

Abstract
We present an implementation of a recently introduced method for estimating the allele-frequency spectrum under the diffusion approximation. For single-nucleotide polymorphism (SNP) frequency data from multiple populations, the method computes numerical solutions to the allele-frequency spectrum (AFS) under a complex model that includes population splitting events, migration, population expansion, and admixture. The solution to the diffusion partial differential equation (PDE) that mimics the evolutionary process is found by means of truncated polynomial expansions. In the absence of gene flow, our computation of frequency spectra yields exact results. The results are compared to those that use a finite-difference method and to forward diffusion simulations. In general, all the methods yield comparable results, although the polynomial-based approach is the most accurate in the weak-migration limit. Also, the economical use of memory attained by the polynomial expansions makes the study of models with four populations possible for the first time. The method was applied to a four-population model of the human expansion out of Africa and the peopling of the Americas, using the Environmental Genome Project (EGP) SNP database. Although our confidence intervals largely overlapped previous analyses of these data, some were significantly different. In particular, estimates of migration among African, European, and Asian populations were considerably lower than those in a previous study and the estimated time of migration out of Africa was earlier. The estimated time of founding of a human population outside of Africa was 52,000 years (95% confidence interval: 36,000–80,800 years).
Keywords: demographic inference, spectral expansions, allele-frequency spectrum (AFS), Wright–Fisher processes
THE study of demographic history from genetic data is important for understanding how populations have diverged and come to be in their present state. In the case of human populations, genetic studies of demographic history can be a great complement to archaeological studies of human prehistory. Having a model of demographic history is also important for facilitating the identification of genomic regions that have been evolving under selective pressures. The inference of signatures of natural selection from DNA sequence data sets requires accounting for different demographic forces that contribute to shaping such patterns (Risch and Merikangas 1996; Nielsen 2001; Goldstein and Chikhi 2002; Shriver et al. 2003; Laberge et al. 2005; Schaffner et al. 2005; Lao et al. 2006; Chen 2012).
The extraction of information from patterns of variation in genetic data requires mathematical models that can capture the diversity and richness of population histories, as well as efficient statistical tools for fitting models to data. The demands on models and on tools are especially great for questions about human populations, which have a complex history and for which large amounts of data can often be brought to bear. An important approach to modeling demographic history and comparing models to genetic data focuses on the allele-frequency spectrum (AFS). Given DNA sequence data from several individuals in K populations, the resulting single-nucleotide polymorphism (SNP) joint AFS is a K-dimensional matrix in which each cell specifies the number of derived alleles that were found at a particular set of K frequencies in the data. Inference of a demographic model of history for a data set consists of finding a model and a set of parameter values that correspond to an expected AFS that closely resembles the AFS that was observed in the data.
In this article we present a software implementation of a recently introduced spectral method (Lukic et al. 2011) for estimating the AFS under the diffusion approximation. The implementation allows for the study of a broad class of demographic models for multiple closely related populations. In this Introduction we review the history of methods for solving these types of problems in population genetics, as well as some of the limitations of the different approaches.
A brief history of the use of diffusion processes for approximating the AFS
The classical approach to computing the AFS was developed by Fisher (1930), Wright (1931), and Kimura (1964), who introduced forward diffusion processes and irreversible mutations in a single population to model the evolutionary process. The theory was extended by Kimura (1969) to study many nucleotide positions by introducing the infinite-sites mutation model. Coalescent models can also be used to develop exact solutions for the AFS under the infinite-sites assumption, and these are particularly amenable for some models with multiple populations (Wakeley and Hey 1997).
Until relatively recently, most applications of classical forward diffusion theory to demographic inference were limited to models that assumed some form of equilibrium (mutation/drift equilibrium, mutation/selection equilibrium, etc.) and could be solved exactly (Sawyer and Hartl 1992; Ewens 2004). However, the recent introduction of numerical methods to integrate arbitrary diffusion equations has allowed for the relaxing of the assumption of equilibrium so that general nonequilibrium scenarios can be studied. In Williamson et al. (2005) a finite-difference scheme was used to numerically solve the equations associated with a model that combined the effects of selection and population-size growth in one population. The finite-difference method is a classical technique for numerically solving partial differential equations in which the density of allele frequencies is approximated by a piecewise linear function, and the derivatives are approximated by means of finite subtractions. The piecewise linear approximation relies upon a grid defined on frequency space, and the accuracy of the method increases as the grid becomes finer. Later in Gutenkunst et al. (2009), these techniques were implemented in the program ∂a∂i and extended to consider models with two and three simultaneous populations that undergo random drift, migration events, arbitrary population size changes, admixture events, and directional selection (e.g., Xing et al. 2010; Albert et al. 2011; Jensen and Bachtrog 2011).
Despite the superficial differences between these approaches to population genetics, it is known that both coalescent and forward diffusion processes are dual processes in an analytic sense (Griffiths and Spano 2010). The connection between these dual processes can be made explicit through the application of orthogonal polynomial theory to Wright–Fisher processes (see Griffiths and Spano 2010 for a recent review on the topic). Previous studies that applied orthogonal polynomial theory to the two-allele neutral Wright–Fisher process made it possible to solve the associated diffusion equations for one population (Myers et al. 2008). Also, a recently introduced spectral expansion makes it possible to exactly integrate the same diffusion equations with arbitrary diploid selection (Song and Steinrücken 2011). In this study, we employ series of orthogonal polynomials to solve multipopulation Wright–Fisher processes (Lukic et al. 2011), and we use these solutions to infer models of demographic history from genetic data. In general, these methods provide approximate solutions for finite numbers of polynomials and approach the exact solution in the limit as the number of polynomials goes to infinity. In the particular case of neutral models wherein each population size is fixed for the duration that a population persists, and there is no gene exchange between populations, our polynomial-based approach provides the exact solution of the AFS, which was first described using a coalescent approach by Wakeley and Hey (1997) (see supporting information, File S1, section 1).
The curse of dimensionality
Another important motivation for the use of orthogonal polynomials is to be able to tackle models in which the high dimensionality of the frequency spectra becomes an important limitation. As the number of variables used to approximate the density of population allele frequencies grows exponentially, the total number of simultaneous populations that one can study becomes limited. In particular, if we use a grid of G grid points per population to approximate the density of allele frequencies φ as a piecewise linear function on the grid, the number of variables used in the approximation is GK, with K the total number of populations.
The use of truncated polynomial expansions to approximate the density of population allele frequencies has two main advantages with respect to the piecewise linear approximations on grids that are used in finite-difference methods:
- First, the contributions to an allele-frequency spectrum built from C haploid genomes sampled per population can be located in the first (C – 2)K terms of lowest degree of a polynomial expansion of the density of population allele frequencies φ. More precisely, given an AFS built from C sampled chromosomes in K populations, a particular demographic scenario θ, and a joint density of population frequencies φ(x|θ) associated with θ, we know that the AFS and the joint density of frequencies are related (Sawyer and Hartl 1992) as
(1) - If φ(x|θ) is expanded in the basis of shifted Jacobi polynomials (see Appendix),
- the following integrals,
(2) vanish for n1 > C − 2 or n2 > C − 2 … or nK > C − 2. Therefore, only the first (C − 2)K terms of lowest degree in the polynomial expansion yield nonzero contributions to the AFS in Equation 1, for 0 < i1 < C, … , 0 < iK < C. This means that the information in φ(x, t) required to compute the AFS can be represented as a vector in the vector space spanned by , i.e., the vector space of polynomials of degree bounded by C − 2 (see Appendix).
Second, it is known that polynomial approximations of smooth functions exhibit exponential convergence (Hesthaven et al. 2007). In particular, the amount of computational resources needed to numerically solve the multipopulation Wright–Fisher equations depends on the number of variables needed to approximate φ(x) (e.g., number of floating-point values). We denote this number as nφ. Any other relevant quantity in the algorithm, such as the number of variables needed to approximate the diffusion operator or the number of operations needed to evaluate the AFS, will be a function of nφ. Therefore, any efficient algorithm that solves the multipopulation Wright–Fisher diffusion process needs to be designed with the goal of minimizing nφ given a fixed bound for the numerical error. If one uses a finite-difference algorithm, then nφ = GK with G the number of grid points per population. On the other hand, if one uses spectral methods, then nφ = (Λ + 2)K − 2. Here, Λ is the number of polynomials per population and the term Λ + 2 comes from the fact that each boundary component, except the ancestral and derived vertices of the K-cube, contributes its own polynomial expansion (Lukic et al. 2011). In both algorithms the diffusion operator is approximated as a sparse matrix of size nφ × nφ, and the number of operations needed to evaluate Equation 1 is the same function of nφ, K, and C. As the polynomial approximations of smooth functions exhibit exponential convergence, we can use lower values of Λ to accurately approximate the solutions of the diffusion equations. This allows us to use a larger number of populations.
As a simple illustration of the convergence properties of polynomial expansions, we approximated the equilibrium density of allele frequencies in one population [φ(x) = dx/x with 1/2 N ≤ x ≤ 1] by means of polynomial expansions φΛ(x). Also, we considered piecewise linear approximations φG(x) on grids of size G. For 0.005 ≤ x ≤ 1, the error function decayed exponentially as
with Λ the number of polynomials in the polynomial approximation of the density of allele frequencies; see Figure 1. However, in the case of piecewise linear approximations, the error function decayed as a power law
with G the number of grid points; see Figure 1. The parameters of the exponential and power law functions were estimated by means of the least-squares method. Therefore, given any fixed truncation nφ = Λ = G, a polynomial expansion gives a much more accurate approximation of the true density φ(x).
Figure 1 .

Decay of the error function between the equilibrium density of allele frequencies and its polynomial approximation and piecewise linear approximation. The horizontal axis denotes the number of polynomials for the lower curve and the number of grid points for the upper curve.
Outline of the article
In this Introduction we have reviewed some of the history of methods for modeling demographic history by means of approximations of the AFS. Additionally, we have described two numerical methods, the finite-difference method and the spectral method, that can be used to solve different diffusion equations that arise in the computation of AFS. The benefits of the latter approach include exact solutions for a large family of multipopulation models and rapidly converging approximations for a larger class of models. In the remainder of the article we describe the method in detail and assess how well it performs both based on simulated data sets and in comparison to a grid-based approach. Finally, we report the analyses of a four-population model of human history, using a SNP data set that has previously been analyzed under three-population models, using the grid-based approach.
Materials and Methods
Background
The observable object that we aim to reproduce theoretically is the allele-frequency spectrum. In this section, we review the definition of the AFS in a multipopulation context and how one can approximate it with numerical solutions of diffusion-based models that use truncated expansions by orthogonal polynomials (Lukic et al. 2011).
The joint AFS is defined as a K-dimensional matrix built from the allele counts observed in a sample of individuals from K different populations. Each value in the matrix is an expected number (in the case of an AFS calculated under a theoretical model) or an observed number (in the case of data) of diallelic polymorphisms that fall into a particular frequency class. We denote as an entry of the observed joint AFS that specifies the number of SNPs in which their derived state occurs i1 ε [0, C1] times in the first population, i2 ε [0, C2] times in the second population, etc. Here, Ca is the total number of chromosomes sampled from the ath population (a = 1, … , K). For simplicity in the notation, we assume Ca = C for all a throughout this article. Here, while denotes an entry of the empirical AFS, we denote as the analogous entry of the theoretical AFS.
The AFS can be seen as an object derived from the distribution of population allele frequencies φ(x) on [0, 1]K. In particular, if the derived allele frequencies of a SNP taken at random consist of a vector , where xa is the frequency of the SNP in population a, independently and identically distributed with respect to the distribution φ(x), the AFS consists of a finite sample of population alleles as defined in Equation 1. In our model-based approach φ(x) is interpreted as a present-time density that has been shaped by a historical Wright–Fisher process on a population tree specified by the parameters θ. We denote the resulting model-dependent joint density by φ(x|θ). The parameters depend on the particular model and usually involve effective population sizes, migration rates, splitting times, admixture coefficients, population growth rates, etc. In the diffusion approximation to multipopulation Wright–Fisher processes exchanging migrants, the time evolution of φ(x, t) obeys a partial differential equation (PDE) of the type
| (3) |
Here, denotes the effective population sizes, denotes the fraction of chromosomes that population a receives from b, and the nonhomogeneous term ρ(x, t) describes the total incoming/outgoing flow of SNPs per generation into the K-cube from different boundary components of the K-cube and from de novo mutations. These boundary conditions are treated in more detail later.
Approximate solutions by means of polynomial expansions:
Our approach to approximate the solutions of Equation 3 assumes that the density solution φ can be expanded in a polynomial basis with time-dependent coefficients that can be determined numerically. The expansion consists of a contribution associated with the bulk of the K-cube and other different contributions associated with each boundary component. The expansion can be expressed compactly as
| (4) |
for a truncation parameter Λ. The set of functions is defined as
| (5) |
| (6) |
| (7) |
where are the classical Jacobi polynomials defined on the interval −1 ≤ z ≤ 1 with weight w(z) = (1 − z)(1 + z), and δ(x) is the Dirac delta function (for more details on the basis of polynomials see the Appendix). When the migration coefficients mab in Equation 3 vanish, and Λ = C − 2 (with C the number of haploid genomes sampled in the definition of the AFS), the truncated expansion Equation 4 yields an exact formula for the AFS (see File S1, section 1 for a detailed derivation). However, in general the coefficients obey a numerically integrable ordinary differential equation, and the truncated polynomial expansion in Equation 4 gives rise to approximations of the AFS that have the potential to become more accurate as the truncation parameter Λ increases.
Some of the most important differences between scenarios with and without migration originate in the delicate dynamics of φ at the boundary of the K-cube. In the following paragraphs we review these boundary conditions. In the next subsection we examine how to deal with numerical artifacts (known as Gibbs phenomena) that appear when we consider approximations with finite Λ.
By the boundary dynamics of φ we mean the dynamics of those SNPs that have an allele fixed in some populations but that remain polymorphic in others and of those new SNPs that arise by the influx of mutations. This class of SNPs is described by those terms that are multiplied by Dirac deltas in Equation 4. For illustrative purposes we examine in detail the particular case of two populations. When K = 2, we can decompose Equation 4 as
| (8) |
The terms that are multiplied by Dirac deltas represent the density of allele frequencies of those SNPs that are localized in the different boundary components. In particular, the A term is localized in the bulk of the square, the four B terms are localized in the edges of the square, and finally the two C terms are localized in the two vertices of the square that are not absorbing. The ancestral vertex (x1 = 0, x2 = 0) and the derived vertex (x1 = 1, x2 = 1) are absorbing and hence do not contribute SNPs to the density φ(x1, x2, t). Now, we can recover Equation 4 if we expand each term in Equation 8, using the basis of shifted Jacobi polynomials Tn(x). In particular, we write the polynomial expansion of each term in Equation 8 as
| (9) |
The inflow/outflow of polymorphisms affects the dynamics of each term differently. For instance, in every generation, de novo mutations contribute a mass 2N1uδ(x1−1/2N1) to and a mass 2N2uδ(x2−1/2N2) to , where u is the mutation rate. This means that a total of 2Nau new SNPs at frequency xa = 1/2Na appear each generation in population a due to de novo mutation events. On the other hand, random drift can fix some variants that were polymorphic in populations 1 and 2. This means that a SNP with allele frequencies distributed as φA(x1, x2, t) becomes a SNP with allele frequencies distributed as , , , or , depending on which frequency class becomes fixed (x2 = 0, x2 = 1, x1 = 0, or x1 = 1). Finally, variants segregating on any of the edges of the square can also become fixed because of random drift. Therefore, the density of SNPs at the edge (x1 = 1, x2 = 0), , receives SNPs that reach the fixed frequency x1 = 1 from and SNPs that reach x2 = 0 from . Similarly, receives SNPs that reach x1 = 0 from and SNPs that reach x2 = 1 from . When the migration coefficients are zero, this dynamic can be integrated exactly (see File S1, section 1).
The boundary dynamics become more complicated when the migration rates are nonzero. This is due to the fact that when a SNP reaches fixation in one population and remains polymorphic in the other, it can become polymorphic again in the first population because of potential migration events. This contrasts with the zero-migration scenario, in which the number of populations where a SNP is polymorphic decreases as a function of time. The contributions due to migration events in the different components of Equation 8 follow a complicated formula that was previously analyzed in the literature. We refer to Lukic et al. (2011) for more detailed information on these terms and how to integrate the full dynamics using a numerical method such as a Runge–Kutta method. Here, for illustrative purposes, we write only the contribution to φA due to migration events,
where Δmφ/Δt denotes the change in the density of SNPs per unit of time due to migration events.
Gibbs phenomena:
In general, it is not possible to work with infinite sums such as the ones introduced in Equation 9. This is why proper truncated expansions such as Equation 4 are used instead. Although truncated polynomial expansions can sometimes yield exact results, in general scenarios with migration we are faced with the task of approximating the influx of mutations 2Nuδ(x − 1/2N), the contributions due to migration events [e.g., ], and the time evolution of the density φ by means of truncated polynomial expansions. Technically, using a truncated polynomial expansion to approximate a generalized function such as a Dirac delta is a far from perfect approximation. In particular, the approximations tend to exhibit oscillatory behaviors and a slow convergence rate. The circle of phenomena associated with imperfect polynomial approximations of nonsmooth functions is known as Gibbs phenomena.
A particular way to deal with this limitation consists of using smooth exponential functions with proper normalization constants, instead of plain Dirac deltas located near the boundary. For instance, the influx of mutations can be approximated by the term ck exp(−k(Λ)x) (see section 3 in File S1 for a derivation of ck), and terms due to migration events such as can be approximated by . Here, k(Λ) is a function of the truncation parameter, and it is defined as the largest real number k that satisfies a bound for the truncation error between exp(−k(Λ)x) and its truncated polynomial approximation.
This treatment of the boundary conditions is superficially different from the conditions used by Gutenkunst et al. (2009). In the case of one population, one can prove that our solution and that of Gutenkunst et al. (2009) converge to the same exact solution (see section 3 in File S1 for a mathematical proof of this statement). The case of two or more populations with migration is significantly more difficult and we could not demonstrate that both treatments of the boundary conditions yield the same exact solution in the limits of infinite Λ and an infinitely fine grid. Our choice of exponential functions to approximate the Dirac deltas associated with migration events at the boundary is inspired by the approximation of δ(x − 1/2N) in the one-population case. Although we demonstrate that this approximation converges to the exact solution in the one-population case, we do not have an equivalent demonstration for the case of the migration events at boundary. Therefore, our use of exponential functions in the case of migration events is justified by a heuristic argument and is not based on a mathematical proof that the associated approximations converge to the exact solution (for evidence using simulated data that both approaches converge to the same solution see Comparison of different diffusion theory-based approaches in Results).
Maximum-likelihood inference:
We used a maximum-likelihood approach to infer the model parameters and a nonparametric bootstrap resampling approach to estimate confidence intervals and confidence regions. In particular, given the theoretical AFS as defined in Equation 1, we used the likelihood function L(θ|x) of a random Poisson field as defined in Sawyer and Hartl (1992),
| (10) |
with the number of counts observed in the specified bin of the empirical AFS. We also used the associated log-likelihood function
| (11) |
Simulations
To compare the different approaches to solving the diffusion equations, we used simulated data. To simulate the AFS we used an algorithm that mimics the forward diffusion process on population trees. Although there are very efficient coalescent-based tools to simulate data, we preferred to use the forward simulation approach because it exactly models the solution to the forward diffusion Equation 3 and because it can be adapted to incorporate the effects of natural selection more easily than coalescent-based models. As a quality check, we compared our Monte Carlo simulations with coalescent-based simulations [we used the program ms (Hudson 2002)]. As expected, both approaches produced nearly identical frequency spectra.
The basic approach is a standard one (Glasserman 2003) and consists of three steps:
-
1.
Specify the sample sizes in the AFS and a demographic model. The model includes a population tree topology and relevant demographic parameters such as effective sizes, migration rates, and splitting times.
-
2.
Generate Π stochastic sampling paths of SNP frequencies. These will yield population allele frequencies associated with Π independent SNPs.
-
3.
Map each population allele frequency to the AFS matrix by using binomial sampling formulas for the target sample sizes and sum over all the Π contributions. The variance of each entry in the simulated AFS is inversely proportional to Π, as is standard in Monte Carlo simulation.
The simulation of a single sampling path begins by generating a random initial condition (i.e., initial time of the sampling path and initial frequency), followed by sampling a sequence of allele frequencies using the Euler approximation
| (12) |
In this approximation we replace Δt with a short time interval and we randomly sample K values from the standard normal distribution N(0, 1) in each time step (Matsumoto and Nishimura 1998).
The space of initial conditions depends on the particular model, and it consists of the following:
The frequencies in the ancestral population at drift–mutation equilibrium at time zero: These frequencies are distributed as 4NAμ/x.
De novo mutations on the different branches of the population tree: In this case, the frequencies are fixed (xi = 1/2Ni with i the population where the mutation event arises, and xj≠1 = 0 for the rest) and the time coordinate is a random variable uniformly distributed on the time interval.
As an example, let us consider a two-population model in more detail (see Figure 2). An ancestral population with size NA increases its size to NB at time t = 0; the population size remains constant and equal to NB up to time t = t1; at time t = t1 the population splits into two populations with sizes N1 and N2; the present time is reached at time t = t2. Time is measured in units of generations. In this model one can define the space of initial conditions as the union of sets
| (13) |
The probability density on this space of initial conditions is
Here, x is the random variable in the first set, and t is the random variable in the remaining three sets. The total number of initial states is
Therefore, a random initial state is a random variable on the space of initial conditions associated with the probability density function
Finally, the simulated AFS nij associated with a sample of C chromosomes per population is
| (14) |
with 0 ≤ i ≤ C, 0 ≤ j ≤ C, 0 < i + j < 2C, and the simulated population allele frequencies.
Figure 2 .

Simulation of two Brownian paths on a two-population tree. The plot illustrates different types of initial conditions for the paths. The initial condition can be either a random allele frequency in the ancestral population at mutation–drift equilibrium (solid lines) or a de novo mutation that arises in one of the populations after the ancestral population leaves the state of equilibrium (shaded lines).
Demographic scenarios:
We simulated data under seven different demographic histories for two and three populations to compare different approaches to forward diffusions. We generated 50,000 population allele frequencies using Monte Carlo simulations in each of the seven demographic scenarios. We sampled 20 chromosomes in the scenarios that involved three populations and 50 chromosomes in the scenarios that involved two populations. For each demographic scenario, we computed the AFS with our polynomial-based approach (MultiPop) and with the finite-difference method (∂a∂i) and compared each AFS with the AFS computed with Monte Carlo simulations. The different models and parameters used in the simulations are described below.
Two populations:
In all the two-population scenarios the ancestral population size NA was 4000. The time from the population splitting event up to the present was T = 150 generations. The population sizes after the splitting event were N1 = 4000 and N2 = 1815.1. Finally, the migration matrices were
for the 2Nm = 0 scenario (model 1),
for the 2Nm < 0.5 scenario (model 2),
for the 2Nm ∼ 1 scenario (model 3), and
for the 2Nm > 1 scenario (model 4).
Three populations:
In all the three-population scenarios the ancestral population size NA was 5000. The time from the first population splitting event up to the second population splitting event was T = 220 generations. The time from the second population splitting event up to the present was T = 130 generations. The population sizes after the first splitting event were N1 = 1815.1 and N2 = 815.1. The second population splitting event occurred in population 2. The population sizes after the second splitting event were N1 = 1815.1, N2 = 315.1, and N3 = 815.1. Finally, the migration matrices were
and
for the 2Nm = 0 scenario (model 5),
and
for the 2Nm ∼ 1 scenario (model 6), and
and
for the 2Nm > 1 scenario (model 7).
Data
We used the Environmental Genome Project (EGP) SNP database (Environmental Genome Project 2010) to determine the observed joint AFS in four human populations. The sampled populations consist of 12 individuals of West African ancestry (YRI), 22 individuals of northern European ancestry (CEU), 24 individuals of East Asian ancestry (CHB), and 22 individuals of Mexican ancestry (MEX). These data are the result of direct Sanger resequencing (with a low error rate) of environmental response genes and have been the subject of several previous studies (Akey et al. 2004; Williamson et al. 2005; Gutenkunst et al. 2009). We used this data set to compare our method with that of Gutenkunst et al. (2009). The number of environmentally responsive genes sequenced as part of the EGP has been steadily increasing since the project started in 2001 (Environmental Genome Project 2010), and the EGP database is now larger than when Gutenkunst et al. (2009) performed their study. However, the number of individuals in the data set has not changed. As we were not able to reconstruct the original set of SNPs used in Gutenkunst et al. (2009), we instead used all the available loci. The difference between the data sets turned out to be small; while Gutenkunst et al. (2009) used 27,824 SNPs of noncoding DNA, we used 28,875 SNPs.
We considered all 28,875 diallelic SNPs located in 4.07 Mb of noncoding DNA resequenced from 197 different autosomal genic regions. Each SNP was ordered into an ancestral and a derived state, using the pairwise alignment from chimpanzee to human available in the UCSC panTro2 draft of the chimpanzee genome (Chimpanzee-Sequencing-Consortium 2005). We computed the context-dependent probability of misidentification of the ancestral state and introduced the associated corrections in the allele-frequency spectrum (see Hernandez et al. 2007 and File S1, section 2). As in Gutenkunst et al. (2009), we assumed a divergence time of 6 million years between human and chimpanzee and a generation time of 25 years. We estimated a mutation rate of μ = 2.35 × 10−8 per site per generation from sequence divergence present in the data. This is identical to the estimate by Gutenkunst et al. (2009) and comparable to other estimates (e.g., Nachman and Crowell 2000).
As the number of chromosomes in the data varies depending on the particular SNP, we projected the AFS down to a fixed number of chromosomes. If the number of chromosomes sampled in each population is the same and equal to C, the total number of bins in the associated AFS will be (C + 1)4 − 2. Also, as the theoretical value of each bin in the AFS is computed as the four-dimensional integral defined by Equation 1, the computational time needed to evaluate a single bin of the theoretical AFS is larger than in the cases of two or three populations. To reduce the computational burden of evaluating Equation 1 many times, we used an adaptive allele-frequency spectrum in which we decomposed the AFS into bins of different sizes, depending on the fraction of SNPs that occupy a particular region of the frequency space. We fixed the bin sizes by sampling 19, 9, or 4 chromosomes per population. We chose 19 as the maximum number of chromosomes because 19 + 1 can be divided twice by two, which allows us to easily build an adaptive histogram using three different bin sizes. To this end we first projected the empirical AFS down to 4 chromosomes per population. In our adaptive construction the coarsest AFS possible has (4 + 1)4 bins, which we further refine by considering bins of size 1/9 and 1/19 within each bin of size 1/4. We computed the fraction of SNPs present in each bin of size 1/4, and if this fraction exceeded a certain bound b, we further refined the adaptive AFS to consider bins of size 1/9. Recursively, we isolated those bins of size 1/9 that contained a fraction of SNPs >b and further refined them into smaller bins of size 1/19. Using all the SNP data and a bound parameter of b = 5 × 10−4, the total number of bins of the adaptive AFS becomes 5078 (452 bins of size 1/4, 2644 bins of size 1/9, and 1982 bins of size 1/19). This is a significant reduction in the total number of bins for a four-population AFS relative to a full representation for 19 chromosomes, which has 204 − 2 = 159,998 bins.
Implementation
In our software implementation we use two different bases of the vector space spanned by polynomials on 0 < x < 1 with degree ≤Λ. We use the shifted Gegenbauer polynomials and the shifted Chebyshev polynomials (see the Appendix for more details). We inject mutations at the boundary, using the term ckexp(−kx). Similarly, the associated drift–mutation equilibrium density that we use as the initial condition in our demographic models is (see section 3 of File S1 for more details)
We evaluated the integrals that appear in Equation 1, such as , by means of the Runge–Kutta four method. In particular, I(y) obeys the ordinary differential equation (ODE)
and I(1) is obtained as the integral of the ODE between 0 and 1.
Population splitting events were modeled by assuming that the distributions of allele frequencies in the two daughter populations are identical; i.e., φ(x, xK+1) = δ(xi − xK+1)φ(x), with i and K + 1 the populations that arise after the divergence of i. The Dirac delta was approximated by a Gaussian function peaked at the diagonal xi = xK+1 with a user-defined standard deviation that we call a thickening parameter in the software implementation. Such a smooth Gaussian approximation allows us to use a truncated polynomial expansion to accurately approximate the density after the splitting event. The larger the truncation parameter used, the smaller the standard deviation that can be used and the closer the approximation will resemble φ(x, xK+1) = δ(xi − xK+1)φ(x).
The computer implementation was written in the C++ language, and the source code is freely available in Google Code (http://www.code.google.com/p/multipop/). We compared the results found using the MultiPop program to those found using a different class of numerical techniques that estimate the time evolution of φ, using grid approximations and a finite-difference method to integrate the PDE. The latter method was implemented in the computer program ∂a∂i (Gutenkunst et al. 2009).
Nonlinear optimization:
When inferring the demographic parameters of a model given the simulated data or the human data set, we have to maximize the likelihood-function Equation 11. Maximizing Equation 11 on a high-dimensional model parameter space is a challenging nonlinear optimization problem. To this end, we used three classical algorithms in nonlinear optimization: simulated annealing, the downhill simplex method, and the Broyden-Fletcher-Goldfarb-Shanno (BFGS) quasi-Newton method. When inferring the parameters from the simulated data, we used only local optimization techniques (downhill simplex). We used the true value as the initial point in the local optimization algorithm. We checked that a local maximum had been reached by numerically evaluating the Hessian of the minus log-likelihood at the critical point and confirmed that its eigenvalues were positive. This technique allowed us to determine the local maximum of the likelihood surface closest to the true value.
In the case of the human data, we performed an initial exploration of the parameter space by means of simulated annealing to find the global maximum for a fixed empirical AFS with all the SNPs. Subsequent maxima of the likelihood function associated with bootstrapped samples were computed by means of local optimization algorithms (e.g., downhill simplex or quasi-Newton). When using a local optimization algorithm, we used as initial seed the global maximum initially determined by means of simulated annealing.
Statistical inference and bootstrap strategy
The theoretical AFS can be considered as both the density of the allele frequency for a single diallelic locus and the expected distribution for a very large set of independent diallelic loci. Therefore, the calculation of the likelihood using Equation 10 is accurate only if each SNP has been sampled independently from other SNPs. This is indeed the approach that we follow with the simulated data, where each SNP is independent from the others.
One approach to dealing with nonindependent (i.e., linked) SNPs is to use Equation 10 with all linked SNPs and treat the result as a composite likelihood. In the case of the human data set, the EGP database consists of just 170 independent loci with 80% of the SNPs occurring in 30% of the loci. As the SNPs are tightly linked within each locus, unusual histories of a DNA segment with many tightly linked SNPs can overrepresent the inferred demographic history when the number of independent loci is small. In other words, although composite-likelihood estimators are known to be consistent (Wiuf 2006), they can be biased when the sample size is small.
Because the human EGP data set consists of a relatively small number of SNPs, many of which lie close to one another, we have tried to minimize the effects of linkage in our bootstrap approach to estimate parameters and confidence intervals. For each sample, 170 SNPs were selected at random, conditioned on their being separated from each other by at least 200 kb. Multiple sets of 170 SNPs were sampled with replacement from the data set, which yielded different maximum-likelihood points in the parameter space. Here, B is the number of bootstrap samples. Ninety-five percent confidence intervals and regions were calculated using the percentile approximation (DiCiccio and Efron 1996). We used the marginal distribution of maximum-likelihood values for each parameter to estimate the confidence intervals by considering the 2.5th percentile and the 97.5th percentile.
Results
Comparison of different diffusion theory-based approaches
To study the biases introduced by the different numerical approximations, we computed the AFS in seven different demographic scenarios with varying numbers of populations and intensities of gene flow (models 1–7). We used spectral methods, finite-difference methods, and Monte Carlo simulations of the forward diffusion process. The different frequency spectra were compared by means of the chi-square statistic (Table 1 and Figures 3 and 4). We found that using 35 polynomials per population gives rise to very good approximations of the true AFS in the seven scenarios. In the limit of very small gene flow, the chi-square statistic associated with the spectral method approach converged much faster to zero as the truncation parameter Λ was increased. This behavior, which was observed in two- and three-population models, is the expected one as the polynomial-based approach yields exact solutions of the AFS in the absence of migration. As the intensity of migration increases, the rate of convergence to the exact AFS in the spectral method approach worsens. This behavior is also the expected one (Lukic et al. 2011), as the polynomial expansion does not yield exact solutions of the diffusion PDEs with nonzero gene flow. Also, it becomes more difficult to implement exactly the boundary conditions because of the emission of polymorphisms to other populations. However, as the truncation parameter Λ increases, the quality of the approximations of the AFS increases in all scenarios.
Table 1 . Comparison of numerical approximations of the AFS and the simulated AFS.
| Model no. | Intensity of migration (2Nm) | MPop vs. Monte Carlo (chi-square statistic) | ∂a∂i vs. Monte Carlo (chi-square statistic) |
|---|---|---|---|
| 1 | 0 | 0.001265 | 0.002479 |
| 2 | <0.5 | 0.00894 | 0.005955 |
| 3 | 1 | 0.01076 | 0.006457 |
| 4 | >1 | 0.01140 | 0.006286 |
| 5 | < 0.5 | 0.007558 | 0.04684 |
| 6 | 1 | 0.01511 | 0.02882 |
| 7 | >1 | 0.02979 | 0.01791 |
Chi-square statistics associated with seven demographic scenarios are shown. The AFS computed by MultiPop (MPop) corresponds to the Λ = 35 AFS, and the AFS computed by ∂a∂i corresponds to a grid size of 40 grid points per population. The frequency spectra were normalized in all the cases such that the total number of SNPs was 1.
Figure 3 .

Decay of the chi-square statistic in MultiPop (top) and ∂a∂i (bottom). Four different demographic scenarios with two simultaneous populations and 50 chromosomes sampled per population are considered. For simplicity, the average scaled migration rate is used to label each scenario. The observed AFS were constructed using Π = 50,000 independent loci produced with Monte Carlo simulations.
Figure 4 .

Decay of the chi-square statistic in MultiPop (top) and ∂a∂i (bottom). Three different demographic scenarios with three simultaneous populations and 20 chromosomes sampled per population are considered. For simplicity, the average scaled migration rate is used to label each scenario. The observed AFS were constructed using Π = 50,000 independent loci produced with Monte Carlo simulations.
Similarly, the finite-difference approach gave rise to approximations of the AFS of a comparable quality to those of the approach that uses polynomial series expansions. In particular, it produced better approximations when the intensity of gene flow was strong. However, the rates of convergence in the two-population models were very different from the ones in the three-population models. In the limit of zero gene flow the rate of convergence in the two-population model was significantly faster than that in the three-population model. There is not a simple way to explain these results, because among other things we do not know how the numerical discretization used in the finite-difference scheme relates to the exact AFS in any subset of the parameter space for a finite grid size.
Although using the chi-square statistics (e.g., Figures 3 and 4) is helpful for quantifying the numerical error in the AFS, this measure of error is not informative enough to estimate the optimal numerical error given a certain level of statistical uncertainty when inferring demographic parameters. This is because the “numerical error” (here, error measures the deviation of the approximated AFS from the exact AFS) can be very different from the “propagated numerical error” (here, error measures the deviation of the numerically approximated likelihood peak from the exact likelihood peak), even if both decay as the truncation parameter increases. For instance, the appearance of nearly flat directions of the likelihood function on the parameter space might amplify the numerical errors that arise in the numerical solution of the PDE, giving rise to large propagated numerical errors. To estimate correctly the optimal error given a certain level of statistical uncertainty, one should compare the location of the maximum-likelihood peaks in the numerical approximation with the true location of the peaks. We perform this analysis in the following subsection.
The inverse problem: maximum-likelihood estimates:
To study these propagated numerical errors we inferred the maxima of the likelihood functions. In this case, the effective population sizes, splitting times, and migration rates were free parameters, and the observed frequency spectra were constructed using Monte Carlo simulations. We computed the maximum-likelihood peaks associated with the polynomial-based and grid-based approximations of the maximum-likelihood function in each demographic model (see Table 2). In our polynomial approximation we used 40 polynomials per population in the two-population scenarios and 35 polynomials per population in the three-population scenarios. For the finite-difference method we used 100 grid points per population in the two-population and three-population scenarios.
Table 2 . Comparison of maximum-likelihood estimates using different numerical approximations.
| Model | Model parameter | True value | θ MPop | θ ∂a∂i |
|---|---|---|---|---|
| 1 | 4NAu | 1 | 1 | 1 |
| 1 | N1/NA | 1 | 1.015 | 1.042 |
| 1 | N2/NA | 0.4538 | 0.4385 | 0.4421 |
| 1 | T/2NA | 0.01875 | 0.01788 | 0.01840 |
| 2 | 4NAu | 1 | 1 | 1 |
| 2 | N1/NA | 1 | 1.08 | 1.042 |
| 2 | N2/NA | 0.4538 | 0.6467 | 0.6379 |
| 2 | T/2NA | 0.01875 | 0.02304 | 0.02720 |
| 2 | 2NAm1→2 | 0.3 | 0.5706 | 3.529 |
| 2 | 2NAm2→1 | 0.88 | 0.8735 | 3.228 |
| 3 | 4NAu | 1 | 1 | 1 |
| 3 | N1/NA | 1 | 1.099 | 1.044 |
| 3 | N2/NA | 0.4538 | 0.6648 | 0.6497 |
| 3 | T/2NA | 0.01875 | 0.02287 | 0.02783 |
| 3 | 2NAm1→2 | 0.8 | 0.5873 | 4.022 |
| 3 | 2NAm2→1 | 1.76 | 0.9469 | 3.965 |
| 4 | 4NAu | 1 | 1 | 1 |
| 4 | N1/NA | 1 | 1.086 | 1.068 |
| 4 | N2/NA | 0.4538 | 0.6712 | 0.6493 |
| 4 | T/2NA | 0.01875 | 0.02258 | 0.02592 |
| 4 | 2NAm1→2 | 1.12 | 0.6190 | 2.574 |
| 4 | 2NAm2→1 | 2.64 | 0.9910 | 3.798 |
| 5 | 4NAu | 1 | 1 | 1 |
| 5 | N1/NA | 0.3630 | 0.3713 | 0.3522 |
| 5 | N2/NA | 0.1630 | 0.1540 | 0.1440 |
| 5 | N3/NA | 0.06302 | 0.05673 | 0.05569 |
| 5 | Ta/2NA | 0.022 | 0.02158 | 0.02041 |
| 5 | Tb/2NA | 0.013 | 0.01225 | 0.01152 |
| 6 | 4NAu | 1 | 1 | 1 |
| 6 | N1/NA | 0.3630 | 0.3713 | 0.3344 |
| 6 | N2/NA | 0.1630 | 0.17855 | 0.1527 |
| 6 | N3/NA | 0.06302 | 0.05950 | 0.06112 |
| 6 | Ta/2NA | 0.022 | 0.02996 | 0.01749 |
| 6 | Tb/2NA | 0.013 | 0.01211 | 0.01249 |
| 6 | 2NAma1→2 | 0.05 | 0.04562 | 0.0001828 |
| 6 | 2NAma2→1 | 5 | 0.02345 | 0.0009895 |
| 6 | 2NAmb1→2 | 2 | 0.002115 | 0.003311 |
| 6 | 2NAmb1→3 | 0.3 | 0.3897 | 1.505 |
| 6 | 2NAmb2→1 | 0.005 | 0.007982 | 0.09358 |
| 6 | 2NAmb2→3 | 0.3 | 0.0007637 | 4.4e-09 |
| 6 | 2NAmb3→1 | 3 | 0.6831 | 3.398 |
| 6 | 2NAmb3→2 | 3 | 1.788 | 2.640 |
| 7 | 4NAu | 1 | 1 | 1 |
| 7 | N1/NA | 0.3630 | 0.3874 | 0.3138 |
| 7 | N2/NA | 0.1630 | 0.1724 | 0.1445 |
| 7 | N3/NA | 0.06302 | 0.05843 | 0.05589 |
| 7 | Ta/2NA | 0.022 | 0.02415 | 0.01296 |
| 7 | Tb/2NA | 0.013 | 0.01155 | 0.01115 |
| 7 | 2NAma1→2 | 0.05 | 0.02982 | 0.7007 |
| 7 | 2NAma2→1 | 12 | 0.007439 | 0.004225 |
| 7 | 2NAmb1→2 | 6 | 0.0007166 | 0.0003699 |
| 7 | 2NAmb1→3 | 0.3 | 0.00090192 | 3.1354 |
| 7 | 2NAmb2→1 | 0.005 | 0.0003413 | 0.0020024 |
| 7 | 2NAmb2→3 | 0.3 | 0.0007713 | 0.0001095 |
| 7 | 2NAmb3→1 | 3 | 0.007362 | 3.232 |
| 7 | 2NAmb3→2 | 3 | 0.4090 | 0.01191 |
Maximum-likelihood estimates of seven different demographic scenarios are shown. Many results are biased due to numerical errors in the calculation of the frequency spectra (see subsection Numerical errors and bias-corrected confidence intervals for a detailed discussion on how numerical errors affect the location of the maximum-likelihood peak). In the case of MultiPop, 40 polynomials were used in the two-population models and 35 polynomials in the three-population models. We used 100 grid points in all models approximated with finite-difference schemes. The observed AFS were constructed using Π = 50,000 independent loci produced with Monte Carlo simulations. The maximum-likelihood peak was found by means of the BFGS method, using the coordinates of the true peak as the initial point in this local optimization algorithm.
We found that in models with few parameters both approaches yield maximum-likelihood peaks that are close to the true values (see Table 2). As a general trend, the finite-difference method tends to overestimate the amount of gene flow while the spectral method tends to overestimate the largest effective population size. We also observe that as the number of model parameters increases, many inferred migration rates deviate significantly from the true values (for instance, see models 6 and 7 in Table 2). As we simulated large sets of SNP allele frequencies for each scenario, the statistical noise was very small and the main source of bias that we observe can be attributed to numerical errors.
These biases are caused by two main factors: the propagation of numerical errors in the evaluation of the likelihood function and the geometry of the likelihood function associated with a particular model. In particular, models in which a set of parameters yields flat directions around the likelihood peak will be particularly prone to propagate small numerical errors toward large errors in the inferred parameters. One can interpret the biases obtained in the migration rates of models 6 and 7 in Table 2 along these lines. Here, the large number of migration parameters introduced in the models gave rise to many flat directions that amplified numerical errors associated with the evaluation of the likelihood function.
Computational performance:
Our current implementation of the spectral method to study demography with diffusion approximations is optimized to use little memory to tackle more than three populations. This economical use of memory is attained by increasing the number of operations in the algorithm and hence reducing its speed. ∂a∂i is optimized to work with two and three populations and is significantly faster than our current implementation (see Table 3).
Table 3 . Comparison of computing time.
| Model | No. polynomials (MPop) | Computing time of MPop (sec) | Grid size (∂a∂i) | Computing time of ∂a∂i (sec) |
|---|---|---|---|---|
| 2 | 15 | 0.27 | 15 | 0.07 |
| 2 | 20 | 0.61 | 50 | 0.10 |
| 2 | 25 | 1.10 | 100 | 0.13 |
| 2 | 30 | 1.86 | 500 | 1.43 |
| 2 | 35 | 2.92 | 1000 | 5.68 |
| 6 | 15 | 3.1 | 15 | 0.16 |
| 6 | 20 | 7.95 | 50 | 0.86 |
| 6 | 25 | 17.31 | 100 | 6.33 |
| 6 | 30 | 33.3 | 200 | 68.41 |
| 6 | 35 | 57.41 | 300 | 253.85 |
Computing times required to evaluate an allele-frequency spectrum using MultiPop and ∂a∂i are shown. The demographic scenarios involved two populations and 50 chromosomes sampled per population or three populations and 20 chromosomes sampled per population. The CPU used to measure the computing times was an Intel Core(TM)2 Duo P8600 with speed 2.40 GHz.
One way to increase the speed of the method is to reconsider the memory model used for cases with two and three populations. In particular, the diffusion operator is a matrix of size [(Λ + 2)K − 2]2, whose storage requires a very large amount of memory. Our present implementation needs only four matrices of size Λ2 for any K, which are used to recover the full diffusion operator at running time by exploiting its tensorial structure. This implementation is very economical from the point of view of memory use. However, it makes the algorithm significantly slower. An implementation that uses sparse matrices to approximate the full diffusion operator will significantly reduce the number of operations and increase the speed of the algorithm when K < 4.
Worldwide human expansion out of Africa and peopling of the Americas
We considered a four-population model with 18 free parameters to model the human expansion out of Africa and peopling of the Americas (see Figure 5). The model is inspired by several studies reported in the literature (e.g., see Gutenkunst et al. 2009; Gravel et al. 2011). The root of the population tree consists of an ancestral human population in Africa at mutation–drift equilibrium. Such a population experiences a sudden increase of its effective population size at some time before the out-of-Africa event. The divergence of non-African populations after the out-of-Africa event is further modeled by population splits with gene flow. These population splits describe the European–Asian split and the bottleneck associated with the peopling of the Americas. An exponential growth model is then used to describe the population growths of Europeans, Asians, and Native Americans after they become independent populations, and recent population admixture is introduced to model high European gene flow into the ancestral Amerindian population associated with the Mexican population. To reduce the number of parameters, we considered symmetric migration rates except during the first stage of the out-of-Africa event (mAF→B ≠ mB→AF). We did not assume that this migration rate was symmetrical because mAF→B might be significantly larger than mB→AF as one indeed infers from the data (see Table 4). We used a basis of polynomials up to degree 20 (Λ = 20) to approximate the density of population frequencies. Taking into account boundary contributions, the dimension of the space of densities was 224 − 2 = 234,254.
Figure 5 .

A graphical representation of a four-population model for the human expansion out of Africa and peopling of the Americas. The nonconstancy of the population sizes of CEU, CHB, and MEX is modeled by means of an exponential growth model with growth rates rEU, rAS, and rMX.
Table 4 . Inference of a four-population model for the human expansion out of Africa and peopling of the Americas.
| Model parameters | θ MPop | 95% C.I. | θ ∂a∂i, 1 | 95% C.I. | θ ∂a∂i, 2 | 95% C.I. |
|---|---|---|---|---|---|---|
| NA | 10,400 | 8,670–12,200 | 7,300 | 4,400–10,100 | ||
| NAF | 17,300 | 10,900–29,300 | 12,300 | 11,500–13,900 | ||
| NB | 2,060 | 346–5,070 | 2,100 | 1,400–2,900 | ||
| NEU0 | 1,710 | 1,030–3,020 | 1,000 | 500–1,900 | 1,500 | 700–2,100 |
| rEU (generation−1) | 0.0055 | 0.00351–0.0103 | 0.004 | 0.0015–0.0066 | 0.0023 | 0.0008–0.0045 |
| NAS0 | 453 | 210–800 | 510 | 310–910 | 590 | 320–800 |
| rAS (generation−1) | 0.016 | 0.0102–0.0301 | 0.0055 | 0.0023–0.0088 | 0.0037 | 0.0016–0.006 |
| mAF→B (×10−5) | 6.06 | 0.0–13.6 | 25 | 15–34 | ||
| mAF→B (×10−5) | 1.63 | 0.0–3.47 | 3.0 | 2.0–6.0 | ||
| mAF→B (×10−5) | 0.487 | 0.0–1.07 | 25 | 15–34 | ||
| mAF→B (×10−5) | 1.54 | 0.0–2.96 | 9.6 | 2.3–17.4 | ||
| TAF (yr) | 125,400 | 54,300–250,000 | 220,000 | 100,000–510,000 | ||
| TB (yr) | 52,400 | 36,000–80,800 | 140,000 | 40,000–270,000 | ||
| TEU→AS (yr) | 29,500 | 23,500–38,000 | 21,200 | 17,200–26,500 | 26,400 | 18,100–43,100 |
| NMX0 | 3,200 | 1,100–6,100 | 800 | 160–1,800 | ||
| rMX (generation−1) | 0.0071 | 0.0043–0.011 | 0.005 | 0.0014–0.0117 | ||
| TMX (yr) | 29,300 | 23,000–37,500 | 21,600 | 16,300–26,900 | ||
| fMX (%) | 20.4 | 3.2–41 | 48 | 42–60 |
Inference of parameters by means of maximum likelihood is shown. Confidence intervals were computed by means of nonparametric bootstrap. The estimated parameters θ with MultiPop correspond to the mean of the bootstrap distribution. The estimates by MultiPop are compared with the estimates by ∂a∂i with two three-population models. ∂a∂i 1 denotes the three-population model for the out-of-Africa event described in Gutenkunst et al. (2009). ∂a∂i 2 denotes the three-population model for the peopling of the Americas studied in Gutenkunst et al. (2009).
The inferred parameters and confidence intervals are shown in Table 4. Our estimate of the time at which the ancestral Amerindian population split from the ancestral East Asian population is earlier than previous estimates for the time of settling of the Americas. This is compatible with the fact that the ancestral population of the people of the Americas should have shared a common ancestor with East Asians some time before the Americas were peopled. The other inferred parameters are consistent with those of many previous studies. For instance, we infer that the human dispersal out-of-Africa event took place ∼52,000 years ago (95% confidence interval: 36–81 KYA) followed by a high migration rate. This agrees with previous studies that infer a separation of Africans and non-Africans ∼60,000 years ago followed by significant genetic exchange up until 20,000−40,000 years ago (see Reich 2001; Keinan et al. 2007; Gravel et al. 2011; Li and Durbin 2011). Our estimates are also in broad agreement with previously reported values using the diffusion approximation in demographic inference. For instance, Gravel et al. (2011) find a time of split between Africans and Eurasians of 51,000 years ago (95% confidence interval: 45–69 KYA) by applying a similar demographic model to the 1000 genomes project data set. In the case of Gutenkunst et al. (2009), the inferred parameters are broadly consistent with our estimates, although our resulting confidence intervals are substantially narrower than the intervals determined by the authors using conventional bootstrap. In particular, Gutenkunst et al. (2009) use a combination of two three-population models very similar to our four-population model and apply it to the EGP data set. In this case the time of the out-of-Africa event was inferred to be 140,000 years ago (95% confidence interval: 40–270 KYA).
The differences in the width of the confidence intervals are due to a combination of different factors. The most important factor is that we use a four-population model instead of two three-population models, and this choice limits the number of polynomials that we can use to solve the diffusion PDEs associated with the model. In particular, we used only 20 polynomials per population. As we discussed in the previous subsection using simulated data (see Figures 3 and 4), for any fixed value of Λ the quality of the approximations of the frequency spectra worsens as the intensity of gene flow increases. Similarly, for any fixed values of the migration rates the quality of the approximations worsens as the number of polynomials used decreases. Therefore, choosing Λ = 20 has given rise to poor approximations of the AFS in regions of the parameter space that involve high intensities of gene flow. The associated distortions of the AFS have yielded artificially low likelihoods in those regions of the parameter space. Hence, this numerical artifact is largely responsible for producing narrower confidence intervals in our study than those in the study by Gutenkunst et al. (2009). For instance, for the time out of Africa we inferred smaller confidence intervals (36–81 KYA) than those in Gutenkunst et al. (2009) (40–270 KYA). While Gutenkunst et al. (2009) infer very large values for the gene flow between populations after the out-of-Africa event, our confidence intervals for the migration rates are significantly smaller and closer to the zero-migration limit. Other differences between this study and the one by Gutenkunst et al. (2009) are in the bootstrap strategy and the particular data set. In our bootstrap strategy we sample one SNP per locus each time, and each pair of SNPs is set to be separated by at least 200 kb. In the analysis by Gutenkunst et al. (2009) the statistical artifacts due to linked SNPs were corrected by considering simulations with linked loci in a parametric bootstrap approach to compute confidence intervals. However, no constraint for the distance between pairs of SNPs was imposed in their nonparametric bootstrap approach. If no constraints are imposed on the minimal distance between any two SNPs in each bootstrap, SNPs from loci with high SNP density will be sampled more often. Therefore, the demographic history of a locus with a high density of SNPs will be overrepresented in the overall inference. Although both bootstrap strategies should converge to the same result in the limit of a large number of loci, in the case of a data set with a small number of loci, such as the EGP data set, the differences might be more important. Indeed, the study by Gravel et al. (2011) inferred significantly narrower confidence intervals by applying ∂a∂i to genome-wide sequence data. Finally, although the EGP data set contains a few more sequenced loci since the study by Gutenkunst et al. (2009) and this fact affects the width of the confidence intervals, this contribution should be small.
One of the inferred parameters that is most different between our study and that of Gutenkunst et al. (2009) is the proportion of European ancestry in Mexicans. We infer an admixture proportion of 20.4% (95% C.I.: 3.2–41%), while the three-population model used by ∂a∂i inferred 48% (95% C.I.: 42–60%) (Gutenkunst et al. 2009). In this case the confidence intervals do not overlap. Again, explanations of this discrepancy range from the presence of more sequence data in the EGP data set since the study by Gutenkunst et al. (2009) was done to small differences in the statistical analyses made. Other studies have pointed out the difficulty of estimating admixture proportions when data of one of the preadmixture ancestral populations are missing (Alexander et al. 2009). Therefore, it is not surprising that slight differences in the data set and in the statistical analyses can yield significant differences in the inferred parameters. In particular, Alexander et al. (2009) found that the European admixture proportion in the individuals of Mexican ancestry genotyped by HapMap III was ∼20% if inferred by the algorithm ADMIXTURE, while it was inferred to be ∼50% by the algorithm STRUCTURE using the same data set.
Discussion
Forward diffusion equations played an important role during the development of classical population genetics, as they were originally introduced by R. Fisher and S. Wright to model the evolutionary process (Kimura 1964). With the arrival of modern DNA sequencing technologies, forward diffusion processes have been applied to the inference of demographic parameters and the effects of natural selection (Williamson et al. 2005; Boyko et al. 2008; Gutenkunst et al. 2009). These studies have been limited to scenarios with one, two, and three simultaneous populations. In this article, we have introduced a different approach to solving the forward diffusion equations by means of truncated polynomial expansions. These methods yield exact solutions of the AFS in the absence of migration; they can be used equally to study demographic models in one, two, and three populations with gene flow, and furthermore they can be applied to study models with four simultaneous populations.
We have applied our method to the study of the human expansion out of Africa and peopling of the Americas by means of a model with four simultaneous populations. Our four-population model can be seen as a combination of two three-population models that were studied before by means of diffusion-theory–based techniques (Gutenkunst et al. 2009). Similarly, we used the Environmental Genome Project SNP database. The demographic parameters that we have inferred in this model are similar to many recent results (see Reich 2001; Keinan et al. 2007; Gravel et al. 2011; Li and Durbin 2011). However, some of our inferred parameters are substantially different from the parameters inferred in another study that used numerical solutions to forward diffusion equations (Gutenkunst et al. 2009).
In this article we have also studied the behavior of different numerical solutions of the diffusion PDEs that approximate the AFS under a specified demographic model. In particular, we have compared the polynomial-based approach introduced in Lukic et al. (2011) with the finite-difference approach implemented in Gutenkunst et al. (2009). Although the methods exhibit comparable behaviors, we found that the polynomial-based approach obtains better results in the regime where it yields exact solutions of the AFS, i.e., in the zero-migration limit (see Table 1 and Figures 3 and 4). Also, we confirmed that the polynomial-based approach allows us to broadly predict the magnitude of the numerical error as a function of the model parameters for a given truncation parameter Λ. The finite-difference approach exhibited a better behavior in the models with strong intensity of migration that we have considered in this work. However, in the case of the method introduced in Gutenkunst et al. (2009) there is not a general theory that allows us to predict how the numerical error behaves as a function of the parameter space.
Numerical errors and bias-corrected confidence intervals
A common limitation that both approaches sometimes exhibit is that small numerical errors in the computation of the AFS can propagate to large biases in the parameter space when we search for the maxima of the numerical approximation of the likelihood function. Hence, even if biases due to numerical errors can be minimized, in some cases small but significant numerical sources of error that affect the statistical accuracy of the inferred demographic parameters will remain. For example, Table 2 exhibits several cases where the bias is very large. These biases are not expected to diminish as the sample size grows, or as the number of SNPs increases, because they are due to numerical artifacts. One could minimize these biases by choosing a larger truncation Λ. However, numerical floating-point errors in the evaluation of polynomials become important sources of numerical error when the degree of the polynomial is large enough. In our implementation, we found that for values of Λ > 40 these sources of numerical error became larger than the truncation error due to a finite choice of Λ.
To minimize the impact of such biases one can either consider models with fewer parameters or introduce statistical corrections to the propagated numerical errors. Here, we apply standard bootstrap methods to correct for bias in the estimators and confidence intervals (see Efron and Tibshirani 1994; DiCiccio and Efron 1996) of some of the models studied above. If is the maximum-likelihood estimator of a demographic model, where Λ denotes the truncation parameter of the numerical approximation, the bias is defined as
Here, is the expected biased estimator and is the expected unbiased estimator. Although we do not know the unbiased estimator, we can estimate the bias by means of the parametric bootstrap. In particular, we simulate a large number of SNP allele frequencies under the estimated parameters and consider the associated AFS for each set of simulated SNPs. We denote by the maximum-likelihood estimate associated with the bth simulated AFS (where 1 ≤ b ≤ B and B is the number of bootstraps). Therefore, the bootstrap estimate of bias is
where is the original maximum-likelihood estimate. This approximation will be valid as long as the bias is small and the number of bootstraps B is large enough. Given , we can now compute the bias-corrected 95% confidence intervals using nonparametric bootstraps (see DiCiccio and Efron 1996) as , where is the 100 ⋅ αth percentile of the nonparametric bootstrap distribution.
As an example, in model 3 we used the parameters inferred by maximum likelihood that are shown in Table 2 to simulate 10,000 SNPs per bootstrap by means of Monte Carlo methods. After estimating the bias with the parametric bootstrap, the 95% bias-corrected confidence intervals for the parameters of model 3 are 0.988−1.04 for 4NAu, 0.722–1.26 for N1/NA, 0.255–0.488 for N2/NA, 0.0136–0.0197 for T/2NA, 0.479–0.83 for 2NAm1→2, and 1.69–2.24 for 2NAm2→1.
Overcoming current limitations
Several limitations exist in the use of joint allele-frequency spectra with many populations. The most important one is that given the joint density of population frequencies , the time needed to compute an AFS grows exponentially with the number of populations. Therefore, the only way to extract demographic information from such a high number of populations requires reducing the number of cells in the AFS to be computed. Because of this, future applications of our method for K > 3 will require the use of either adaptive frequency spectra or projections of high-dimensional AFS into triplets or couplets of populations. For instance, by integrating out populations we can compute the joint density of frequencies associated with every triplet of populations from the higher joint density as
The associated three-population AFS is derived from Equation 1 and the density .
A second question that remains to be explored concerns the bias of the estimator. For a given observed AFS, our method yields a sequence of maximum-likelihood peaks labeled by the number of polynomials Λ used. The convergence of the numerical approximation to the exact AFS in the limit of an infinite number of polynomials guarantees that is an unbiased estimator. It is important to understand the asymptotic behavior of the sequence of peaks to estimate given a few finite values of the sequence . In this study we computed different estimators for several values of Λ, to confirm that our estimators were converging toward the unbiased true estimator. In practice, this is an elaborate approach that requires running nonlinear optimization algorithms for different values of the parameters used in the approximation. A better understanding of this asymptotic behavior will allow the simplification of the analysis of propagated numerical errors associated with finite values of Λ. Similarly, it is important to identify the maximum sample size (number of SNPs) used in each bootstrap that allows us to estimate accurate confidence intervals for a given Λ and a demographic model. The importance of choosing the right sample size for a given Λ lies in that statistical error decreases as sample size increases and propagated numerical error decreases as Λ increases. Therefore, for any given Λ there exists a large enough sample size that, if used to estimate confidence intervals, will yield significantly biased intervals that are difficult to correct via standard statistical methods. This is due to the fact that the numerical error produced by a given Λ will be significantly larger than the statistical error produced by a large enough sample size. In our four-population model we used only 170 SNPs per bootstrap, which yields a conservative estimate of the confidence intervals as we confirmed in simulations. However, in the general case when more data are available and it is not clear what sample size to use in the bootstrap for a given Λ, one can combine parametric and nonparametric bootstrap techniques to estimate the sample size and the bias. In the parametric bootstrap, one generates simulated data with specified parameters that one uses to estimate the bias due to numerical errors. Also, one can determine which sample sizes are small enough to produce larger statistical errors than numerical errors. Then one can use this knowledge to estimate accurate confidence intervals by applying corrections for bias in the nonparametric bootstrap. This approach, although elaborate, will help to estimate accurate confidence intervals for any value of Λ in a wide variety of models.
Supplementary Material
Acknowledgments
We thank K. Chen, A. Grelaud, R. Gutenkunst, A. Naqvi, A. Sengupta, V. Sousa, and S. Sunyaev for critical comments and the Kavli Institute of Theoretical Physics for supporting the visit of one author (S.L.), during which time part of this project was completed. This work was partially supported by the National Institutes of Health (grant R01GM078204 to J.H.) and the Rita Allen Foundation (Institute for Advanced Study fellowship to S.L.).
Appendix
Here, we describe some basic relationships between the orthogonal polynomials that we use in our implementation of the spectral method. In our numerical solutions of the diffusion equations, we make great use of the vector space of polynomials on the interval [0, 1] with degree bounded by the truncation parameter Λ. In this Appendix, we denote such a vector space as VΛ. We use two different orthonormal bases on VΛ: the basis of Gegenbauer polynomials and the basis of Chebyshev polynomials. In particular, the vector space spanned by the Gegenbauer polynomials of degree ≤Λ and the vector space spanned by the Chebyshev polynomials of degree ≤Λ is the same vector space VΛ. They are different orthonormal bases with respect to different inner products. This implies that any truncated expansion in terms of Gegenbauer polynomials can be exactly written as a truncated expansion of Chebyshev polynomials bounded by the same degree. More precisely, if denotes the normalized Gegenbauer polynomials and denotes the normalized Chebyshev polynomials on the interval [0, 1], the corresponding orthonormality relationships are
One can write any Gegenbauer polynomial Tn(x) in the basis of Chebyshev polynomials as the linear combination
with the coefficients for all i > n. Analogously, one can write any Chebyshev polynomial Cn(x) in the basis of Gegenbauer polynomials as the linear combination
with the coefficients for all i > n. We can summarize the changes of basis by introducing the square matrices M and L, defined as
and
being both related as MT = L−1 (i.e., the transpose matrix of M equals the inverse matrix of L), as is expected. As an example, the first 6 × 6 coefficients of M and L are
and
The reason we use two different bases of the same vector space is for the sake of convenience. The basis of Gegenbauer polynomials is convenient because the diffusion operator is diagonal in such a basis and the integrals that define the AFS become zero when the degree of the polynomial is larger than the number of chromosomes that define the AFS, as defined in Equation 2. The basis of Chebyshev polynomials is convenient to project a given density such as the mutation density μ(x) = exp(−kx) or the associated equilibrium density described in Lukic et al. (2011), onto the vector space VΛ. The advantage of using Chebyshev polynomials to project the mutation density rather than the Gegenbauer polynomials is that the weight associated with the Chebyshev polynomials is very high near the boundaries x = 0, 1, where most of the information contained in the mutation density is located. On the other hand, the weight associated with the Gegenbauer polynomials w = x(1−x) vanishes on the boundaries. There exist several bases of orthogonal polynomials associated with weights that are high at the boundaries x = 0, 1; however, we chose the basis of Chebyshev polynomials because of the simplicity associated with their implementation.
Being more precise, the two different L2 products that we use allow us to project the mutation density μ(x) onto VΛ in two different ways:
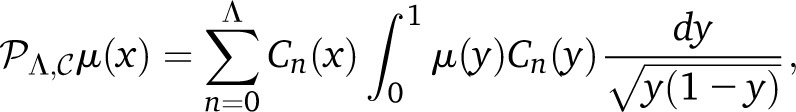 |
and
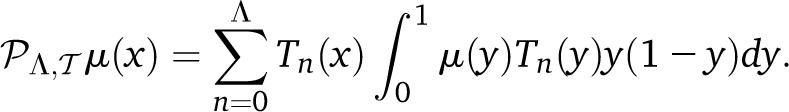 |
Generically, 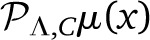 and
and 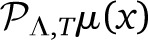 are two different polynomials in VΛ. Even if μ(x) is a smooth function, and |
are two different polynomials in VΛ. Even if μ(x) is a smooth function, and |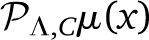 −
− 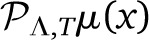 | → 0 in the limit of large Λ, both finite polynomials will be different. In our work we use the projection
| → 0 in the limit of large Λ, both finite polynomials will be different. In our work we use the projection 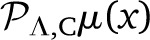 , which is implemented by means of the Gauss–Chebyshev quadrature. The implementation of the Gauss quadrature also illustrates the benefits of using the Chebyshev projection. In particular, Gauss quadratures are evaluated by means of the roots {xi} of an orthogonal polynomial of high degree and a set of weights {wi}:
, which is implemented by means of the Gauss–Chebyshev quadrature. The implementation of the Gauss quadrature also illustrates the benefits of using the Chebyshev projection. In particular, Gauss quadratures are evaluated by means of the roots {xi} of an orthogonal polynomial of high degree and a set of weights {wi}:
Given a degree d, the roots of the Chebyshev polynomial are closer to the boundaries x = 0, 1 than the roots of the Gegenbauer polynomial. For instance, given d = 10, the roots of C10(x) are 0.994, 0.946, 0.854, 0.727, 0.578, 0.422, 0.273, 0.146, 0.054, and 0.006, while the roots of T10(x) are 0.972, 0.910, 0.816, 0.699, 0.568, 0.432, 0.3, 0.184, 0.090, and 0.028.
Footnotes
Communicating editor: Y. S. Song
Literature Cited
- Akey J., Eberle M., Rieder M., Carlson C., Shriver M., 2004. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2: e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert F., Hodges E., Jensen J., Besnier F., Xuan Z., et al. , 2011. Targeted resequencing of a genomic region influencing tameness and aggression reveals multiple signals of positive selection. Heredity 107: 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander D., Novembre J., Lange K., 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19: 1655–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyko A. R., Williamson S. H., Indap A. R., Degenhardt J. D., Hernandez R. D., et al. , 2008. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 4: e1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., 2012. The joint allele frequency spectrum of multiple populations: a coalescent theory approach. Theor. Popul. Biol. 81: 179–195 [DOI] [PubMed] [Google Scholar]
- Chimpanzee-Sequencing-Consortium, 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437: 69–87 [DOI] [PubMed] [Google Scholar]
- DiCiccio T., Efron B., 1996. Bootstrap confidence intervals. Stat. Sci. 11: 189228 [Google Scholar]
- Efron B., Tibshirani R., 1994. An Introduction to the Bootstrap. Chapman & Hall/CRC, New York [Google Scholar]
- Environmental Genome Project, 2010. NIEHS Environmental Genome Project. National Institute of Environmental Health Sciences, Research Triangle Park, NC
- Ewens W., 2004. Mathematical Population Genetics. I. Theoretical Introduction, Interdisciplinary Applied Mathematics, Vol. 27. Springer-Verlag, Berlin/Heidelberg, Germany/New York
- Fisher R., 1930. The distribution of gene ratios for rare mutations. Proc. R. Soc. Edinb. 50: 205–220 [Google Scholar]
- Glasserman P., 2003. Monte Carlo Methods in Financial Engineering, Stochastic Modelling and Applied Probability Edition. Springer-Verlag, Berlin/Heidelberg, Germany/New York
- Goldstein D. B., Chikhi L., 2002. Human migrations and population structure: what we know and why it matters. Annu. Rev. Genomics Hum. Genet. 3: 129–152 [DOI] [PubMed] [Google Scholar]
- Gravel S., Henn B., Gutenkunst R., 2011. Demographic history and rare alleles sharing among human populations. Proc. Natl. Acad. Sci. USA 108: 11983–11988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths R., Spano D., 2010. Diffusion Processes and Coalescent Trees, London Mathematical Society Lecture Notes Series Probability and Mathematical Genetics: Papers in Honour of Sir John Kingman. Cambridge University Press, Cambridge, UK
- Gutenkunst R., Hernandez R., Williamson S., Bustamante C., 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5: e100695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez R. D., Williamson S. H., Bustamante C. D., 2007. Context dependence, ancestral misidentification, and spurious signatures of natural selection. Mol. Biol. Evol. 24: 1792–1800 [DOI] [PubMed] [Google Scholar]
- Hesthaven J. S., Gottlieb S., Gottlieb D., 2007. Spectral Methods for Time-Dependent Problems (Cambridge Monographs on Applied and Computational Mathematics No. 21). Cambridge University Press, Cambridge/London/New York
- Hudson R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338 [DOI] [PubMed] [Google Scholar]
- Jensen J., Bachtrog D., 2011. Characterizing the influence of effective population size on the rate of adaptation: Gillespie’s Darwin domain. Genome Biol. Evol. 3: 687–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinan A., Mullikin J., Patterson J., Reich D., 2007. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat. Genet. 39: 1251–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M., 1964. Diffusion models in population genetics. J. Appl. Probab. 1: 177–232 [Google Scholar]
- Kimura M., 1969. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61: 893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge A.-M., Michaud J., Richter A., Lemyre E., Lambert M., et al. , 2005. Population history and its impact on medical genetics in Quebec. Clin. Genet. 68: 287–301 [DOI] [PubMed] [Google Scholar]
- Lao O., van Duijn K., Kersbergen P., de Knijff P., Kayser M., 2006. Proportioning whole-genome single-nucleotide-polymorphism diversity for the identification of geographic population structure and genetic ancestry. Am. J. Hum. Genet. 78: 680–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R., 2011. Inference of human population history from individual whole-genome sequences. Nature 475: 493–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukic S., Hey J., Chen K., 2011. Non-equilibrium allele frequency spectra via spectral methods. Theor. Popul. Biol. 79: 203–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M., Nishimura T., 1998. Mersenne twister: a 623-dimensionally equidistributed uniform pseudo-random number generator. ACM Trans. Model. Comput. Simul. 8: 3–30 [Google Scholar]
- Myers S., Fefferman C., Patterson N., 2008. Can one learn history from the allelic spectrum? Theor. Popul. Biol. 73: 342–348 [DOI] [PubMed] [Google Scholar]
- Nachman M., Crowell S., 2000. Estimate of the mutation rate per nucleotide in humans. Genetics 156: 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R., 2001. Statistical tests of selective neutrality in the age of genomics. Heredity 86: 641–647 [DOI] [PubMed] [Google Scholar]
- Reich D., 2001. Linkage disequilibrium in the human genome. Nature 411: 199–204 [DOI] [PubMed] [Google Scholar]
- Risch N., Merikangas K., 1996. The future of genetic studies of complex human diseases. Science 273: 1516–1517 [DOI] [PubMed] [Google Scholar]
- Sawyer S., Hartl D., 1992. Population genetics of polymorphism and divergence. Genetics 132: 1161–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner S. F., Foo C., Gabriel S., Reich D., Daly M. J., et al. , 2005. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 15: 1576–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver M. D., Parra E. J., Dios S., Bonilla C., Norton H., et al. , 2003. Skin pigmentation, biogeographical ancestry and admixture mapping. Hum. Genet. 112: 387–399 [DOI] [PubMed] [Google Scholar]
- Song Y. S., Steinrücken M., 2011. A simple method for finding explicit analytic transition densities of diffusion processes with general diploid selection. Genetics 190: 1117–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley J., Hey J., 1997. Estimating ancestral population parameters. Genetics 145: 847–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson S., Hernandez R., Fledel-Alon A., Zhu L., Nielsen R., 2005. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc. Natl. Acad. Sci. USA 102: 7882–7887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiuf C., 2006. Consistency of estimators of population scaled parameters using composite likelihood. J. Math. Biol. 53: 821–841 [DOI] [PubMed] [Google Scholar]
- Wright S., 1931. Evolution in Mendelian populations. Genetics 16: 97–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J., Watkins W., Hu Y., Huff C., Sabo A., et al. , 2010. Genetic diversity in India and the inference of Eurasian population expansion. Genome Biol. 11: R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.