

Abstract
Astrocyte elevated gene-1 (AEG-1) is a key contributor to hepatocellular carcinoma (HCC) development and progression. To enhance our understanding of the role of AEG-1 in hepatocarcinogenesis, a transgenic mouse with hepatocyte-specific expression of AEG-1 (Alb/AEG1) was developed. Treating Alb/AEG-1, but not Wild type (WT) mice, with N-nitrosodiethylamine (DEN), resulted in multinodular HCC with steatotic features and associated modulation of expression of genes regulating invasion, metastasis, angiogenesis and fatty acid synthesis. Hepatocytes isolated from Alb/AEG-1 mice displayed profound resistance to chemotherapeutics and growth factor deprivation with activation of pro-survival signaling pathways. Alb/AEG-1 hepatocytes also exhibited marked resistance towards senescence, which correlated with abrogation of activation of a DNA damage response. Conditioned media (CM) from Alb/AEG-1 hepatocytes induced marked angiogenesis with elevation in several coagulation factors. Among these factors, AEG-1 facilitated association of Factor XII (FXII) mRNA with polysomes resulting in increased translation. siRNA-mediated knockdown of FXII resulted in profound inhibition of AEG-1-induced angiogenesis.
Conclusion
We uncover novel aspects of AEG-1 functions, including induction of steatosis, inhibition of senescence and activation of coagulation pathway to augment aggressive hepatocarcinogenesis. The Alb/AEG-1 mouse provides an appropriate model to scrutinize the molecular mechanism of hepatocarcinogenesis and to evaluate the efficacy of novel therapeutic strategies targeting HCC.
Keywords: Astrocyte elevated gene-1 (AEG-1), transgenic, hepatocellular carcinoma (HCC), senescence, angiogenesis
Astrocyte elevated gene-1 (AEG-1), also known as metadherin (MTDH) and lysine-rich CECAM-1 co-isolated protein (LYRIC), is now established as a positive regulator of tumorigenesis (1). Using immunohistochemistry in a large cohort of patient samples, elevated AEG-1 protein expression has been documented in a variety of cancers (1). AEG-1 expression gradually increases with disease progression and displays a negative correlation with patient survival. The AEG-1 gene is located in human chromosome 8q22, which is amplified in breast and liver cancers (2, 3). AEG-1 is a downstream gene in the Ha-Ras signaling pathway that activates PI3K/Akt and leads to transcriptional upregulation of AEG-1 by c-Myc (4). AEG-1 is a target of miRNA-375, a tumor suppressor in diverse cancers (5). Thus AEG-1 expression might be increased by a variety of mechanisms during carcinogenesis.
Gain- and loss-of-function studies in diverse cell lines confirm the importance of AEG-1 in the development and progression of cancer. In multiple cancer cell lines that express low levels of AEG-1 and are poorly aggressive, AEG-1 overexpression results in a significant increase in in vitro proliferation, anchorage-independent growth, migration and invasion and in vivo tumorigenesis, metastasis and angiogenesis in nude mice xenograft models (1). As a corollary, RNAi-mediated inhibition of AEG-1 in aggressive cell lines expressing high levels of AEG-1 significantly inhibits aforementioned in vitro and in vivo oncogenic phenotypes. AEG-1 overexpression results in activation of multiple pro-survival signal transduction pathways, and profoundly contributes to chemoresistance and tumor angiogenesis, major hallmarks of aggressive cancers (1). Thus AEG-1 plays a fundamental role in aggressive progression of the carcinogenic process.
The molecular mechanism by which AEG-1 induces these profound changes is gradually being clarified. AEG-1 is a 582 amino acids protein with a transmembrane domain and multiple nuclear localization signals (NLS) (1). In cancer cells, AEG-1 is detected in the cytoplasm as well as on the cell membrane and in the nucleus (2). Depending upon location, AEG-1 interacts with different protein complexes regulating diverse functions. AEG-1 interacts with NF-κB and CBP promoting NF-κB-mediated transcription (6) while it interacts with YY1 along with CBP to repress transcription (7). In the cytoplasm, AEG-1 is a component of the RNA-induced silencing complex (RISC) and assists oncomiR-mediated degradation of tumor suppressor mRNAs (8). AEG-1 facilitates translation of specific mRNAs, such as the mRNA for the multidrug resistance gene (MDR1), which contributes to chemoresistance (9). The membrane-located AEG-1 promotes interaction of cancer cells with lung endothelium thus augmenting metastasis (3). The identification of the diverse interacting partners indicates that AEG-1 may be a scaffold protein mediating formation of multi-protein complexes in different intracellular compartments.
AEG-1 plays an important role in hepatocarcinogenesis (2). AEG-1 mRNA and protein overexpression as well as amplification of AEG-1 gene was detected in a large percentage of Hepatocellular carcinoma (HCC) patients (2). To better comprehend the role of AEG-1 in hepatocarcinogenesis and to decipher the underlying molecular mechanism(s) in an in vivo context, we have generated a transgenic mouse with hepatocyte-specific expression of AEG-1 (Alb/AEG-1). We document that compared to wild-type (WT) mice, the hepatocarcinogenic process is significantly amplified in Alb/AEG-1 mice. We unravel novel aspects of AEG-1, including induction of steatosis, protection from senescence and activation of coagulation pathways, which contribute to its tumor promoting functions. This is the first study analyzing AEG-1 function in vivo and Alb/AEG-1 mouse provides a useful model to further understand the hepatocarcinogenic process and evaluate emerging novel therapies for this invariably fatal disease.
Materials & Methods
Generation of Alb/AEG-1 mouse and induction of chemical carcinogenesis
Alb/AEG-1 mouse was generated by directing the expression of human AEG-1 under an upstream enhancer region (−10400 to −8500) fused to the 335-bp core region of mouse albumin promoter (10). Microinjection and manipulation procedures were performed according to standard procedures in the VCU Massey Cancer Center Transgenic/Knockout Mouse Core. For induction of chemical carcinogenesis, a single i.p. injection of 10 µg/gm body weight of N-nitrosodiethylamine (DEN) was given at 14 days of age to male WT and Alb/AEG-1 mice (11).
Cells and culture condition
Primary mouse hepatocytes were isolated from WT and Alb/AEG-1 mice as described (12). Primary human hepatocytes were obtained from the Liver Tissue Cell Distribution System (LTCDS) (NIH contract #N01-DK-7-0004 / HHSN267200700004C) and were cultured in Hepatocyte Culture Medium (HCM) containing the supplements (Lonza). Human umbilical vein endothelial cells (HUVEC) were obtained from Lonza and were cultured according to the provided protocol.
Polysome purification, total RNA extraction, real time PCR and microarray assay
Purification of polysomal fractions from WT and Alb/AEG-1 hepatocytes was performed as described (9). Total RNA was extracted from each polysomal fraction and from WT and Alb/AEG-1 livers using Qiagen miRNAeasy mini kit (Qiagen). Real time PCR was performed using an ABI ViiA7 fast real time PCR system and Taqman gene expression assays according to the manufacturer’s protocol (Applied Biosystems). An Affymetrix oligonucleotide microarray (GeneChip® Mouse Genome 430A 2.0 Array representing approximately 14,000 well-characterized mouse genes) analysis was performed to compare gene expression between DEN-treated WT and Alb/AEG-1 liver samples as described (2).
Mass spectrometric analysis of conditioned media
Conditioned media from WT and Alb/AEG-1 hepatocytes were collected one day after isolation and subjected to mass spectrometric analysis as described (8). Peptide samples were fractionated by reverse phase liquid chromatography, and analyzed by a high performance LC_MS/MS using an LTQ OrbiTrap XL mass spectrometer (Thermo Electron) and utilizing a label-free approach. Two independent replicate MS analysis was carried out per sample.
Statistical analysis
Data were represented as the mean ± Standard Error of Mean (S.E.M) and analyzed for statistical significance using one-way analysis of variance (ANOVA) followed by Newman-Keuls test as a post hoc test. A P value of < 0.05 was considered as significant.
Results
Generation and characterization of Alb/AEG-1 mice
We have created a transgenic mouse in a B6/CBA background with hepatocyte-specific expression of human AEG-1 (Alb/AEG-1) by using the mouse albumin promoter/enhancer element to drive AEG-1 expression. This particular strain of mouse was chosen since it is very sensitive to hepatocarcinogenesis induced by N-nitrosodiethylamine (DEN) (11). The human AEG-1 has a C-terminal HA-tag. The expression of AEG-1 in the liver of Alb/AEG-1 mice was confirmed by Western blot analysis using anti-HA antibody (Fig. 1A). Two founder lines were characterized initially revealing no significant differences. We, therefore, pursued further characterization employing one founder line.
Fig. 1. Induction of HCC by DEN in Alb/AEG-1 mice.

A. AEG-1 expression was detected in the indicated tissues of Wild-type (WT) and Alb/AEG-1 (TG) mice using anti-HA antibody by Western blotting. B. Photomicrograph of livers of mice treated with DEN at 28 weeks of age. The arrows indicate nodules. C. Graphical representation of number of liver nodules in WT (n = 11) and Alb/AEG-1 (n = 17) mice. D. Ratio of liver weight versus body weight of WT and Alb/AEG-1 mice. The data represent mean ± SEM. *: p<0.01.
Alb/AEG-1 mice develop HCC
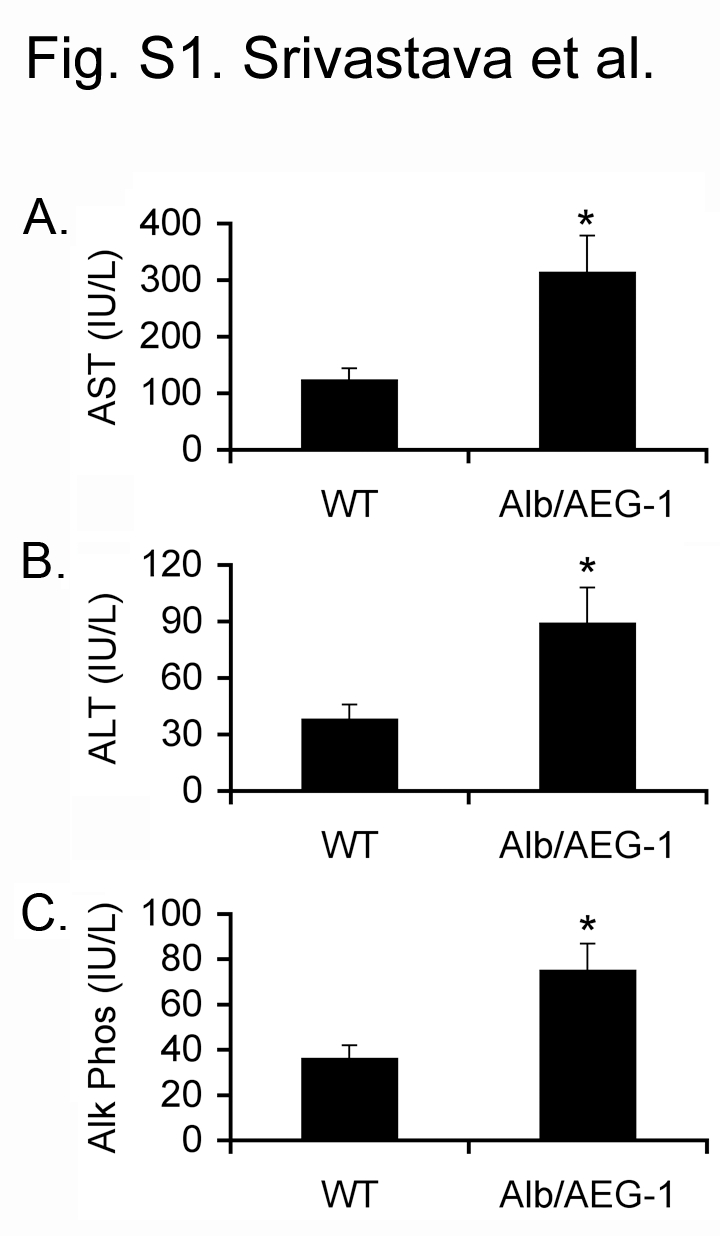
Male WT and Alb/AEG-1 littermates were given a single intraperitoneal injection of DEN (10 µg/gm) at 14 days of age and were monitored every four weeks starting at 20 weeks. At 28 weeks of age, only two out of 11 WT animals showed a few very small nodules in the liver, whereas all of the 17 Alb/AEG-1 mice livers harbored numerous nodules of different sizes (arrows in Fig. 1B and Fig. 1C). There was a significant increase in liver weight to body weight ratio in Alb/AEG-1 mice when compared to that in WT (Fig. 1D). Histological analysis of the liver of WT mice showed a few dysplatic, hyperchromatic nuclei (arrow in Fig. 2A) indicating that with time HCC would eventually develop. In Alb/AEG-1 mice, a marked increase in dysplastic, hyperchromatic nuclei was observed both in the nodules as well as in the adjacent normal liver (arrows in Fig. 2B and 2C). The most striking feature was observed in the hepatic nodules of Alb/AEG-1 mice, showing profound steatotic phenotypes with large lipid droplets in the hepatocytes (Fig. 2C). A moderate level of steatosis was also observed in the adjacent normal liver in Alb/AEG-1 mice. There was a significant increase in hepatic enzymes in the sera of Alb/AEG-1 mice versus the sera of WT mice (Fig. S1). At 32 weeks of age, the WT mice developed hepatic nodules, however, the nodules developed in Alb/AEG-1 mice were markedly larger (Fig. S2). These findings indicate that AEG-1 significantly accelerated the hepatocarcinogenic process in DEN-treated animals. The WT and Alb/AEG-1 mice were followed for one year without any DEN treatment. Although AEG-1-induced steatosis was profoundly evident, overt nodular HCC did not develop at this time point.
Fig. 2. Alb/AEG-1 mice develop hepatic nodules with steatosis.

Histological analysis of livers of WT (A) and Alb/AEG-1 mice (B & C) treated with DEN at 28 weeks of age. The nodule in Alb/AEG-1 mice shows marked steatotic changes. The arrows indicate hyperchromatic nuclei.
Modulation of global gene expression in Alb/AEG-1 mice
To define AEG-1-induced gene expression changes potentially leading to an oncogenic phenotype we compared DEN-treated WT and Alb/AEG-1 livers by performing oligonucleotide microarray experiments using biological triplicates in each animal group. We used samples at 28 weeks, since this is the earliest time point at which nodules are observed in Alb/AEG-1 mice. Using a 2-fold cut-off and a p-value of <0.05 we identified 25 AEG-1 regulated genes that might contribute to AEG-1 function (Table S1). A supervised gene cluster analysis is shown in Fig. S3. These genes include HCC marker α-feto protein (Afp); invasion and metastasis-associated genes Tspan8 and Lcn2; several genes associated with fat metabolism, such as, Scd2, Lpl, Apoa4 and Apoc2; and genes regulating angiogenesis, such as TFF3 and Meox2. The mRNA and protein expression levels in WT and Alb/AEG-1 mice were validated by real-time PCR and immunohistochemistry, respectively, using five animals per group (Fig. S4). A significant increase in CD31, a marker for micro-vessels, was observed in Alb/AEG-1 mice when compared to WT mice supporting pro-angiogenic properties of AEG-1 (Fig. S4).
AEG-1 protects from chemotherapeutics and growth factor deprivation
To understand what properties of AEG-1 promote the hepatocarcinogenic process, we isolated and characterized hepatocytes from WT and Alb/AEG-1 mice. The overexpression of AEG-1 was confirmed in the hepatocytes by Western blot analysis using both anti-AEG-1 and anti-HA antibodies (Fig. S5). One profound phenotype conferred by AEG-1 is chemoresistance (3, 9, 13). Indeed, Alb/AEG-1 hepatocytes demonstrated marked resistance to doxorubicin (DOX) and 5-fluorouracil (5-FU) treatment when compared to their WT littermates (Fig. 3A & B). Primary mouse hepatocytes, cultured in the presence of growth factors, do not divide and show decreasing viability after ~4 days as they enter senescence. The viability of Alb/AEG-1 hepatocytes in complete growth media was significantly higher than that of WT hepatocytes, as monitored by standard MTT assay over a seven day period (Fig. 3C). Upon removal of growth factors, the WT hepatocytes started losing viability within 1 day and by 3 days more than 50% of the cells were dead (Fig. 3C). In contrast, Alb/AEG-1 hepatocytes were significantly resistant to removal of growth factors and even after 7 days in basal media the cell viability was only reduced by 20% (Fig. 3C). These observations indicate that AEG-1 might autonomously activate growth factor-induced signaling and might inhibit pathways mediating senescence. Indeed, Alb/AEG-1 hepatocytes exhibited higher levels of activated (phosphorylated) ERK, Akt and p38 MAPK and anti-apoptotic proteins, Bcl-2 and Mcl-1 but not Bcl-xL, when compared to the WT hepatocytes (Fig. 3D).
Fig. 3. Alb/AEG-1 hepatocytes demonstrate chemoresistance and resistance to growth factor deprivation.

WT and Alb/AEG-1 hepatocytes were treated either with Doxorubicin (DOX; 5 nM) (A) or with 5-fluorouracil (5-FU; 100 nM) (B) and cell viability was analyzed by standard MTT assay. C. WT and Alb/AEG-1 hepatocytes were cultured either in complete media or in basal media devoid of growth factors and cell viability was analyzed by standard MTT assay. For A-C, cell viability at day 0 (D0) was considered as 100%. The data represent mean ± SEM of three independent experiments. # and *: p<0.01 comparing WT and Alb/AEG-1 of the same treatment groups. D. The expression of the indicated proteins was analyzed by Western blot using lysates from WT and Alb/AEG-1 hepatocytes.
AEG-1 inhibits senescence
WT and Alb/AEG-1 hepatocytes were cultured for 7 days and senescence was monitored by senescence-associated β-galactosidase (SA β-gal) assays. At day 7, the WT hepatocytes became large and vacuolated and ~55% cells were positive for SA β-gal while only 3% Alb/AEG-1 hepatocytes stained positive for SA β-gal (Fig. 4A). Similarly, the number of senescence-associated heterochromatin foci (SAHF) was ~4-times more per nucleus in WT hepatocytes when compared to Alb/AEG-1 hepatocytes (Fig. 4B). Senescence might be induced by activation of Rb/p16 pathway or by activation of a DNA damage response pathway leading to activation of p53 and p21 (14). We did not observe any change in activation of Rb (data not shown). However, in WT hepatocytes at day 7 there was significant activation of ATM and ATR, their downstream kinases CHK1 and CHK2 leading to p53 phosphorylation and increase in p53 and p21 levels (Fig. 4C). In Alb/AEG-1 hepatocytes there was a marked dampening of the activation of DNA damage response at day 7 indicating that AEG-1 significantly protects from a DNA damage response thereby nullifying the anti-cancer process of senescence. To investigate the mechanism of DNA damage response, we measured reactive oxygen species (ROS) levels in WT and Alb/AEG-1 hepatocytes. During the initial period of culture, such as at day 1, there was a significant increase in total ROS level in WT hepatocytes when compared to that in Alb/AEG-1 hepatocytes (Fig. 4D). At day 7, when the WT hepatocytes have become metabolically inactive due to senescence, the ROS level decreased significantly by ~90%, while the basal ROS level was higher in Alb/AEG-1 hepatocytes, demonstrating ~75% decrease, indicating more metabolically active cells. The protection from senescence by AEG-1 was also substantiated in primary human hepatocytes (Fig. S6).
Fig. 4. Alb/AEG-1 hepatocytes are resistant to induction of senescence.

A. Photomicrograph of WT and Alb/AEG-1 hepatocytes stained for senescence-associated β-galactosidase (SA-β-gal) after 1 week of culture. Graphical representation of quantification of SA-β-gal positive cells. B. Photomicrograph of WT and Alb/AEG-1 hepatocytes stained for γ-H2AX. Graphical representation of quantification of γ-H2AX foci/cell. For A and Bthe data represent mean ± SEM of three independent experiments. *: p<0.01. C. Analysis of expression level of the members of DNA damage response pathway in WT and Alb/AEG-1 hepatocytes by Western blotting. D. Total ROS level in WT and Alb/AEG-1 (TG) hepatocytes at day 1 and day 7 of culture. The data represent mean ± SEM of four independent experiments. *: p<0.01.
AEG-1 profoundly augments angiogenesis
We next investigated the effect of AEG-1 on angiogenesis, another hallmark of cancer. Human umbilical vein endothelial cells (HUVEC) were treated for 2 days with conditioned media (CM) collected from WT and Alb/AEG-1 hepatocytes. Addition of CM from WT hepatocytes to HUVEC cultured in basal media failed to induce capillary-like structures while CM from Alb/AEG-1 hepatocytes induced differentiation (Fig. 5A). The pro-angiogenic property of AEG-1 was further characterized in 9-day old chick embryos by treating chicken chorioallantoic membrane (CAM) with CM from WT and Alb/AEG-1 hepatocytes. CM from Alb/AEG-1 hepatocytes induced a marked angiogenic response while CM from WT hepatocytes failed to do so (Fig. 5B). To identify the AEG-1-induced pro-angiogenic secreted factors we analyzed the CM from WT and Alb/AEG-1 hepatocytes by mass spectrometry. Interestingly, we identified upregulation of several components of the coagulation pathway, including Fibrinogen α and β chains, Factor XII (FXII), Plasminogen and Prothrombin, that are known to play significant roles in cancer angiogenesis, metastasis and invasion (Table S2) (15). Overexpression of Fibrinogen and FXII in the CM of Alb/AEG-1 hepatocytes was confirmed by Western blot analysis (Fig. 5C).
Fig. 5. Conditioned media (CM) from Alb/AEG-1 hepatocytes induce angiogenesis.

A. Human umbilical vein endothelial cells (HUVEC) were treated with CM from WT and Alb/AEG-1 hepatocytes and tube formation was photographed. Graphical representation of tube formation by HUVEC. The data represents mean ± SEM of three independent experiments. *: p<0.01. B. Chicken chorioallantoic membrane (CAM) was treated with CM from WT and Alb/AEG-1 hepatocytes and neovascularization was photographed. The numbers indicate percentage of new blood vessels arising from the existing blood vessels in naïve CAM when VEGF-treated CAM (used as positive control) was considered as 100%. The data represents mean ± SEM of three independent experiments. *: p<0.01. C. Confirmation of overexpression of Fibrinogen and FXII in CM from Alb/AEG-1 hepatocytes by Western blotting. Similar intensity of the non-specific bands indicate equal loading. The panels represent results from one representative set of mice which were confirmed in two additional sets.
We focused on TFF3 and FXII on their ability to regulate AEG-1-induced angiogenesis. TFF3 and FXII were knocked down by siRNA in WT and Alb/AEG-1 hepatocytes (Fig. S7) and the CM were subjected to HUVEC differentiation and CAM assays. While control siRNA did not affect angiogenesis induced by CM from Alb/AEG-1 hepatocytes, inhibition of either TFF3 or FXII resulted in marked inhibition of angiogenesis (Fig. 6A & B). It should be noted that although inhibition of either TFF3 or FXII abrogated sprouting of small vessels in a similar magnitude, knocking down FXII exhibited more pronounced inhibition in the growth of larger blood vessels suggesting a pivotal role of FXII in mediating AEG-1-induced angiogenesis.
Fig. 6. TFF3 and FXII mediate AEG-1-induced angiogenesis.

Alb/AEG-1 hepatocytes were transfected with control siRNA (siCon) or siRNA for TFF3 (siTFF3) or FXII (siFXII) and after 48 h the CM was subjected to HUVEC differentiation assay (A) and CAM assay (B). The data represent mean ± SEM of three independent experiments. *: p<0.01. C. HUVEC were treated with CM from Alb/AEG-1 hepatocytes, transfected with siCon or siFXII, for 1 to 4 h and lysates were subjected to Western blot analysis using the indicated antibodies.
FXII cross-talks with EGFR activating MAPK and Akt signaling to promote proliferation and differentiation of endothelial cells. We treated HUVEC with CM from Alb/AEG-1 hepatocytes transfected with either control siRNA or FXII siRNA. While CM from control siRNA treated HUVEC maintained activation of EGFR, Akt, ERK and p38 MAPK, absence of FXII in the CM form FXII knock-down cells significantly abrogated the activation of EGFR, Akt, ERK and p38 MAPK in HUVEC (Fig. 6C). These findings further support that FXII plays an important role in AEG-1-induced proliferation and differentiation of endothelial cells.
AEG-1 upregulates FXII translation
We analyzed FXII mRNA level in WT and Alb/AEG-1 hepatocytes and observed only modest changes (data not shown) indicating that AEG-1 may preferentially increase FXII at the protein level. In WT hepatocytes, AEG-1 is expressed at low levels and predominantly localized in the nucleus (Fig. 7A). In contrast, in Alb/AEG-1 hepatocytes, AEG-1 is almost exclusively contained in the cytoplasm (Fig. 7A). We hypothesized that cytoplasmic AEG-1 might augment translation of FXII mRNA by facilitating its association with polysomes. Polysomal fractions were collected from WT and Alb/AEG-1 hepatocytes, RNA was extracted from each fraction and an equal amount of RNA from each fraction was subjected to cDNA synthesis and Taqman real-time PCR for FXII. The mean cycle threshold (CT) value for FXII amplification was significantly lower in Alb/AEG-1 hepatocytes compared to WT hepatocytes indicating that AEG-1 preferentially helps FXII mRNA associate with polysomes and thereby facilitates translation (Fig. 7B). In addition to increased polysomal association of FXII mRNA, miRNA-mediated regulation might also be involved in increased protein level of FXII. One known miRNA targeting FXII mRNA is miR-181a (16). We analyzed the expression levels of mature miR-181a in WT and Alb/AEG-1 hepatocytes and did not observe any difference (Fig. S8). Thus miRNA-mediated regulation might not be a major mechanism of FXII induction by AEG-1.
Fig. 7. AEG-1 facilitates association of FXII mRNA with polysomes.

A. WT and Alb/AEG-1 hepatocytes were stained for AEG-1 and with DAPI and the images were analyzed using a confocal laser scanning microscope. B. RNA was extracted from fractions of WT and Alb/AEG-1 hepatocytes centrifuged through a sucrose gradient and subjected to FXII mRNA level analysis by real-time PCR. Fractions 9–16 typically represent the polysomal fractions. CT: cycle threshold. The data represent mean ± SEM of three independent experiments. *: p<0.05.
Discussion
The oncogenic properties of AEG-1 have been validated through numerous studies employing human patient samples, in vitro cell culture system and nude mice xenograft models (1, 17). When overexpressed in normal immortal human cells AEG-1 significantly protects from serum starvation-induced apoptosis (18). Overexpression of AEG-1 in normal immortal cloned rat embryo fibroblasts (CREF) results in aggressive tumor formation in nude mice (19). These studies indicate that AEG-1 could confer transforming properties to normal immortal cells. However, whether AEG-1 alone might induce transformation and evoke an explicit carcinogenic phenotype was not clear. Our studies comparing WT and Alb/AEG-1 mice for a period of up to one year of age did not identify overt dysplastic changes in Alb/AEG-1 mice indicating that a pre-carcinogenic initiating event, such as mutagenesis by DEN, might be necessary before AEG-1 might contribute to the tumorigenesis process. However, these transgenic mice need to be followed for a longer timeframe, such as 18 months to 2 years, to test whether prolonged steatohepatitis, induced by AEG-1, might ultimately induce frank HCC.
Our studies unravel the striking observation that AEG-1 provides strong protection from senescence, a phenomenon that may not be explicit in immortal normal cells. AEG-1 provided a strong inhibition to the DNA damage response induced in the hepatocytes as they age and protected them from senescence. We observed that during the initial culture period, the endogenous ROS level was ~30% lower in Alb/AEG-1 hepatocytes versus WT hepatocytes. This initial level of increased endogenous ROS in WT hepatocytes might be sufficient to trigger the DNA damage response resulting in senescence. Senescence is a potential anti-cancer mechanism (20) and by blocking senescence AEG-1 may further promote the carcinogenic process.
We demonstrate an intriguing aspect of AEG-1 when it is overexpressed resulting in translational upregulation of coagulation factors. AEG-1 induces marked upregulation of FXII protein, while modestly affecting the mRNA level, by increasing association of FXII mRNA with polysomes, a phenomenon also evident for MDR1 mRNA (9). FXII displays angiogenic activity, which is independent of its function in coagulation. FXII binds to uPAR or crosstalks with EGFR on HUVEC membranes leading to activation of MAPK and Akt with subsequent proliferation and differentiation (21). siRNA-mediated knockdown of FXII resulted in profound inhibition of AEG-1-induced angiogenesis indicating a central role of FXII in this process. The question is does AEG-1 also regulate FXII under normal condition? Will AEG-1 knock-out mice suffer from clotting deficiencies? The answer is most likely not. In primary mouse hepatocytes AEG-1 is predominantly localized in nucleus/nucleolus. However, when overexpressed, AEG-1 is most abundantly detected in the cytoplasm, a phenomenon also observed in human HCC patients as well as human HCC cells stably overexpressing AEG-1 (2, 22). In prostate cancer cells AEG-1 is monoubiquitinated resulting in its stabilization and cytoplasmic sequestration (23). A similar mechanism might also be applicable to HCC cells as well as in Alb/AEG-1 hepatocytes and monoubiquitination of overexpressed AEG-1 was confirmed (Fig. S9). The promiscuous accumulation of AEG-1 in the cytoplasm might facilitate interaction with the translational machinery and loading of selective mRNAs to the polysome. Indeed, ribosomal proteins as well as eukaryotic translation initiation factors were identified as potential AEG-1-interacting proteins indicating a potential direct role of AEG-1 in regulating translation (8). It is intriguing that AEG-1 facilitates translation of multiple members of the coagulation pathway, all of which are known mediators of tumor growth, metastasis and angiogenesis, and this particular aspect of AEG-1 function might play a pivotal role in promoting tumor progression and metastasis. Plasma FXII analysis thus might be a potential biomarker for HCC. We observe that knocking down either FXII or TFF3 results in marked inhibition of AEG-1-induced angiogenesis. Interestingly, both FXII and TFF3 interacts with EGFR on endothelial cells to augment proliferation and differentiation, hence angiogenesis (21, 24). Thus, there might be a key role of endothelial EGFR in mediating AEG-1 function, a hypothesis that needs to be experimentally validated.
One novel aspect of AEG-1 function is induction of steatosis. Non-alcoholic fatty liver disease (NAFLD) is one of the precursors leading to non-alcoholic steatohepatitis (NASH) and HCC (25). It will be interesting to check whether AEG-1 is also overexpressed in NAFLD patients thus contributing to eventual hepatocarcinogenesis. Apart from significant increases in expression of some components of fatty acid metabolism our gene expression network analysis did not identify modulation of any major adipogenic/lipogenic pathway such as PPARγ, LXR or PXR pathways. This observation argues that rather than affecting a network, AEG-1 overexpression might lead to promiscuous increases in distinct regulators of fat metabolism resulting in steatosis. The significant increase in SCD2 expression by AEG-1 alone might contribute to steatosis. Induction in SCD2 has also been observed in TGF-α/c-myc transgenic mouse model of HCC (26). SCDs are crucial lipogenic enzymes for monounsaturated fatty acid biosynthesis. SCD1 expression is induced after weaning in mouse liver, while SCD2 expression is detected in livers of mouse embryos and neonates (27). There is a significant reduction in liver and plasma triglycerides in neonatal SCD2 knockout mice (27). The increased SCD2 expression by AEG-1 suggests a shift towards embryonic gene expression pattern, another hallmark of cancer. Crossing SCD2 knockout mice with Alb/AEG-1 mice might provide insight into the importance of SCD2 in mediating the AEG-1-induced steatotic phenotype. Additionally, similar to FXII, AEG-1 might also lead to changes in specific adipogenic/lipogenic factors only at the protein level. Indeed, our preliminary studies reveal overexpression of fatty acid synthase and acetyl co-A carboxylase, two important enzymes of fatty acid synthesis, in livers of Alb/AEG-1 mice (Fig. S10). A detailed study analyzing the molecular mechanism of AEG-1-induced steatosis is currently undergoing.
In summary, the Alb/AEG-1 mouse uncovers several novel aspects of AEG-1 function that might not be possible using in vitro models and nude mice xenograft studies. Characterization of this model facilitates identification of potential biomarkers that might be further validated in HCC patient samples. This mouse model might also be valuable in evaluating novel therapeutic approaches targeted towards NAFLD and HCC.
Supplementary Material
{kind=link}
Acknowledgments
Financial Support
The present study was supported in part by grants from the James S. McDonnell Foundation and National Cancer Institute Grant R01 CA138540 (DS), the Samuel Waxman Cancer Research Foundation (SWCRF) Grant (DS and PBF) and NIH grant R01 CA134721 (PBF). DS is the Harrison Endowed Scholar in Cancer Research and a Blick scholar. PBF holds the Thelma Newmeyer Corman Chair in Cancer Research and is a SWCRF Investigator.
Abbreviations
- AEG-1
Astrocyte elevated gene-1
- DEN
N-nitrosodiethylamine
- Alb/AEG-1
Transgenic mouse with hepatocyte-specific expression of AEG-1
- WT
Wild-type
- SA-β-gal
Senescence associated β-galactosidase
- SAHF
Senescence-associated heterochromatin foci
- HUVEC
Human vascular endothelial cells
- CAM
Chicken chorioallantoic membrane
- FXII
Coagulation factor XII
References
- 1.Yoo BK, Emdad L, Lee S-G, Su Z-Z, Santhekadur PK, Chen D, Gredler R, et al. Astrocyte elevated gene-1 (AEG-1): a multifunctional regulator of normal and abnormal physiology. Pharmacology & Therapeutics. 2011;130:1–8. doi: 10.1016/j.pharmthera.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoo BK, Emdad L, Su ZZ, Villanueva A, Chiang DY, Mukhopadhyay ND, Mills AS, et al. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J Clin Invest. 2009;119:465–477. doi: 10.1172/JCI36460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu G, Chong RA, Yang Q, Wei Y, Blanco MA, Li F, Reiss M, et al. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell. 2009;15:9–20. doi: 10.1016/j.ccr.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SG, Su ZZ, Emdad L, Sarkar D, Fisher PB. Astrocyte elevated gene-1 (AEG-1) is a target gene of oncogenic Ha-ras requiring phosphatidylinositol 3-kinase and c-Myc. Proc Natl Acad Sci U S A. 2006;103:17390–17395. doi: 10.1073/pnas.0608386103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He XX, Chang Y, Meng FY, Wang MY, Xie QH, Tang F, Li PY, et al. MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene. 2011 doi: 10.1038/onc.2011.500. [DOI] [PubMed] [Google Scholar]
- 6.Sarkar D, Park ES, Emdad L, Lee SG, Su ZZ, Fisher PB. Molecular basis of nuclear factor-kappaB activation by astrocyte elevated gene-1. Cancer Res. 2008;68:1478–1484. doi: 10.1158/0008-5472.CAN-07-6164. [DOI] [PubMed] [Google Scholar]
- 7.Lee SG, Kim K, Kegelman TP, Dash R, Das SK, Choi JK, Emdad L, et al. Oncogene AEG-1 promotes glioma-induced neurodegeneration by increasing glutamate excitotoxicity. Cancer Res. 71:6514–6523. doi: 10.1158/0008-5472.CAN-11-0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoo BK, Santhekadur PK, Gredler R, Chen D, Emdad L, Bhutia SK, Pannell L, et al. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocelllular carcinoma. Hepatology. 2011;53:1538–1548. doi: 10.1002/hep.24216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoo BK, Chen D, Su Z-Z, Gredler R, Yoo J, Shah K, Fisher PB, et al. Molecular mechanism of chemoresistance by Astrocyte Elevated Gene-1 (AEG-1) Cancer Res. 2010;70:3249–3258. doi: 10.1158/0008-5472.CAN-09-4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramirez MI, Karaoglu D, Haro D, Barillas C, Bashirzadeh R, Gil G. Cholesterol and bile acids regulate cholesterol 7 alpha-hydroxylase expression at the transcriptional level in culture and in transgenic mice. Mol Cell Biol. 1994;14:2809–2821. doi: 10.1128/mcb.14.4.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murakami H, Sanderson ND, Nagy P, Marino PA, Merlino G, Thorgeirsson SS. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993;53:1719–1723. [PubMed] [Google Scholar]
- 12.Bissell DM, Guzelian PS. Degradation of endogenous hepatic heme by pathways not yielding carbon monoxide. Studies in normal rat liver and in primary hepatocyte culture. J Clin Invest. 1980;65:1135–1140. doi: 10.1172/JCI109767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoo BK, Gredler R, Vozhilla N, Su ZZ, Chen D, Forcier T, Shah K, et al. Identification of genes conferring resistance to 5-fluorouracil. Proc Natl Acad Sci U S A. 2009;106:12938–12943. doi: 10.1073/pnas.0901451106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nierodzik ML, Karpatkin S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell. 2006;10:355–362. doi: 10.1016/j.ccr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Teruel R, Corral J, Perez-Andreu V, Martinez-Martinez I, Vicente V, Martinez C. Potential role of miRNAs in developmental haemostasis. PLoS ONE. 6:e17648. doi: 10.1371/journal.pone.0017648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarkar D, Emdad L, Lee SG, Yoo BK, Su ZZ, Fisher PB. Astrocyte elevated gene-1: far more than just a gene regulated in astrocytes. Cancer Res. 2009;69:8529–8535. doi: 10.1158/0008-5472.CAN-09-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SG, Su ZZ, Emdad L, Sarkar D, Franke TF, Fisher PB. Astrocyte elevated gene-1 activates cell survival pathways through PI3K-Akt signaling. Oncogene. 2008;27:1114–1121. doi: 10.1038/sj.onc.1210713. [DOI] [PubMed] [Google Scholar]
- 19.Emdad L, Lee SG, Su ZZ, Jeon HY, Boukerche H, Sarkar D, Fisher PB. Astrocyte elevated gene-1 (AEG-1) functions as an oncogene and regulates angiogenesis. Proc Natl Acad Sci U S A. 2009;106:21300–21305. doi: 10.1073/pnas.0910936106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LaRusch GA, Mahdi F, Shariat-Madar Z, Adams G, Sitrin RG, Zhang WM, McCrae KR, et al. Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood. 2010;115:5111–5120. doi: 10.1182/blood-2009-08-236430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu K, Dai Z, Pan Q, Wang Z, Yang GH, Yu L, Ding ZB, et al. Metadherin Promotes Hepatocellular Carcinoma Metastasis through Induction of Epithelial-Mesenchymal Transition. Clin Cancer Res. 2011;17:7294–7302. doi: 10.1158/1078-0432.CCR-11-1327. [DOI] [PubMed] [Google Scholar]
- 23.Thirkettle HJ, Girling J, Warren AY, Mills IG, Sahadevan K, Leung H, Hamdy F, et al. LYRIC/AEG-1 is targeted to different subcellular compartments by ubiquitinylation and intrinsic nuclear localization signals. Clin Cancer Res. 2009;15:3003–3013. doi: 10.1158/1078-0432.CCR-08-2046. [DOI] [PubMed] [Google Scholar]
- 24.Rodrigues S, Van Aken E, Van Bocxlaer S, Attoub S, Nguyen QD, Bruyneel E, Westley BR, et al. Trefoil peptides as proangiogenic factors in vivo and in vitro: implication of cyclooxygenase-2 and EGF receptor signaling. FASEB J. 2003;17:7–16. doi: 10.1096/fj.02-0201com. [DOI] [PubMed] [Google Scholar]
- 25.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 26.Griffitts J, Tesiram Y, Reid GE, Saunders D, Floyd RA, Towner RA. In vivo MRS assessment of altered fatty acyl unsaturation in liver tumor formation of a TGF alpha/c-myc transgenic mouse model. J Lipid Res. 2009;50:611–622. doi: 10.1194/jlr.M800265-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyazaki M, Dobrzyn A, Elias PM, Ntambi JM. Stearoyl-CoA desaturase-2 gene expression is required for lipid synthesis during early skin and liver development. Proc Natl Acad Sci U S A. 2005;102:12501–12506. doi: 10.1073/pnas.0503132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.