

Abstract
Elevated Src expression correlates with malignant potential and metastatic disease in many tumors including pancreas cancer. We sought to characterize the molecular effects of Src kinase inhibition with dasatinib (BMS-354825) a novel, multi-targeted kinase inhibitor that targets Src family kinases, in pancreas ductal adenocarcinoma (PDA). We identified sensitive and resistant PDA cell lines to dasatinib treatment and tested the molecular effects of Src inhibition in vitro and in vivo. We show for the first time that cellular localization of Src expression impacts survival in patients with PDA. Pancreas tumors with increased membranous expression of Src result in decreased survival compared with tumors that have increased cytoplasmic Src expression. Src kinase inhibition with dasatinib markedly inhibits cell proliferation, migration, invasion, cell cycle progression and anchorage independent growth and stimulates apoptosis. This is accompanied by decreased phosphorylation of Src, FAK, paxillin, AKT, STAT3, ERK, JNK and MAPK, as well as decreased cyclinD1 expression in a time and concentration-dependent manner. Furthermore, siRNA to Src results in significant decrease in cell proliferation, invasion and migration of pancreas cancer cells. Dasatinib treatment also inhibits in vivo pancreas tumor growth. Mechanisms of resistance to Src inhibition appear to be related to a lack of inhibition of STAT3 and MAPK signaling. These results establish a mechanistic rationale for Src inhibition with dasatinib as a therapeutic target in the treatment of pancreas cancer and identify potential biomarkers of resistance to Src inhibition.
Keywords: Src family kinase, Dasatinib, Pancreas cancer, Therapeutic target, Tumor growth
Introduction
Pancreas cancer remains one of the most lethal forms of human cancer, with a 5 year survival rate of only 3-5% (1). Cytotoxic chemotherapy based on the purine analogue, gemcitabine, remains the standard approach in the adjuvant and palliative setting, but results in minimal responses in the majority of patients. The failure of conventional chemotherapeutic regimes to produce any meaningful impact on survival in patients with pancreas cancer highlights a desperate need for novel treatment strategies.
The proto-oncogene c-Src (Src) encodes a non-receptor tyrosine kinase whose expression and activity are correlated with cancer progression, advanced malignancy and poor prognosis in a variety of human cancers including pancreas cancer (2-6). Src family kinases (SFK) are involved in regulating important mechanisms of receptor tyrosine kinases, G-protein-coupled receptors, and focal adhesion kinase (FAK), thereby influencing many aspects of tumor cell behavior, including proliferation, survival, angiogenesis, adhesion, invasion and metastasis (7-11).
Src is an integrator of divergent signals, facilitating the action of other signaling proteins, making it an attractive target for the treatment of human tumors. It is able to channel phosphorylation signals through Ras/Raf/extracellular signal-regulated kinase (ERK) 1/2 and in certain cells, phosphatidylinositol 3-kinase (PI3K)/AKT pathways (3). Phosphorylation of FAK Tyr397 creates a binding site for Src, indicating that Src may also regulate cell adhesion (12). Paxillin is a substrate for the FAK-Src complex that functions as an adaptor molecule for various signaling and structural proteins in adhesions (13, 14). The mitogen-activated protein kinase (MAP kinase)/ERK cascade is a well-known target of FAK-Src signaling (15) and its activation can be facilitated by association with paxillin (16). Therefore, Src transmits multiple signals including PI3K/AKT, c-Myc/cyclin D1 and FAK/p130CAS/paxillin to induce tumor growth and survival.
Several lines of evidence suggest that Src inhibition is effective as a therapeutic target in pre-clinical models of pancreas cancer. The well-characterized Src inhibitor pyrazolopyrimidine (PP2) and the quinazoline compound AZM475271 have shown activity in orthotopic models for pancreas cancer (5, 17). Src inhibition has also been shown to decrease proliferation, microvessel density, and decrease metastasis in an orthotopic implant mouse model (5, 18). However, most SFKs have been shown to have limited single-agent activity in the clinical setting, therefore, understanding their molecular mechanisms of action remains critical to the successful clinical implementation of these inhibitors.
This study provides the first characterizations of the molecular effects of Src kinase inhibition with dasatinib, (BMS-354825), a multi-targeted kinase inhibitor of Bcr-Abl and SFK signaling (19) in pancreas ductal adenocarcinoma (PDA) in vitro and in vivo. We show for the first time that cellular localization of Src expression impacts survival in patients with pancreas cancer. We have identified sensitive and resistant pancreas cancer cell lines to Src inhibition, allowing us to further elucidate the signaling pathways affected by dasatinib treatment and identify potential biomarkers of resistance to Src inhibition.
Materials and Methods
Materials
Mouse monoclonal antibodies against STAT3 and Src; rabbit monoclonal antibodies against MAPK, pSrc (Tyr416), pAKT (Ser473) and pSTAT3 (Ser727); rabbit polyclonal antibodies against pFAK (Tyr925), pPaxillin (Tyr118), pAKT (Ser473), pMAPK (Thr202/Tyr204), AKT, paxillin and FAK were purchased from Cell Signaling Technology (Beverly, MA). Mouse polyclonal antibodies directed against pJNK, cyclin D1, and also Rabbit polyclonal antibodies directed against JNK were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). The Rabbit polyclonal antibody against pFAK (Y397) was purchased from Abcam Inc. (Cambridge, MA). The secondary antibodies for western blots (anti-mouse and anti-rabbit IgG antibodies) were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA).
Dasatinib (BMS-354825) was kindly provided by Richard Smykla from Bristol-Myers Squibb Oncology (Princeton, NJ).
Cell culture and animals
Human pancreas cancer cell lines BxPC3, PANC1, MiaPaca2, AsPC1, CEPAC, Capan1, Capan2, SW1990 and HPAC were obtained from American Type Culture Collection (ATCC). Tumor cells were maintained according to the ATCC procedures.
Female Athymic Nude mice – Foxn1 nu/nu (4-5 weeks old) were purchased from Harlan Sprague Dawley, Inc, (Indianapolis, IN) and maintained at the Vanderbilt University School of Medicine animal facility under protocols approved by the Vanderbilt Institutional Animal Care and Use Committee (IACUC).
Tissue microarray (TMA)
TMA was constructed as previously described (20). TMA slides were concurrently evaluated by 2 of the authors (M.K.W. and N.B.M.). Nuclear and cytoplasmic staining was scored as follows: the staining index was considered as the sum of the intensity score (0, no staining; 1+, weak; 2+, moderate; 3+, strong) and the distribution score (0, no staining; 1+, staining of <33% of cells; 2+, between 33% and 66% of cells; and 3+, staining of >66% of cells). Staining indices were classified as follows: 3+ or higher, strong staining; 1+ to 2+, weak staining; and 0, negative staining. c-Src was scored as positive if any detectable membranous or cytoplasmic staining was present.
Src gene knockdown by shRNA
Open Biosystems pGIPZ-based short hairpin RNA (shRNA) lentiviral vectors were used to deplete Src expression. Human pancreas cancer cell line BxPC3 was cultured in RPMI containing 10% FBS. Lentiviral shRNA vector pGIPZ with either targeting sequences for knocking-down human Src (Clone IDs: V2LHS_262793 and V2LHS_70230) or non-silencing control sequence was obtained from Vanderbilt University Microarray Core and transfected into BxPC3 cells with FuGENE 6 transfection reagent (Roche, Indianapolis, IN) following the manufacturer’s instruction.
Proliferation assay
Cells were treated with DMSO or dasatinib (0-5000 nmol/L) for 48 hours, and cell viability was determined by MTT (Sigma, St. Louis, MO) assay according to the manufacturer’s direction. IC50 was calculated using Prism software package.
Apoptosis assay
Apoptosis was determined using a luminescence assay method that quantifies caspase 3 and 7 activity. Caspase-3/7-activity was measured using the Caspase Glo 3/7 assay kit (Promega) according to the manufacturer’s protocol and as previously described (21).
Cell cycle analysis
Cells were harvested, washed, and re-suspended as described before (22). Cells were stained with propidium iodide for 30 minutes. Cell fluorescence signals were determined using a FACSCaliber flowcytometer (BD Biosciences, San Jose, CA) and analyzed with its Cell Quest software. The percentage of cells present in different phases of the cell cycle was measured and analyzed.
Migration and invasion assay
The upper chamber of 8-μM pore transwells were overlaid with 50 μl (~100 μg) of diluted matrigel (BD Biosciences, San Diego, CA) solution for invasion. 3×104 cells were seeded into the upper chamber of 8-μM pore transwells coated with collagen for migration. Medium containing 10% FBS and DMSO or dasatinib (1-1000 nmol/L) was used as a chemo attractants in the lower chamber as indicated. The vector control and Src-shRNA cells were also plated as above with or without DMSO or dasatinib. Cells were allowed to migrate for 5 hours or invade the matrigel for 24 hours. Migrated or invaded cells were fixed with 4% paraformaldehyde, stained with 1% crystal violet and counted from six random fields for each membrane at ×20 magnifications and averaged.
Soft-agar colony formation
BxPC3 and PANC1cells (1.5×104) were suspended in 1ml of 0.4% sea plaque agarose containing10% FBS and then plated on top of 1ml of semisolid 0.8% agarose in 35-mm plates. Cells were treated with DMSO or dasatinib (1-1000 nmol/L) for every 48 hours for 2 weeks. Colonies grown on soft agarose were counted in 10 random fields per well.
Western blot analysis
Western blot analyses were done using standard methods (21). Cells were grown in complete media overnight and then treated with dasatinib as required in each assay. Membranes were probed with total and phosphorylated antibodies as detailed above in materials.
Immunofluorescence assay
Cells were grown and treated with dasatinib (5 nmol/L) for 12 hours, fixed and stained with anti-cyclin D1 or c-myc. The anti-cyclin D1 staining was detected with a Cy3-conjugated donkey anti-mouse antibody (Jackson Immuno-Research, West Groove, PA) and c-myc with FITC-green. Slides were prepared using Vecta Shield mounting media for fluorescence with DAPI (Vector Laboratories, Inc, Burlingame, CA), and imaged with a Zeiss Axiophot microscope (Carl Zeiss, Inc., Thornwood, NY). Images were merged using NIH Image J software.
In vivo tumorigenicity assay
Tumors were established by injecting 5×106 BxPC3 or PANC1 cells into the flank of 6-week-old female athymic nude mice Fox1-nu/nu mice (n=5, in each group). Dasatinib (25 mg/kg/day) or citrate buffer (vehicle) was administered by oral gavage and tumor volume (V) was determined by caliper measurements obtained every two days and calculated by the equation V = L × W2 ×0.5, where L is length and W is width of a tumor. At the end of the study, animals were sacrificed and their primary tumors were excised for further analysis. Growth curves for tumors were plotted as the mean volume ± SD of tumors of mice from each group. All experiments were performed in compliance with the Vanderbilt IACUC guidelines.
Immunohistochemistry (IHC)
Mice were euthanized and tumor tissues were collected for immunohistochemical analysis. Tissues were fixed and immunostained using antibodies against cleaved caspase-3, pSrc (Tyr416), pAKT (Ser473) and Ki67 (Biocare, Concord, CA). Cleaved caspase-3, Ki67, pSrc and pAKT were evaluated by an expert pathologist (M.K.W).
For Ki67, caspase 3, pSrc and pAKT staining quantification, positive staining was quantified by using NIH image analysis software, Image J, and is reported as percent area of positive staining.
Statistical analysis
Statistics including mean values and SD were calculated using Microsoft Excel and Prism software (Graphpad, La Jolla, CA). All data represent at least three independent experiments and are expressed as the means ± SD unless otherwise indicated. ANOVA was used to assess the differences between experimental groups unless otherwise indicated.
Survival analysis was performed by using SPSS PC-package (SPSS Inc., Chicago, IL). Overall survival time was calculated from the date of diagnosis until death or the last follow-up contact. The effect of Src expression on survival was assessed using the Kaplan-Meier method and compared using the log-rank test. Multivariate analysis (Cox model) was performed using tumor grade with survival time and median Src-expression score (>1 or ≤1).
Results
Src expression in human pancreas tissues
A TMA consisting of cores of 25 normal pancreas ductal epithelium, 13 chronic pancreatitis and PDAs of 13 well differentiated, 25 moderately differentiated and 30 poorly differentiated tumors was stained for Src expression.
The staining indices of cytoplasmic Src and membranous Src varied significantly between normal pancreas, chronic pancreatitis and PDA tissues. Analysis confirmed a step-wise progression of cytoplasmic and membranous staining of Src from normal pancreas to chronic pancreatitis to advancing tumor grade of PDAs (Figs. 1A and 1B). These results indicate that Src expression increases with progression of pancreatic neoplasia.
Figure 1. Prevalence of Src staining in human pancreas tissue.

(A) There is a step-wise progression of Src expression from normal pancreas to chronic pancreatitis to advancing grade of pancreatic adenocarcinoma in both the cytoplasmic and membranous compartments. (B) The percentage of cases with negative staining (IHC staining index=0), weak staining (1+ to 2+), or strong staining (3+) in each diagnostic group are displayed. (C) Kaplan-Meier survival curves for overall survival of patients with adenocarcinoma according to cytoplasmic and membranous staining for Src are shown. The discontinuous line represents tumors that showed an immunoreactivity score < 2+ staining (n=24). Continuous line represents tumors with an immunoreactivity score > 2+ staining (n=36). Increased membranous Src staining results in decreased overall survival (p=0.001) while increased cytoplasmic Src staining results in improved overall survival (p=0.001).
Although increased Src expression and activity are correlated with cancer progression and advanced malignancy, these results also suggest that increased Src expression also occurs at an earlier neoplastic stage associated with inflammation in patients with chronic pancreatitis.
Furthermore, an increase in Src expression with increasing tumor grade of PDA was also seen on an independent analysis of Src staining of tumor grade compared to normal ducts (p<0.01, Fisher’s exact test).
Of the 68 PDAs represented on the TMA (median patient age was 66 years (range, 37-84 years), there were 36 AJCC stage IIB, 25 stage IIA, 2 stage III, and 1 case each was stage IA and IB. Median duration of follow-up was 19 months during which time 51 patients died (Supplementary Table S1). Median cytoplasmic and membranous Src expression score was determined and overall survival analysis was performed for scores above and below the median expression score. Analysis of the impact of low or high Src-expression composite score on overall survival by cytoplasmic or membranous Src expression is shown in Fig. 1C. Increased membranous Src expression resulted in a significantly lower overall survival (p=0.001), whereas survival outcomes were significantly improved when Src expression was higher in the cytoplasmic compartment (p=0.001).
Src inhibition attenuates pancreas cancer cell proliferation, induces cell cycle arrest and enhances apoptosis
Nine human pancreas cell lines were tested for sensitivity to dasatinib in vitro using a proliferation assay. These cell lines were derived from different stages of tumor and different genetic backgrounds including the oncogenic K-ras mutations (in >95% of analyzed tumors), the loss of the tumor suppressor p53 (~75%) and DPC4/smad4 (50%). The IC50 was calculated after treatment in complete medium for 48 hours (Table 1). In general, dasatinib showed antiproliferative activity at low nanomolar concentrations in all cell lines studied. BxPC3 and HPAC (IC50: 2.7-2.8 nmol/L) cell lines were markedly more sensitive than any other cell line studied. SW1990, Capan1 and Capan2 (IC50: 4.8-9.6 nmol/L) were less sensitive when compared to BxPC3 and HPAC cells, but more sensitive compared to MiaPaca2, PANC1, AsPC1 and CEPAC (IC50: 24.9-51.2 nmol/L) cell lines which were least sensitive.
Table 1.
Pancreas cancer cell lines characteristics and their dasatinib IC50
| Cell Line | Derivation | Genetic background | IC50 (nmol/L) |
|---|---|---|---|
| MiaPaca2 | Primary | Mutant K-ras, mutant p53, wt smad4 | 51.28 |
| PANC1 | Primary | Mutant K-ras, mutant p53, wt smad4 | 45.68 |
| AsPC1 | Metastasis (ascites) | Mutant K-ras, wt p53, wt smad4 | 44.97 |
| CEPAC1 | Metastasis (liver) | Mutant K-ras, wt p53, mutant smad4 | 24.99 |
| Capan1 | Metastasis (liver) | Mutant K-ras, ND p53, mutant smad4 | 9.604 |
| SW1990 | Metastasis (spleen) | Mutant K-ras, wt p53, ND smad4 | 5.106 |
| Capan2 | Primary | Mutant K-ras, wt p53, wt smad4 | 4.872 |
| HPAC | Primary | Mutant K-ras, wt p53, wt smad4 | 2.791 |
| BxPC3 | Primary | Wt K-ras, mutant p53, mutant smad4 | 2.809 |
WT: wild type; ND: not determined
BxPC3 and PANC1 cells were treated with dasatinib resulted in a decreased percentage of cells in G2-M and S phases and an increased percentage of cells in the sub-G0 population, consistent with cell cycle blockade in the G2-S transition and induction of apoptosis. Dasatinib treatment resulted in decreased percent of cells in S-phase in both BxPC3 (most sensitive) and PANC1 (least sensitive) cell lines (Fig. 2A). This concentration-dependent decrease was significant (p<0.001) starting at 10 nmol/L of dasatinib in BxPC3 cells and starting at 50 nmol/L in PANC1 cells consistent with the sensitivity of BxPC3 cells to dasatinib and the resistant nature of PANC1 cells to dasatinib.
Figure 2. Effect of Src inhibition results on apoptosis and cell cycle regulation.

(A) There was a concentration-dependent decrease in percentage of cells in G2-M and S phases starting at 10 nmol/L of dasatinib in BxPC3 cells and starting at 50 nmol/L in PANC1 cells. (B) There was a concentration-dependent induction of apoptosis by Caspase-Glo 3/7 assay starting at 5 nmol/L in BxPC3 cells and starting at 100 nmol/L in PANC1 cells. Each condition was assayed in triplicate and the means ± SD are presented. Standard error: *p<0.05; **p<0.01; ***p<0.001; ns=p>0.05.
To further investigate the mechanism of growth inhibition by dasatinib, we examined its effect on the induction of apoptosis (Fig. 2B). BxPC3 cells were highly apoptotic at any concentration studied when compared with more resistant PANC1 cells.
Src inhibition attenuates the metastatic potential of pancreas cancer cells
There was a concentration-dependent reduction in cell motility (Supplementary Fig. S1A) and migration (Fig. 3A) in pancreas cancer cells treated with dasatinib. BxPC3 cells showed a significant decrease in motility and migration starting at dasatinib concentrations as low as 1 to 2 nmol/L. BxPC3 cells were significantly more sensitive to dasatinib when compared to more resistant PANC1 cells. Cells were allowed to recover by removal of dasatinib after wounding. PANC1 cells recovered faster than BxPC3 cells at any concentration of dasatinib treatment (Supplementary Fig. S1B).
Figure 3. Effect of Src inhibition on migration, invasion and colony formation.

Concentration-dependent decrease in cell migration (A), invasion (B) and colony formation (C) were seen. BxPC3 cells were significantly more sensitive at any concentration studied when compared with more resistant PANC1 cells. Individual data point represent the mean ± SD of three independent experiments. (D) Inhibition of Src expression by shRNA decreases migration and invasion in BxPC3 cells. Each data point represents the mean ± SD of triplicate plates. *p<0.05; **p<0.01; ***p<0.001; ns=p>0.05.
Inhibition of BxPC3 cell migration increased with the concentration of dasatinib to a maximum at 10 nmol/L, while in PANC1 cells, the decrease was significant starting at 100 nmol/L (Fig. 3A). There was also a concentration-dependent decrease in cell invasion with dasatinib treatment in BxPC3 and PANC1 cells (Fig. 3B). BxPC3 cell invasion was significantly inhibited with as little as 5 nmol/L dasatinib (p<0.05). Dasatinib at100 nmol/L showed a 22 fold decrease in BxPC3 cell invasion compared with untreated cells whereas only a 1.5 fold decrease was observed compared to untreated cells in PANC1 cell invasion at the same dose.
To determine whether dasatinib affects cell transformation, we performed a soft-agar colony formation assay. Dasatinib inhibited anchorage-independent growth of BxPC3 and PANC1 cells (Fig. 3C). With the addition of dasatinib, BxPC3 and PANC1 cells formed fewer colonies in soft agar compared with DMSO control (BxPC3 control, 96.53 ± 12.76 colonies versus dasatinib 100 nmol/L, 6.37 ± 2.83 colonies, p<0.001; PANC1 control, 76.32 ± 10.3 colonies versus dasatinib 100 nmol/L, 57.68 ± 5.92 colonies, p<0.05).
Since dasatinib is a multi-targeted kinase inhibitor, we sought to confirm that these effects of dasatinib were specific to its activity on Src kinase inhibition. We stably inhibited the protein expression levels of Src in BxPC3 cells using small interfering RNA (Lentiviral-shRNA vector, Supplementary Fig. S2A). Blocking Src expression resulted in a significant reduction in proliferation (p<0.001) (Supplementary Fig. S2B), migration (p<0.001) and invasion (P<0.01) (Fig. 3D) as compared to control cells. These data further confirms that dasatinib inhibits migration and invasion through Src signaling regardless of the antiproliferative or apoptotic effects of the drug.
Src inhibition blocks activation of multiple downstream signaling pathways in pancreas cancer cells
To explore the potential regulatory mechanisms mediating the growth inhibitory effects of dasatinib, we tested its ability to inhibit kinases important in survival, angiogenesis, proliferation, migration and invasion in a concentration- and time-dependent manner in BxPC3 (sensitive) and PANC1 (least sensitive) cells. Src kinase activity was significantly inhibited in all cell lines, while total Src levels were not affected. Phosphorylation of Src, FAK, Paxillin, AKT, STAT3, JNK and MAPK were decreased in BxPC3 cells treated with dasatinib compared to control cells in a concentration-dependent manner (Fig. 4A). There was a similar inhibition of Src, FAK and AKT phosphorylation but at much higher concentrations of dasatinib in PANC1 cells, while no significant effects were noted in phosphorylation of paxillin, STAT3 or MAPK (Fig. 4A). STAT3 and MAPK are involved in cell survival and angiogenesis, while JNK and paxillin are involved in motility/migration and invasion of cancer cells. The lack of inhibition of phosphorylation of paxillin, STAT3 and MAPK even at higher concentrations of dasatinib in PANC1 cells, suggests that some of the resistance of dasatinib in certain pancreas cancer cell lines is related to its limited activity in targeting certain pathways related to cell survival, angiogenesis (STAT3 and MAPK) and cell adhesion (paxillin). Dasatinib did markedly inhibit AKT activation, suggesting a role for dasatinib in targeting both cell survival and proliferation pathways in pancreas cancer cells.
Figure 4. Effects of Src inhibition with dasatinib on cell signaling.

Cells from BxPC3 (A), and PANC1 (B) were treated with DMSO or dasatinib (0-1000 nmol/L) for 12 h, lysed, and analyzed by Western blotting with indicated antibodies. Blots are representative of at least two separate experiments with similar results. There is a lack of inhibition of pSTAT3 and pMAPK signaling with dasatinib in PANC1 cells. (C) BxPC3 cells were treated with dasatinib (5 nmol/L) for 12 h. There was a significant decrease in expression of c-Myc and cyclin-D1 by immunofluorescence.
The effect of dasatinib on G1/S transition prompted us to study its effect on c-Myc, a Src target gene (23) and cyclin D1, the rate limiting factor for cellular proliferation (24). Both c-Myc and cyclin D1 were down regulated with dasatinib treatment (Figs. 4A, 4B and 4C). A more striking correlation was found between the cell cycle regulatory effect of dasatinib and the modulation of cyclin D1 expression.
Dasatinib inhibits pancreas tumor progression in vivo
Having shown the biologic effects and the cellular responses to Src inhibition with dasatinib in vitro, we sought to determine its efficacy in vivo using a mouse xenograft model implanting BxPC3 (Fig. 5A) and PANC1 (Fig. 5B) cells. Mice were treated with citrate buffer (control or vehicle) or dasatinib (25 mg/kg) by oral gavage for 14 days. Tumor volumes of untreated BxPC3 xenografts increased 62% compared with a 17% (1.96 fold) decrease in tumor volume in dasatinib treated animals at 10 days of treatment (p<0.001). Tumor volumes of untreated PANC1 xenografts increased174% at 10 days, compared with only a 50% increase in tumor volume of dasatinib treated animals (p<0.001). These in vivo data show tumor growth inhibition even in less sensitive PANC1 cells, indicating that dasatinib is a promising agent for inhibiting pancreas tumor progression.
Figure 5. Src inhibition with dasatinib effectively inhibits tumor growth in vivo.
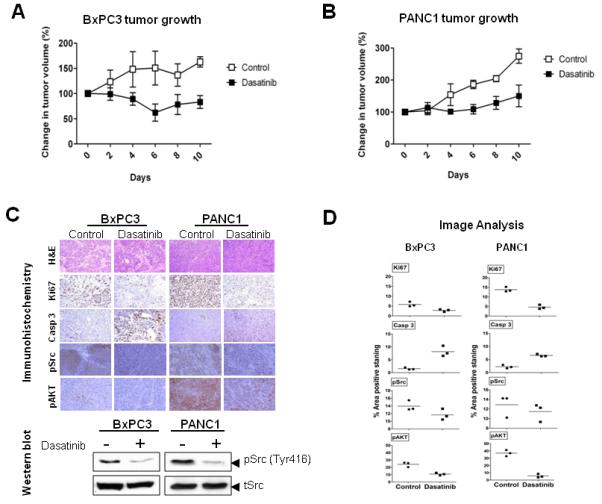
Mice with subcutaneously established tumors from BxPC3 (A) or PANC1 (B) cells were treated with dasatinib (n = 5) at 25 mg/kg or vehicle (n = 5) daily by oral gavage for 10 days. Growth curves for tumors are presented as the mean ± SD of five tumors in each data point. (C) Representative examples of immunohistochemical analysis of BxPC3 and PANC1 tumor tissues stained with H&E, Ki67, cleaved caspase 3, pSrc and pAKT. There is no evidence of treatment-induced necrosis on H&E staining. Compared with vehicle treated controls, Ki-67, pSrc and pAKT staining is markedly decreased, and cleaved caspase 3 staining is markedly elevated in dasatinib treated animals, showing successful target inhibition by dasatinib. Magnification ×20. Tumor tissue was analyzed for the expression of pSrc by Western blotting. (D) The percent area positive staining was determined using Image J image analysis software for Ki67, cleaved caspase 3, Phospho Src (pSrc) and phospho AKT (pAKT) stained immunohistochemistry data from BxPC3 and PANC1 tumor xenografts. Individual data points represent the mean ± SD of three independent tissue samples analyzed in each treatment.
Inhibition of Src activity correlates with in vivo inhibition of proliferation and increased apoptosis in pancreas tumors
To correlate our in vitro findings to in vivo, immunohistochemical analysis of dasatinib treated tumor xenografts of BxPC3 and PANC1 relative to vehicle-treated controls at the end of the treatment period was measured. Dasatinib treated tumors exhibited increased apoptosis (cleaved caspase 3) and decreased proliferation (Ki67) as well as a significant inhibition of pSrc Tyr416 and pAKT Ser473 relative to controls (Figs. 5C and 5D).
Discussion
The role of the proto-oncogene c-Src in the progression of many tumors is well documented (19). However, overexpression of wild-type Src is weakly oncogenic on its own (25) and activating mutations are rare, limiting our understanding of Src in the development, maintenance, and progression of cancer (26-30). Our results show that Src inhibition in pancreas cancer cells treated with dasatinib result in inhibition of cell proliferation, migration, invasion and anchorage independent growth, which directly correlate with a significant reduction in tumor growth in vivo. We identified sensitive and more resistant pancreas cancer cell lines to dasatinib treatment to further elucidate the molecular mechanisms involved with Src inhibition. Src kinase inhibition with dasatinib blocks the activation of multiple downstream signal transduction pathways known to promote survival, angiogenesis, proliferation, motility, migration and invasion of tumor cells (31, 32) including AKT, STAT3, MAPK, JNK, FAK and paxillin, in sensitive cell lines (BxPC3).
Targeting signal transduction pathways is an effective strategy in many tumor types. However, activation of parallel signaling pathways can limit the efficacy of this approach, therefore, understanding the mechanisms of resistance to targeted agents remains critical to enhance their clinical usefulness. Resistance to dasatinib treatment appears to be related to lack of inhibition of STAT3 and MAPK signaling. STAT3 phosphorylation is inhibited with dasatinib treatment in sensitive pancreas cancer cell lines, but not in the more resistant PANC 1 cell line. Consistent with this finding, recently, reactivation of STAT3 signaling after Src inhibition has been shown in head and neck cancer (33) and non-small cell lung cancer (34). Somewhat selective to SFKs is their ability to activate STAT3, which leads to the activation of downstream targets including Bcl-XL, c-Myc (35, 36) and cyclin D1 (37), resulting in increased cell survival, proliferation and tumor growth. Our results suggest that this compensatory pathway may account for the resistance to Src inhibition and implicates elevated STAT3 expression as a potential biomarker of this resistance. Furthermore, this data provides rationale to combine STAT3 inhibitors with dasatinib to improve clinical response in pancreas cancer.
Additionally, basal pAKT levels were also noted to be markedly elevated in the more resistant PANC1 cell line, as well as other less sensitive cell lines such as MiaPaca2 and AsPC1(data not shown), when compared with BxPC3 sensitive cells. Although, dasatinib treatment resulted in significant inhibition of pAKT activation in both PANC1 and BxPC3 cells, this increased basal pAKT levels may be another contributing factor towards the resistant nature of PANC1 cells and requires further investigation.
We also show for the first time that Src expression increases with progression of pancreas neoplasia from normal pancreas to chronic pancreatitis to PDA. In addition, the increase in Src expression with increasing tumor grade of PDAs is consistent with the role of Src activity playing an important role in cancer progression and metastasis (19, 38). Our finding that survival outcomes are significantly improved when Src expression is lower in the membranous compartment compared to the cytoplasmic compartment, suggests that cytoplasmic Src is weakly oncogenic and requires membranous localization to become activated and regulate important mechanisms of specific receptor pathways, stimulating oncogenesis. We have previously shown that activation of Src results in its translocation from the cytoplasm to the cell membrane where it activates TNF-α converting enzyme (TACE) to induce shedding EGFR ligands from the cell membrane and activate EGFR signaling (21, 39).
Interestingly, we also found that Src expression is increased in patients with chronic pancreatitis. Increased Src expression and activity are generally correlated with cancer progression and advanced malignancy. However these results implicate activation of Src at an earlier stage of pancreatic neoplasia and may suggest that patients with chronic pancreatitis may have a higher propensity toward malignant transformation depending on their levels of Src activation.
Dasatinib is a multi-targeted kinase inhibitor and it is likely that not all of its biological and molecular effects are due to Src inhibition alone. However, several lines of evidence suggest that the effects of dasatinib in pancreas cancer are related to Src kinase inhibition. Phosphorylation of Tyr416 in the kinase domain is a critical activation step in the regulation of Src tyrosine kinase activity (10). We show Src phosphorylation at Tyr416 is inhibited with addition of dasatinib. In addition to our results with Src shRNA cells, these findings suggest that the predominant effects of dasatinib treatment is related to inhibition of Src kinase activity.
Inhibition of Src signaling has been shown to restore sensitivity to gemcitabine in cell lines derived from pancreas tumors. Similarly, silencing of FAK expression in PANC1 cells restores sensitivity to gemcitabine treatment (4, 5, 40). Furthermore, inhibition of Src also has been shown to revert chemoresistance against 5-fluorouracil in human pancreas carcinoma cells (41). Therefore, these results clearly implicate Src inhibition in restoring chemosensitivity and suggest a novel approach using dasatinib in combination with gemcitabine for the treatment of pancreas cancer.
There were no known genetic mutations that influenced cell sensitivity to dasatinib (Table 1). Inactivating p53 mutations, activating Ras mutations (predominantly K-ras) and Smad4/DPC4 mutations are commonly found in PDAs. We did not find any relationship between p53 or K-ras or smad4 status and sensitivity to dasatinib which is consistent with other studies in head and neck squamous cell carcinoma and non-small cell lung carcinoma (NSCLC) cells (42).
In conclusion, we show that dasatinib inhibits pancreas tumor growth both in vitro and in vivo. Our results, for the first time, demonstrate that dasatinib is effective in inhibiting multiple signaling pathways involved in tumor growth, proliferation, angiogenesis, survival, motility, migration and invasion in pancreas cancer, while resistance to Src inhibition appears to be related to activation of parallel signaling pathways including STAT3. Furthermore, dasatinib inhibits the metastatic potential of pancreas cancer by inhibiting cell migration, invasion and anchorage-independent growth. The in vivo effects of dasatinib on pancreas tumor growth correlate with significant inhibition of Src and AKT phosphorylation, demonstrating biological and translational relevance.
Supplementary Material
Acknowledgements
We thank Mr. Jason M. Herndon for his excellent technical assistance with the animal experiments.
Grant Support. P50 95103 GI Special Program of Research Excellence Grant (SPORE Project 1), 5P30DK058404-08 Digestive Disease Research Center (DDRC) Translational Award and Vanderbilt-Ingram Cancer Center Support Grant 5P30 CA068485-1, Core services performed through Vanderbilt University Medical Center’s DDRC supported by NIH grant P30DK058404 (N.B. Merchant). Special thanks to Mrs. Ginny Auschwitz and family for provision of funds to N.B. Merchant.
Footnotes
Supplementary material for this article is available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
References
- 1.Strimpakos A, Saif MW, Syrigos KN. Pancreatic cancer: from molecular pathogenesis to targeted therapy. Cancer Metastasis Rev. 2008;27:495–522. doi: 10.1007/s10555-008-9134-y. [DOI] [PubMed] [Google Scholar]
- 2.Allgayer H, Boyd DD, Heiss MM, Abdalla EK, Curley SA, Gallick GE. Activation of Src kinase in primary colorectal carcinoma - An indicator of poor clinical prognosis. Cancer. 2002;94:344–51. doi: 10.1002/cncr.10221. [DOI] [PubMed] [Google Scholar]
- 3.Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008;27:6365–75. doi: 10.1038/onc.2008.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Inhibition of Src tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells. Clinical Cancer Research. 2004;10:2307–18. doi: 10.1158/1078-0432.ccr-1183-3. [DOI] [PubMed] [Google Scholar]
- 5.Yezhelyev MV, Koehl G, Guba M, et al. Inhibition of Src tyrosine kinase as treatment for human pancreatic cancer growing orthotopically in nude mice. Clinical Cancer Research. 2004;10:8028–36. doi: 10.1158/1078-0432.CCR-04-0621. [DOI] [PubMed] [Google Scholar]
- 6.Serrels B, Serrels A, Mason SM, et al. A novel Src kinase inhibitor reduces tumour formation in a skin carcinogenesis model. Carcinogenesis. 2009;30:249–57. doi: 10.1093/carcin/bgn278. [DOI] [PubMed] [Google Scholar]
- 7.Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–42. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- 8.Yeatman TJ. A renaissance for SRC. Nature Reviews Cancer. 2004;4:470–80. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 9.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer and Metastasis Reviews. 2003;22:337–58. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 10.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annual Review of Cell and Developmental Biology. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 11.Chang Q, Jorgensen C, Pawson T, Hedley DW. Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. British Journal of Cancer. 2008;99:1074–82. doi: 10.1038/sj.bjc.6604676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Webb DJ, Donais K, Whitmore LA, et al. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nature Cell Biology. 2004;6:154. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 13.Bellis SL, Miller JT, Turner CE. Characterization of Tyrosine Phosphorylation of Paxillin in-Vitro by Focal Adhesion Kinase. Journal of Biological Chemistry. 1995;270:17437–41. doi: 10.1074/jbc.270.29.17437. [DOI] [PubMed] [Google Scholar]
- 14.Turner CE. Paxillin and focal adhesion signalling. Nature Cell Biology. 2000;2:E231–E6. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- 15.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Progress in Biophysics & Molecular Biology. 1999;71:435–78. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 16.Ishibe S, Joly D, Zhu XL, Cantley LG. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol Cell. 2003;12:1275–85. doi: 10.1016/s1097-2765(03)00406-4. [DOI] [PubMed] [Google Scholar]
- 17.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Inhibition of Src Tyrosine Kinase Impairs Inherent and Acquired Gemcitabine Resistance in Human Pancreatic Adenocarcinoma Cells. Clin Cancer Res. 2004;10:2307–18. doi: 10.1158/1078-0432.ccr-1183-3. [DOI] [PubMed] [Google Scholar]
- 18.Trevino JG, Summy JM, Lesslie DP, et al. Inhibition of Src expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. American Journal of Pathology. 2006;168:962–72. doi: 10.2353/ajpath.2006.050570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist. 2009;14:667–78. doi: 10.1634/theoncologist.2009-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cates JM, Byrd RH, Fohn LE, Tatsas AD, Washington MK, Black CC. Epithelial-mesenchymal transition markers in pancreatic ductal adenocarcinoma. Pancreas. 2009;38:e1–6. doi: 10.1097/MPA.0b013e3181878b7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merchant NB, Voskresensky I, Rogers CM, et al. TACE/ADAM-17: a component of the epidermal growth factor receptor axis and a promising therapeutic target in colorectal cancer. Clin Cancer Res. 2008;14:1182–91. doi: 10.1158/1078-0432.CCR-07-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagaraj NS, Vigneswaran N, Zacharias W. Hypoxia inhibits TRAIL-induced tumor cell apoptosis: involvement of lysosomal cathepsins. Apoptosis. 2007;12:125–39. doi: 10.1007/s10495-006-0490-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barone MV, Courtneidge SA. Myc but not Fos rescue of PDGF signalling block caused by kinase-inactive Src. Nature. 1995;378:509–12. doi: 10.1038/378509a0. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe G, Howe A, Lee RJ, et al. Induction of cyclin D1 by simian virus 40 small tumor antigen. Proc Natl Acad Sci U S A. 1996;93:12861–6. doi: 10.1073/pnas.93.23.12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biscardi JS, Ishizawar RC, Silva CM, Parsons SJ. Tyrosine kinase signalling in breast cancer: epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000;2:203–10. doi: 10.1186/bcr55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Irby RB, Mao W, Coppola D, et al. Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet. 1999;21:187–90. doi: 10.1038/5971. [DOI] [PubMed] [Google Scholar]
- 27.Sugimura M, Kobayashi K, Sagae S, et al. Mutation of the SRC gene in endometrial carcinoma. Jpn J Cancer Res. 2000;91:395–8. doi: 10.1111/j.1349-7006.2000.tb00958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang NM, Yeh KT, Tsai CH, Chen SJ, Chang JG. No evidence of correlation between mutation at codon 531 of src and the risk of colon cancer in Chinese. Cancer Lett. 2000;150:201–4. doi: 10.1016/s0304-3835(99)00398-5. [DOI] [PubMed] [Google Scholar]
- 29.Nilbert M, Fernebro E. Lack of activating c-SRC mutations at codon 531 in rectal cancer. Cancer Genet Cytogenet. 2000;121:94–5. doi: 10.1016/s0165-4608(00)00226-0. [DOI] [PubMed] [Google Scholar]
- 30.Laghi L, Bianchi P, Orbetegli O, Gennari L, Roncalli M, Malesci A. Lack of mutation at codon 531 of SRC in advanced colorectal cancers from Italian patients. Br J Cancer. 2001;84:196–8. doi: 10.1054/bjoc.2000.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Summy JM, Gallick GE. Treatment for advanced tumors: Src reclaims center stage. Clinical Cancer Research. 2006;12:1398–401. doi: 10.1158/1078-0432.CCR-05-2692. [DOI] [PubMed] [Google Scholar]
- 32.Furukawa T. Molecular targeting therapy for pancreatic cancer: current knowledge and perspectives from bench to bedside. Journal of Gastroenterology. 2008;43:905–11. doi: 10.1007/s00535-008-2226-1. [DOI] [PubMed] [Google Scholar]
- 33.Sen B, Saigal B, Parikh N, Gallick G, Johnson FM. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009;69:1958–65. doi: 10.1158/0008-5472.CAN-08-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byers LA, Sen B, Saigal B, et al. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin Cancer Res. 2009;15:6852–61. doi: 10.1158/1078-0432.CCR-09-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bowman T, Broome MA, Sinibaldi D, et al. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:7319–24. doi: 10.1073/pnas.131568898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furstoss O, Dorey K, Simon V, Barila D, Superti-Furga G, Roche S. c-Abl is an effector of Src for growth factor-induced c-myc expression and DNA synthesis. Embo Journal. 2002;21:514–24. doi: 10.1093/emboj/21.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steiner MS, Anthony CT, Lu Y, Holt JT. Antisense c-myc retroviral vector suppresses established human prostate cancer. Hum Gene Ther. 1998;9:747–55. doi: 10.1089/hum.1998.9.5-747. [DOI] [PubMed] [Google Scholar]
- 38.Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 2004;6:209–14. doi: 10.1016/j.ccr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Voskresensky I, Lee W, Rogers C, Trivedi B, Coffey R, Merchant N. QS284. Secondary Bile Acids Activate the Epidermal Growth Factor Receptor (EGFR) Through a Src Kinase-Mediated Release of EGFR Ligands in Colon Cancer Cells. Journal of Surgical Research. 2008;144:379–80. [Google Scholar]
- 40.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. siRNA directed against c-Src enhances pancreatic adenocarcinoma cell gemcitabine chemosensitivity. Journal of the American College of Surgeons. 2004;198:953–9. doi: 10.1016/j.jamcollsurg.2004.01.037. [DOI] [PubMed] [Google Scholar]
- 41.Ischenko I, Camaj P, Seeliger H, et al. Inhibition of Src tyrosine kinase reverts chemoresistance toward 5-fluorouracil in human pancreatic carcinoma cells: an involvement of epidermal growth factor receptor signaling. Oncogene. 2008;27:7212–22. doi: 10.1038/onc.2008.326. [DOI] [PubMed] [Google Scholar]
- 42.Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clinical Cancer Research. 2005;11:6924–32. doi: 10.1158/1078-0432.CCR-05-0757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.