this may become more ordered and contribute additional β-sheet structure. Regardless of the precise secondary structure composition, CD spectropolarimetry indicated that the CHAPK protein has an αβ fold, which is consistent with the predicted 3D model.
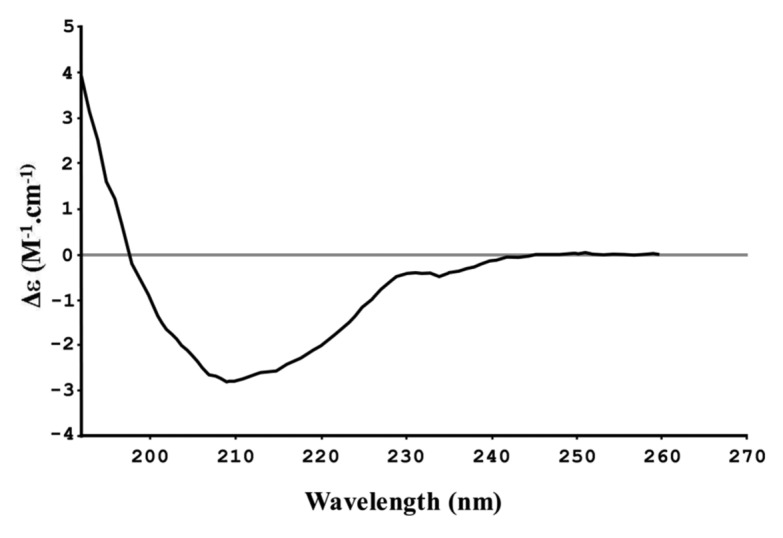
Figure 6. CD spectropolarimetry spectrum for CHAPK.
Table 1. Secondary structure composition based on CD spectropolarimetry.
| SELCON | CONTINLL | CDSSTR | |
|---|---|---|---|
|
Secondary structure |
|
|
|
| α-helix (%) |
17.5 |
15.1 |
6 |
| β-sheet (%) | 33.9 | 35.2 | 39 |
Both target and template proteins have highly-conserved cysteine and histidine residues with Cys54-His117 residues of CHAPK (magenta in Fig. 7) located approximately in the same position as the Cys57-His109 residues in the active site of 2K3A (cyan in Fig. 7). The distance between the Cys57 and His109 catalytic residues of 2K3A was found to be 3.3 Ångströms (Å) (Fig. 7), which is within hydrogen bond distance.33 This distance is a critical feature in imparting enzymatic activity to CHAP proteins. Typically, the presence of a substrate in the Cys-His active site, triggers the deprotonation of cysteine by the histidine residue, resulting in a nucleophilic attack by the deprotonated cysteine on a substrate acyl carbonyl carbon, releasing a fragment of the substrate with an N-terminus. This leads to the formation of a tetrahedral ionic intermediate, restoration of the histidine residue to its deprotonated form and finally the release of the carboxylic acid moiety from the cysteine residue via hydrolysis. This results in the regeneration of the Cys-His active site.47,48 The distance of 3.7 Å between the corresponding Cys54-His117 active site residues in the CHAPK model are comparable with the active site of the 2K3A template (Fig. 7). It is therefore plausible that the CHAPK Cys54-His117 highly conserved residues have a similar catalytic mode of action. When localized to their staphylococcal peptidoglycan substrate, they initiate a rapid nucleophilic attack on the peptide bond between D-alanine and the first glycine in the pentaglycine cross-bridge, resulting in cleavage of the bacterial cell wall and rapid cell death. To further investigate the proposed role of the conserved Cys54-His117 active-site residues, a QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies Irealand Ltd) was used to create point mutations where a change from Cys54 to alanine (Ala) or His117 to Ala resulted in loss of enzymatic activity (data not shown). This strongly indicates that these two residues play a crucial role in CHAPK catalytic activity.

Figure 7. Close-up stereoview of highly conserved Cys-His active site residues of CHAPK (magenta) and 2K3A (cyan) superimposed. CHAPK Cys54 (orange) and His117 (light blue), 2K3A Cys57 (yellow), His 109 (blue). Broken lines with labels indicate distance between residues, measured in Ångströms (Å).
In total, eight conserved residues, Cys54, His117, Glu134, Asn 136, Gly116, Gly74, Thr59 and Gln55 at or near the surface of the predicted CHAPK model may be essential for enzymatic activity. These residues coordinate the formation of a broad and shallow cleft which encompasses the active site of the enzyme (Fig. 8). This was consistent with the 2K3A template structure.33 In the 2K3A protein, active site residues Cys57-His109-Glu126 are arranged in a viable proteolytic triad both in a sequence order and a topology typical of cysteine peptidases, with Asn128 acting as an additional stabilizing group.33 Analysis of the CHAPK model revealed corresponding highly conserved residues constituted by a Cys54-His117-Glu134 charge relay and an oxyanion hole (stabilizing) residue Asn136, near the surface of the active site cleft.
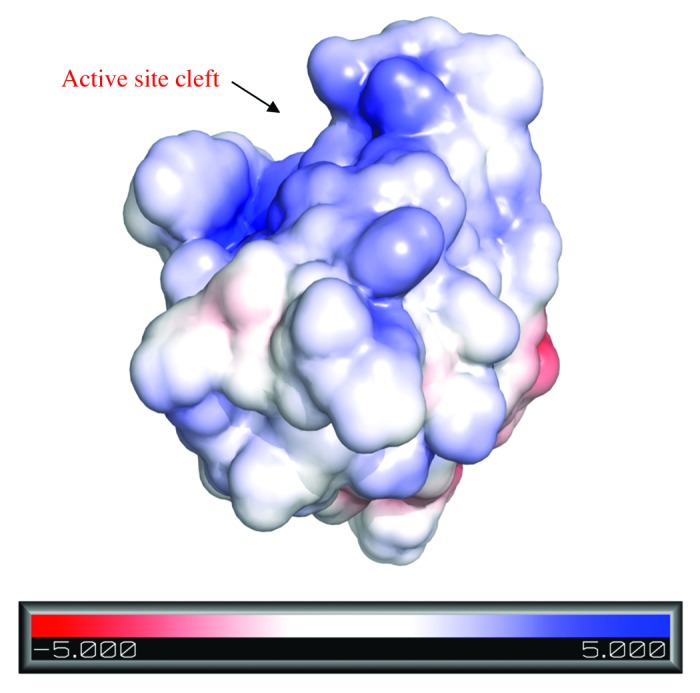
Figure 8. Electrostatic surface representation of CHAPK with the active site cleft indicated by a black arrow. The protein surface is colored according to its electrostatic potential from regions of red (negatively charged) to blue (positively charged). Model created in PyMOL using the Adaptive Poison Boltzmann Solver (APBS) program.
The exact mechanism of binding of CHAPK to its peptidoglycan substrate remains unknown. However, examination of the electrostatic surface potential of the active site of CHAPK revealed an overall electropositive charge (Fig. 8) indicating electrostatic complementarity with the electronegative character of its peptidoglycan substrate. This combination of attractive and repulsive forces may help guide the substrate into the correct position for binding before contacting the active site of CHAPK. In addition, it was suggested that in the case of 2K3A, two exposed tyrosine aromatic groups, Tyr107 and Tyr129, either side of the active site possibly function as rails to anchor the substrate in position.33 This may also be the case for CHAPK as it has two exposed tyrosine residues (Tyr112 and Tyr140) on either side of its active site (Fig. 3A) and whose location corresponds closely to the position of Tyr107 and Tyr129 in the 2K3A template structure.
Conclusion
In the present study, comparative modeling was used to construct a 3D model of the phage derived peptidase, CHAPK. Analysis of the 3D model provided a structural basis for CHAPK activity and presented a probable mechanism involving the highly conserved Cys54-His117-Glu134-Asn136 active site residues. These residues have been identified in other lysins as the key catalytic residues involved in the hyrolysis of a D-Ala-Gly substrate, in the pentaglycine cross-bridge of staphylococcal cell wall peptidoglycan. The proposed nucleophilic-attack mechanism of the conserved Cys54 residue on the peptide bond in an acid-base reaction is thought to be the most likely catalytic mechanism for all members of the CHAP NplC/P60 family of proteases.
Further exploration and investigation of phage lysins and their various domains, particularly at a structural and bioinformatic level, can provide important information for understanding biochemical function and specific substrate interaction mechanisms. This in turn may contribute to specifically engineered designer lysins with diverse and improved applications for development as novel antibacterials. Additional research is currently underway to experimentally resolve the crystal structure of CHAPK.
Methods
The following web-based servers, suites, molecular analysis and visualization programs were used in the comparative modeling process for CHAPK structure prediction. Basic Local Alignment Search Tool (BLAST) (www.ncbi.nlm.nih.gov/BLAST/), T-Coffee (www.tcoffee.org), MEGA4,35 SWISS-MODEL (www.swissmodel.expasy.org),36 iterative threading assembly refinement (I-TASSER),38 PyMOL,40 PROCHECK,39 CNS_SOLVE41 LSQMAN42 and DSSP.43,44 For CD spectropolarimetry The CHAPK domain was dialysed extensively against 10 mM potassium phosphate buffer, pH 7.0. Protein was diluted to 0.019 mg/mL in the same buffer for analysis. Data was collected from 260 nm to 180 nm on a Chirascan instrument (Applied Photophysics). Data was converted to Δε values prior to analysis. Determination of the secondary structure from the CD spectrum was performed using the CDPro45,46 software package.
Acknowledgments
This work was financed by Science Foundation Ireland Ref 06/RF/BIM004. We thank Dr. Todd Kagawa for his valuable input in the analysis of CD data.
Footnotes
Previously published online: www.landesbioscience.com/journals/bacteriophage/article/18245
References
- 1.Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–32. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.European Centre for Disease Prevention and Control. Antimicrobial resistance surveillance in Europe 2009. Antimicrobial Resistance Surveillance Network (EARS-Net). Stockholm: ECDC; 2010. ISBN 978-92-9193-227-6. [Google Scholar]
- 3.Loessner MJ. Bacteriophage endolysins–current state of research and applications. Curr Opin Microbiol. 2005;8:480–7. doi: 10.1016/j.mib.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Fischetti VA. Bacteriophage lysins as effective antibacterials. Curr Opin Microbiol. 2008;11:393–400. doi: 10.1016/j.mib.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fenton M, Ross P, McAuliffe O, O'Mahony J, Coffey A. Recombinant bacteriophage lysins as antibacterials. Bioeng Bugs. 2010;1:9–16. doi: 10.4161/bbug.1.1.9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischetti VA. Bacteriophage lytic enzymes: novel anti-infectives. Trends Microbiol. 2005;13:491–6. doi: 10.1016/j.tim.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Borysowski J, Weber-Dabrowska B, Gorski A. Bacteriophage endolysins as a novel class of antibacterial agents. Exp Biol Med (Maywood) 2006;231:366–77. doi: 10.1177/153537020623100402. [DOI] [PubMed] [Google Scholar]
- 8.García P, Garcia JL, Garcia E, Sanchez-Puelles JM, Lopez R. Modular organization of the lytic enzymes of Streptococcus pneumoniae and its bacteriophages. Gene. 1990;86:81–8. doi: 10.1016/0378-1119(90)90116-9. [DOI] [PubMed] [Google Scholar]
- 9.Loessner MJ, Kramer K, Ebel F, Scherer S. C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol Microbiol. 2002;44:335–49. doi: 10.1046/j.1365-2958.2002.02889.x. [DOI] [PubMed] [Google Scholar]
- 10.O'Flaherty S, Coffey A, Meaney W, Fitzgerald GF, Ross RP. The recombinant phage lysin LysK has a broad spectrum of lytic activity against clinically relevant staphylococci, including methicillin-resistant Staphylococcus aureus. J Bacteriol. 2005;187:7161–4. doi: 10.1128/JB.187.20.7161-7164.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokoi KJ, Kawahigashi N, Uchida M, Sugahara K, Shinohara M, Kawasaki K, et al. The two-component cell lysis genes holWMY and lysWMY of the Staphylococcus warneri M phage varphiWMY: cloning, sequencing, expression, and mutational analysis in Escherichia coli. Gene. 2005;351:97–108. doi: 10.1016/j.gene.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Sass P, Bierbaum G. Lytic activity of recombinant bacteriophage phi11 and phi12 endolysins on whole cells and biofilms of Staphylococcus aureus. Appl Environ Microbiol. 2007;73:347–52. doi: 10.1128/AEM.01616-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rashel M, Uchiyama J, Ujihara T, Uehara Y, Kuramoto S, Sugihara S, et al. Efficient elimination of multidrug-resistant Staphylococcus aureus by cloned lysin derived from bacteriophage phi MR11. J Infect Dis. 2007;196:1237–47. doi: 10.1086/521305. [DOI] [PubMed] [Google Scholar]
- 14.Obeso JM, Martinez B, Rodriguez A, Garcia P. Lytic activity of the recombinant staphylococcal bacteriophage PhiH5 endolysin active against Staphylococcus aureus in milk. Int J Food Microbiol. 2008;128:212–8. doi: 10.1016/j.ijfoodmicro.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Gu J, Xu W, Lei L, Huang J, Feng X, Sun C, et al. A Novel Bacteriophage Lysin, LysGH15, Protects a Murine Bacteremia Model Efficiently against Lethal Methicillin-Resistant Staphylococcus aureus Infection. J Clin Microbiol. 2011;49:111–7. doi: 10.1128/JCM.01144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jun SY, Jung GM, Son JS, Yoon SJ, Choi YJ, Kang SH. Comparison of the Antibacterial Properties of Phage Endolysins SAL-1 and LysK. Antimicrob Agents Chemother. 2011;55:1764–7. doi: 10.1128/AAC.01097-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Navarre WW, Ton-That H, Faull KF, Schneewind O. Multiple enzymatic activities of the murein hydrolase from staphylococcal phage phi11. Identification of a D-alanyl-glycine endopeptidase activity. J Biol Chem. 1999;274:15847–56. doi: 10.1074/jbc.274.22.15847. [DOI] [PubMed] [Google Scholar]
- 18.Becker SC, Dong S, Baker JR, Foster-Frey J, Pritchard DG, Donovan DM. LysK CHAP endopeptidase domain is required for lysis of live staphylococcal cells. FEMS Microbiol Lett. 2009;294:52–60. doi: 10.1111/j.1574-6968.2009.01541.x. [DOI] [PubMed] [Google Scholar]
- 19.Rigden DJ, Jedrzejas MJ, Galperin MY. Amidase domains from bacterial and phage autolysins define a family of gamma-D,L-glutamate-specific amidohydrolases. Trends Biochem Sci. 2003;28:230–4. doi: 10.1016/S0968-0004(03)00062-8. [DOI] [PubMed] [Google Scholar]
- 20.Horgan M, O'Flynn G, Garry J, Cooney J, Coffey A, Fitzgerald GF, et al. Phage lysin LysK can be truncated to its CHAP domain and retain lytic activity against live antibiotic-resistant staphylococci. Appl Environ Microbiol. 2009;75:872–4. doi: 10.1128/AEM.01831-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anantharaman V, Aravind L. Evolutionary history, structural features and biochemical diversity of the NlpC/P60 superfamily of enzymes. Genome Biol. 2003;4:R11. doi: 10.1186/gb-2003-4-2-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bateman A, Rawlings ND. The CHAP domain: a large family of amidases including GSP amidase and peptidoglycan hydrolases. Trends Biochem Sci. 2003;28:234–7. doi: 10.1016/S0968-0004(03)00061-6. [DOI] [PubMed] [Google Scholar]
- 23.Hermoso JA, Monterroso B, Albert A, Galan B, Ahrazem O, Garcia P, et al. Structural basis for selective recognition of pneumococcal cell wall by modular endolysin from phage Cp-1. Structure. 2003;11:1239–49. doi: 10.1016/j.str.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Low LY, Yang C, Perego M, Osterman A, Liddington RC. Structure and lytic activity of a Bacillus anthracis prophage endolysin. J Biol Chem. 2005;280:35433–9. doi: 10.1074/jbc.M502723200. [DOI] [PubMed] [Google Scholar]
- 25.Korndörfer IP, Danzer J, Schmelcher M, Zimmer M, Skerra A, Loessner MJ. The crystal structure of the bacteriophage PSA endolysin reveals a unique fold responsible for specific recognition of Listeria cell walls. J Mol Biol. 2006;364:678–89. doi: 10.1016/j.jmb.2006.08.069. [DOI] [PubMed] [Google Scholar]
- 26.Porter CJ, Schuch R, Pelzek AJ, Buckle AM, McGowan S, Wilce MC, et al. The 1.6 A crystal structure of the catalytic domain of PlyB, a bacteriophage lysin active against Bacillus anthracis. J Mol Biol. 2007;366:540–50. doi: 10.1016/j.jmb.2006.11.056. [DOI] [PubMed] [Google Scholar]
- 27.Korndörfer IP, Kanitz A, Danzer J, Zimmer M, Loessner MJ, Skerra A. Structural analysis of the L-alanoyl-D-glutamate endopeptidase domain of Listeria bacteriophage endolysin Ply500 reveals a new member of the LAS peptidase family. Acta Crystallogr D Biol Crystallogr. 2008;64:644–50. doi: 10.1107/S0907444908007890. [DOI] [PubMed] [Google Scholar]
- 28.Fenton M, Casey PG, Hill C, Gahan CGM, Ross RP, McAuliffe O, et al. The truncated phage lysin CHAPk eliminates Staphylococcus aureus in the nares of mice. Bioeng Bugs. 2010;1:404–7. doi: 10.4161/bbug.1.6.13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–6. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 30.Claverie J-M, Notredame C. Bioinformatics for dummies 2nd ed.–For dummies. 2007, Hoboken, N.J.: Wiley Pub. xviii, 436 p. [Google Scholar]
- 31.Martí-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 32.Ginalski K. Comparative modeling for protein structure prediction. Curr Opin Struct Biol. 2006;16:172–7. doi: 10.1016/j.sbi.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Rossi P, Aramini JM, Xiao R, Chen CX, Nwosu C, Owens LA, et al. Structural elucidation of the Cys-His-Glu-Asn proteolytic relay in the secreted CHAP domain enzyme from the human pathogen Staphylococcus saprophyticus. Proteins. 2009;74:515–9. doi: 10.1002/prot.22267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armougom F, Moretti S, Poirot O, Audic S, Dumas P, Schaeli B, et al. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006;34(Web Server issue):W604-8. doi: 10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 36.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37(Database issue):D387–92. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 40.DeLano WL. The PyMOL Molecular Graphics System 2002, DeLano Scientific, San Carlos, CA, USA. [Google Scholar]
- 41.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- 42.Novotny M, Madsen D, Kleywegt GJ. Evaluation of protein fold comparison servers. Proteins. 2004;54:260–70. doi: 10.1002/prot.10553. [DOI] [PubMed] [Google Scholar]
- 43.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 44.Joosten RP, te Beek TA, Krieger E, Hekkelman ML, Hooft RW, Schneider R, et al. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011;39(Database issue):D411–9. doi: 10.1093/nar/gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sreerama N, Woody RW. On the analysis of membrane protein circular dichroism spectra. Protein Sci. 2004;13:100–12. doi: 10.1110/ps.03258404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sreerama N, Woody RW. Computation and analysis of protein circular dichroism spectra. Methods Enzymol. 2004;383:318–51. doi: 10.1016/S0076-6879(04)83013-1. [DOI] [PubMed] [Google Scholar]
- 47.Lau EY, Bruice TC. Consequences of breaking the Asp-His hydrogen bond of the catalytic triad: effects on the structure and dynamics of the serine esterase cutinase. Biophys J. 1999;77:85–98. doi: 10.1016/S0006-3495(99)76874-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin CH, Kwon DS, Bollinger JM, Jr., Walsh CT. Evidence for a glutathionyl-enzyme intermediate in the amidase activity of the bifunctional glutathionylspermidine synthetase/amidase from Escherichia coli. Biochemistry. 1997;36:14930–8. doi: 10.1021/bi9714464. [DOI] [PubMed] [Google Scholar]