

Abstract
The first complete genome sequence of a phage infecting Weissella cibaria (Weissella kimchii) is presented. The bacteriophage ϕYS61 was isolated from kimchi, a Korean fermented vegetable dish. Bacteriophages are recognized as a serious problem in industrial fermentations; however, ϕYS61 differed from many virulent phages associated with food fermentations since it was difficult to propagate and was very susceptible to resistance development. Sequence analysis revealed that ϕYS61 resembles Podoviridae of the subfamily Picovirinae. Within the subfamily Picovirinae, the ϕ29-like phages have been extensively studied, and their terminal protein-primed DNA replication is well characterized. Our data strongly suggest that ϕYS61 also replicates by a protein-primed mechanism. Weissella phage ϕYS61 is, however, markedly different from members of the Picovirinae with respect to genome size and morphology. Picovirinae are characterized by small (approximately 20-kb) genomes which contrasts with the 33,594-bp genome of ϕYS61. Based on electron microscopy analysis, ϕYS61 was classified as a member of the Podoviridae of morphotype C2, similar to the ϕ29-like phages, but its capsid dimensions are significantly larger than those reported for these phages. The novelty of ϕYS61 was also emphasized by the low number of open reading frames (ORFs) showing significant similarity to database sequences. We propose that the bacteriophage ϕYS61 should represent a new subfamily within the family Podoviridae.
INTRODUCTION
Kimchi, a traditional Korean dish, is manufactured by fermentation of vegetables such as Chinese cabbage and radish. Hundreds of kimchi varieties are produced by the addition of different seasonings, such as scallions, powdered chili peppers, garlic, ginger, and fermented seafood. Lactic acid produced during fermentation contributes to preservation and gives kimchi its characteristic sour taste. Proper ripening and preservation are ensured by a 2 to 5% (wt/vol) salt content and anaerobic fermentation at low temperatures. Kimchi is traditionally prepared by spontaneous fermentation by lactic acid bacteria (LAB) indigenous to the vegetable ingredients. However, starter cultures have been developed in order to better control the fermentation and thus improve quality, safety, and shelf life of the fermented product (15, 16, 20, 24). Several LAB species have been identified as likely contributors in kimchi fermentations, including species of the genera Leuconostoc, Lactobacillus, Lactococcus, Pediococcus, and Weissella (6, 18, 19, 32, 35, 36, 38–41, 51, 53, 57). Weissella species isolated from kimchi include Weissella confusa, Weissella kimchii, and Weissella koreensis. These bacteria are abundant late in the fermentation process and can continue to grow during storage at low temperatures (−1°C). Weissella species have thus been associated with the excessive acidic taste of overripened kimchi products. The species Weissella kimchii, first described in 2002 (19), was reclassified as Weissella cibaria in 2004 (9, 23).
The kimchi fermentation process is characterized by an initial heterofermentative phase, followed by a homofermentative phase, and production of high-quality kimchi relies on proper succession of the different LAB species. Bacteriophages have been reported to affect the bacterial community successions in sauerkraut fermentation (42) and are apparently responsible for the variability observed in such vegetable fermentations (8). In a recent study, a high abundance of phage DNA was found in kimchi fermentation (32). This indicates that bacteriophages most probably also affect kimchi production. Bacteriophage infection is a well-recognized problem in industrial food fermentations, and a wide range of countermeasures are employed for their control (50). More knowledge about how bacteriophages affect industrial food fermentations will be valuable for the improvement of phage countermeasures.
Recently, a bacteriophage infecting a Weissella cibaria starter culture strain used in the fermentation of Thai Nham sausage was reported (54). Bacteriophages likely infecting the genus Weissella were also reported in a sauerkraut fermentation, but the bacterial isolate was not conclusively identified (42). Here, we report on a novel Weissella cibaria bacteriophage, ϕYS61, isolated from kimchi fermentation and present the first complete genome sequence of a phage infecting the genus Weissella.
MATERIALS AND METHODS
Bacteriophage isolation, growth, and purification.
Bacteriophage ϕYS61 was isolated from a commercial Chinese cabbage kimchi purchased at a Korean hypermarket 1 week after manufacture. For bacteriophage isolation and determination of phage titers, log-phase host strain Weissella cibaria YS61 cells were inoculated (≈3 × 107 CFU/ml) in deMann-Rogosa-Sharpe ([MRS] Oxoid, Basingstoke, Hampshire, United Kingdom) soft agar supplemented with 5 mM CaCl2 (MRS-C) and cast on MRS-C agar slants. Phage suspensions were spotted on top, and plaques were isolated and/or counted after overnight incubation at 30°C. Bacteriophage amplification was carried out at 30°C in MRS-C medium. Cells were grown to an optical density at 600 nm (OD600) of 0.3 to 0.4 before being infected with a 1/20 volume of filtered (0.45-μm pore size) phage lysate and incubated until lysis occurred (approximately 2 h). The multiplicity of infection (MOI) was approximately 10. Lysis was completed by the addition of 0.5% chloroform and 1 M NaCl. The mix was incubated on ice for 1 h before chloroform and cell debris were removed by centrifugation. Phage particles were precipitated with polyethylene glycol (PEG) and purified on cesium chloride (CsCl) gradients as described elsewhere (12), except that all centrifugation steps before ultracentrifugation in CsCl gradients were carried out at 5,000 × g and 4°C. Purified phage particles were dialyzed against TM buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 10 mM CaCl2) and stored at 4°C. Single-step growth curve analysis was performed essentially as described elsewhere (31).
DNA purification.
Phage precipitated with PEG was treated with 1 μg/ml DNase I and 10 μg/ml RNase A for 1 h at 37°C and then incubated for 1 h at 65°C with 25 mM EDTA, 0.5% sodium dodecyl sulfate (SDS), and 200 μg/ml proteinase K (Qiagen). After removal of residual PEG by chloroform extraction, standard phenol-chloroform extraction and ethanol precipitation were used to obtain phage DNA.
Sequencing and sequence analysis.
A shotgun library was made in pUC19 (62) after partial digestion with Sau3A. Insert sizes ranged from 0.3 to 5 kb, with an approximate average of 1.6 kb. Clones were sequenced using BigDye, version 3.1, chemistry (Applied Biosystems, Foster City, CA) and standard M13 primers. Gaps were filled by primer walking on PCR-amplified genomic DNA. Sequence assembly, bioinformatic analyzes, and genome annotation were done using the CLC Main Workbench, version 6.1.1 (CLC bio, Aarhus, Denmark). Open reading frames (ORFs) and ribosomal binding sites (RBSs) were identified using the Prodigal (30) online tool (http://prodigal.ornl.org), and homology searches were done using BLASTP and PSI-BLAST build 2.2.25+ (3, 4) at www.ncbi.nlm.nih.gov (accessed June 2012). Conserved domains were found by searching the Conserved Domains Database (CDD) (44–46) at www.ncbi.nih.gov (accessed June 2012). Phylogenetic trees were produced at www.phylogeny.fr (version 2) (17, 21, 28). Promoters were predicted by manual inspection of intergenic regions and by use of the Prokaryote Promoter Prediction tool (http://bioinformatics.biol.rug.nl/websoftware/ppp) (63). Putative transcription terminators were found through manual inspection of RNA secondary structures predicted using the CLC software. The genome comparison figure (see Fig. 5) was produced using Easyfig (60).
Fig 5.

Genome comparison of ϕYS61 and its two closest relatives, Bacillus phage ϕ29 (NC_011048) and Streptococcus phage Cp-1 (NC_001825). Genes are indicated by arrows and colored according to gene function, as indicated. Comparisons were done by tBLASTx, and similarities with E values lower than 0.01 were plotted as gray lines. Darker gray fills indicate lower E values, as shown below the figure.
Analysis of structural proteins.
Purified phage particles were denatured at 100°C for 10 min in Laemmli buffer (37) and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were stained with Coomassie brilliant blue R250 (Bio-Rad). Visible protein bands were excised, trypsin treated, and extracted as previously described (56). Extracted peptides were desalted with C18 Stage tips (55). The peptides were eluted with 70% acetonitrile before being mixed with an equal volume of matrix solution (15 mg/ml alpha-cyano-4-hydroxy cinnamic acid in ethanol-acetonitrile at 1:1) and applied to a matrix-assisted laser desorption ionization (MALDI) target plate (Bruker Daltonik GmbH, Bremen, Germany). Peptide mass fingerprinting (PMF) and tandem mass spectrometry (MS/MS) were performed on an Ultra Flex MALDI-tandem time of flight (MALDI-TOF/TOF) instrument (Bruker Daltonik). The mass range for MALDI-TOF/MS was 800 to 4,000 Da, with a mass accuracy of 50 ppm. The mass range for MALDI-TOF/MS data acquisition was 800 to 4,000 Da, and the spectra were externally calibrated using a peptide calibration mixture (Bruker Daltonik) ranging from 757 to 3,147 Da. Protein identification was carried out using Mascot software (Matrix Science Ltd., London, United Kingdom) with searches against the NCBI nonredundant (nr) database and a database containing all predicted ORF sequences (≥50 amino acids) from the genome sequence of ϕYS61.
Electron microscopy.
Purified phage samples were negatively stained with 2% (wt/vol) phosphotungstic acid (pH 7.2) on a carbon-Formvar membrane grid and examined by transmission electron microscopy (TEM). TEM analysis was performed on an EF-TEM Leo 912AB instrument (Carl Zeiss Inc., Germany) at an accelerating voltage of 120 kV. Electron micrographs were taken at a magnification of ×200,000 at the Korea Basic Science Institute in Chuneheon. The phage sizes were determined from the average of five independent measurements.
Transcription analysis.
Seventy-two ϕYS61-specific primers with binding sites interspersed in the ϕYS61 genome (see Table S1 in the supplemental material) were used to identify ϕYS61 transcript boundaries. The primers were used to set up reverse transcriptase PCR amplification of intergenic regions using RNA from infected host bacteria as the template. Lack of amplification indicated discontinuation of the RNA template. Genomic DNA from the phage was used as a positive control. Start and stop sites for transcription were deduced from putative promoters and terminators found in the genomic sequence. The methods used to synchronize infections, purify RNA, and produce cDNA are described below.
The host bacterium was grown to an OD600 of 0.3 in 50 ml MRS-C medium, infected with phages at an MOI of 1 to 2, incubated on ice for 30 min, and then rapidly brought to 30°C in a water bath. After 10 and 36 min, 10-ml samples were taken, and cells were washed twice in ice-cold Tris-EDTA (TE) buffer (pH 7.4) and quickly frozen in ethanol and ice. Cell disruption in a FP120 FastPrep bead beater (Bio101/Savant) and total RNA isolation by use of an RNeasy Minikit (Qiagen) were performed as described elsewhere (59). To avoid residual DNA carryover, an additional digest with RNase-Free DNase I (Qiagen) in RDD buffer (Qiagen) was done at 37°C for 30 min. DNase was removed by phenol-chloroform extraction, and RNA was precipitated with ethanol at −20°C overnight. The RNA was washed once in 70% (vol/vol) ethanol in diethyl pyrocarbonate (DEPC)-treated water and once in 96% ethanol, dried for 5 min at 45°C in an SPD 2010 SpeedVac concentrator (Savant), and dissolved in RNase-free water. RNA concentrations were measured on an ND-1000 spectrophotometer (NanoDrop Technologies), and RNA integrity was assessed using an RNA 600 Nano LabChip kit and an Agilent 2100 Bioanalyzer (Agilent Technologies) according to the manufacturer's instructions. cDNA was synthesized by use of random hexamer primers and a SuperScript III Reverse Transcriptase kit (Invitrogen) as instructed by the manufacturer.
Nucleotide sequence accession number.
The genome sequence of phage ϕYS61 has been deposited in the GenBank database under accession number JQ341413.
RESULTS
Bacteriophage ϕYS61 infecting Weissella cibaria YS61 was isolated from a 1-week-old vegetable kimchi purchased at a Korean hypermarket. To better understand bacteriophages infecting the genus Weissella and possibly gain insight into the dynamic microbial populations of kimchi fermentation, the virulence, morphology, and genome of ϕYS61 were explored.
Virulence and resistance development.
The infection characteristics of ϕYS61 were assessed by single-step growth analysis. After a latent period of about 40 min, the lytic life cycle ended with the release of approximately 100 progeny phage. When spotted on host strain YS61 in MRS-C soft-agar, ϕYS61 produced small plaques (approximately 0.5 mm in diameter) with slightly diffuse edges. Growth of phage-resistant bacteria was observed during phage amplification. To investigate this, YS61 and mixtures of YS61 and ϕYS61 (MOI of 0.01) were inoculated on MRS-C plates. Colony counts for phage-resistant mutants were only 10,000-fold lower than counts of uninfected cells, demonstrating the high frequency of resistance development against ϕYS61. Twelve phage-insensitive colonies (YS61-R1 to -R12) were isolated, cast in soft agar, and challenged with a 10-fold dilution series of ϕYS61. Compared to YS61, two of the isolates showed wild-type phage sensitivities in this assay; however, plaques produced on these isolates differed from those on YS61 as they were diffuse and hard to identify. Seven isolates displayed similar diffuse plaque morphologies and showed a 2- to 3-log reduction in plaque counts compared to the YS61 strain. One isolate (YS61-R10) showed a 3-log reduction in plaque counts but with clear plaque morphology, similar to that displayed by the wild-type YS61. Two isolates (YS61-R2 and -R3) showed complete resistance to ϕYS61 infection. All but two isolates retained their reduced phage sensitivity after repeated subculturing without phage challenge. YS61-R10 reverted to a wild-type phage-sensitive phenotype while the more persistent YS61-R3 isolate displayed only slight growth inhibition when challenged with an undiluted phage suspension (2 × 108 PFU/ml).
Morphology.
Electron microscopy (Fig. 1) showed that phage ϕYS61 has a moderately elongated capsid (85 by 36 nm) and a short noncontractile tail. This identified ϕYS61 as a member of the Podoviridae of the C2 morphotype (1). The distinct baseplate (28 nm wide) has six appendages (6 nm long) and a central spike. The distance from the head-tail interface to the spike tip was measured to 29 nm.
Fig 1.

Transmission electron micrograph of a ϕYS61 particle taken at a magnification of ×200,000.
The genome of ϕYS61.
The complete genome sequence of ϕYS61 was obtained through a combination of shotgun sequencing and primer walking with an average 6.3-fold sequencing coverage. The linear double-stranded DNA genome was 33,594 bp long, with a DNA base composition of 43.9% G+C. A total of 48 open reading frames (ORFs) were predicted (Table 1 and Fig. 2). Forty-one of the ORFs had a putative ribosomal binding site (RBS) upstream of the predicted start codon, and one ORF's translation was initiated by a frameshift event (described below). The predicted ORFs comprised 86% of the genome. Also present were 25-bp inverted terminal repeats, which are characteristic for the Picovirinae subfamily that include the ϕ29-like phages (43).
Table 1.
Open reading frames of ϕYS61 examined in this study
| ORFa | Ribosomal binding site and start codon sequenceb | Position (nt)c |
Predicted protein |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size (aa) | Mol mass (kDa) | pI | Putative functiond | Closest homologe |
Reference and/or accession no. | ||||||||
| Start | Stop | Description | Extent | Identity (%) | % positives | % gaps | |||||||
| 1 | AGGAGGTAAAAATTTATG | 129 | 257 | 42 | 5.0 | 9.35 | None | ||||||
| 2 | AGGAGGGTAATTGAATATG | 287 | 1048 | 253 | 29.5 | 9.00 | None | ||||||
| 3 | AAGGAGAAACAAAATG | 1032 | 1817 | 261 | 30.0 | 10.06 | Terminal protein | None | This study | ||||
| 4 | AGGAGAAACAAATG | 1829 | 3904 | 691 | 79.6 | 5.04 | DNA polymerase (PHA02563, 1E−09) | Streptococcus phage Cp-1, p05 | 523/568 | 18 | 32 | 14 | NP_044817 |
| 5 | GAAAGGTTTATAAACCATG | 3994 | 5082 | 362 | 42.2 | 8.88 | DNA encapsidation protein (PHA00149, 2E−51) | Bacillus phage B103, gene 16 | 303/321 | 33 | 53 | 11 | NP_690650 |
| 6 | AGGGTGGCATTGACTTGTG | 5113 | 5214 | 33 | 3.8 | 6.74 | None | ||||||
| 7 | AGGAGGATAACAATG | 5330 | 6061 | 243 | 27.3 | 8.75 | NMN transporter (PF04973, 1E−18) | Lactococcus phage KSY1, gp052 | 229/249 | 53 | 73 | 0 | YP_001469050 |
| 8 | GAAAGGACGGTCAATTATG | 6393 | 6854 | 153 | 16.5 | 4.79 | None | ||||||
| 9 | AGGAGTCTAATAATG | 6960 | 7304 | 114 | 12.0 | 3.74 | Structural protein | None | This study | ||||
| 10 | AGGAGAAACACAATG | 7447 | 7821 | 124 | 14.4 | 4.81 | None | ||||||
| 11 | GGAGGAATTGAATAACAATG | 7811 | 8200 | 129 | 14.8 | 4.18 | None | ||||||
| 12A | AGGAGAAATATTATG | 8383 | 9618 | 411 | 45.4 | 5.02 | Major capsid protein (PHA00144, 1E−37) | Enterococcus phage EF62ϕ, ORF40 | 421/469 | 31 | 52 | 5 | This study, ADX81364 |
| 12B | AGGAGAAATATTATG | 8383 | 9881 | 500 | 53.9 | 5.31 | Major capsid protein with bacterial Ig-like domain (PF02368, 1E−6) | Bacillus phage ϕ29, major head protein | 483/448 | 25 | 42 | 17 | This study, YP_002004536 |
| 13 | TGGAACATCAAGCACTTATG | 10160 | 10669 | 169 | 18.2 | 5.42 | None | ||||||
| 14 | AGGGGCACAGCATG | 10680 | 10931 | 83 | 10.0 | 6.58 | None | ||||||
| 15 | AGGAGGAATTATG | 10975 | 11172 | 65 | 7.7 | 4.47 | None | ||||||
| 16 | AGTAGAAAGGAGATG | 11208 | 11768 | 186 | 21.1 | 9.95 | None | ||||||
| 17 | AGGAGGCCATACAAAATG | 11847 | 12458 | 203 | 21.1 | 6.76 | None | ||||||
| 18 | GAAAGGAAATAGATTAAATG | 12513 | 13400 | 295 | 32.7 | 6.40 | Lysin, N-acetyl-l-alanine amidase (PF01510, 7E−4) | Weissella paramesenteroides ATCC 33313, LytA | 270/305 | 50 | 61 | 12 | ZP_04782183 |
| 19 | AGGAGAACAAACATG | 13440 | 13967 | 175 | 17.1 | 9.48 | None | ||||||
| 20 | GGAGGAAAATAATTTTG | 14126 | 15472 | 448 | 49.4 | 4.75 | Tail lysin | Bacillus phage B103, gene 13 | 303/365 | 28 | 44 | 13 | NP_690647 |
| 21 | AGGAGAAATTCAAATG | 15535 | 17730 | 731 | 82.1 | 5.84 | Tail protein (PHA00380, 3E−16) | Bacillus phage PZA, gp9 | 731/599 | 23 | 39 | 24 | P07534 |
| 22 | ATTTTGCAAGCAAAATTATG | 18052 | 19422 | 456 | 49.8 | 4.69 | Upper collar connector (PHA00147, 9E−27) | Enterococcus faecalis T2, EFBG_02899 | 328/333 | 33 | 53 | 12 | ZP_05423975 |
| 23 | AGGAGGAGAAATGTAATG | 19422 | 20330 | 302 | 34.5 | 4.46 | Lower collar protein (PHA01077, 6E−13) | Enterococcus phage EF62ϕ, ORF33 | 245/255 | 32 | 51 | 12 | ADX81357 |
| 24 | AAACCGCAAATAATTCAATG | 20396 | 21760 | 454 | 47.5 | 4.96 | Tail fiber protein (PF01391, 9E−9) | Mycobacterium phage Bxz2, gp4 | 108/344 | 62 | 72 | 10 | NP_817595 |
| 25 | GCATTATTTAGCGCCTGATG | 21812 | 22120 | 102 | 11.8 | 6.40 | None | ||||||
| 26 | GCGGTATACAGCACATG | 22333 | 24312 | 659 | 74.6 | 5.01 | Structural protein | Escherichia coli MS 60-1, HMPREF9533_04376 | 419/712 | 24 | 41 | 6 | This study, EGB80821 |
| 27 | AGGAGAGAAAATAGTG | 24328 | 24681 | 117 | 13.0 | 5.65 | Phage holin 4 (PF05105, 8E−11) | Leuconostoc phage 1-A4, LM1A4_024 | 106/123 | 33 | 55 | 0 | ADD71747 |
| 28 | AGGAGGCAGAGCATG | 24800 | 25087 | 95 | 11.0 | 6.72 | None | ||||||
| 29 | AGAGGGTCTTTTTTG | 25059 | 25550 | 163 | 18.8 | 5.10 | None | ||||||
| 30 | AGGAGACTAAGGAATG | 25581 | 25820 | 79 | 9.2 | 4.78 | Lactobacillus acidophilus 30SC, LAC30SC_06460 | 66/67 | 36 | 50 | 0 | YP_004292355 | |
| 31 | AGGAGGGTTTTAAATTATG | 25824 | 26291 | 155 | 17.3 | 5.81 | None | ||||||
| 32 | TTGCTTTATCGTGCGAGGTG | 26263 | 26526 | 87 | 10.2 | 8.77 | None | ||||||
| 33 | AGGAGGCGGCTTATG | 26516 | 26689 | 57 | 6.5 | 4.11 | None | ||||||
| 34 | AGGAGGCAGGCGTTTAATG | 26823 | 26933 | 36 | 4.0 | 5.99 | None | ||||||
| 35 | AGGGGAAAGATAATG | 26933 | 27208 | 91 | 10.3 | 4.11 | None | ||||||
| 36 | GGAGGACAAGTAACATG | 27201 | 27518 | 105 | 12.0 | 4.28 | None | ||||||
| 37 | AGGAGGACAAGTAACATG | 27520 | 28491 | 323 | 36.4 | 6.36 | None | ||||||
| 38 | GAAAGGATTTTATAATG | 28493 | 29017 | 174 | 19.9 | 5.30 | Listeria phage B025, ORF62 | 169/212 | 24 | 38 | 25 | YP_001468701 | |
| 39 | AGGAGGCTTGACATG | 29091 | 29186 | 31 | 3.6 | 5.89 | None | ||||||
| 40 | GAAAGGAAGCATAATG | 29300 | 29998 | 232 | 24.3 | 4.63 | None | ||||||
| 41 | AGGAGGATATAACATG | 29998 | 30321 | 107 | 11.5 | 9.00 | Lactobacillus acidophilus 30SC, LAC30SC_00610 | 98/227 | 29 | 41 | 1 | YP_004286453 | |
| 42 | GAGAGGAAATGCAATG | 30504 | 30986 | 160 | 18.8 | 10.04 | HNH endonuclease | Enterococcus phage EFRM31, gp11 | 120/173 | 38 | 60 | 4 | YP_004306639 |
| 43 | GGAACGCTATG | 30983 | 31102 | 39 | 4.8 | 9.58 | None | ||||||
| 44 | GGGGTGGAAATTATG | 31125 | 31334 | 69 | 8.1 | 4.23 | None | ||||||
| 45 | GAGGTGGAAATTATG | 31487 | 31648 | 53 | 6.2 | 6.71 | None | ||||||
| 46 | GAGGTATGGAAATATG | 31887 | 32144 | 85 | 9.7 | 4.71 | Lactobacillus phage Lb338-1, ORF13 | 57/102 | 35 | 65 | 4 | YP_002790692 | |
| 47 | GAGGAAATTATCATG | 32233 | 32577 | 114 | 13.4 | 3.79 | None | ||||||
Structural proteins identified by MS analysis are shown in boldface.
Predicted ribosomal binding sites and start codons are underlined. nt, nucleotide.
ORF positions on the complement strand are shown in italics.
Best hits in the Conserved Domains Database. Accession numbers and E values are shown in parentheses. PHA, CDD accession; PF, Pfam accession.
Closest homolog determined by BLAST searches. Extent, alignment length/hit sequence length (aa). Percentages of identical positions, positive positions, and gap positions in each BLAST alignment are shown in separate columns.
Fig 2.

Graphic representation of the linear ϕYS61 dsDNA genome. (A) The relative position and direction of each ORF are indicated by a white arrow. Structural proteins identified by MS in this study are shown as black arrows. Putative functions are indicated in gray letters: tp, terminal protein; dnap, DNA polymerase; enc, encapsidation; nmn, NMN transporter; sp, structural protein; mcp, major capsid protein; lys, lysin; taa, tail assembly; tap, tail protein; ucc, upper collar connector; lcp, lower collar protein; taf, tail fiber; hol, holin; hnh, HNH endonuclease. Predicted functional modules are shown at the top with regions of uncertain function shown as dotted lines. The 25-bp inverted terminal repeats (ITR) are also indicated. Scale bars mark genome positions at 5,000-bp intervals. (B) Early (light gray bars) and late (dark gray bars) transcripts are shown at their relative positions on the upper or lower strand. Predicted terminators (○) and promoters (→) located between transcripts are indicated on tall stems or on short stems when found within transcript regions.
Transcription.
To identify transcript borders, RNA was purified from infected YS61 cells and analyzed by reverse transcription-PCR amplification of intergenic regions. Four early and five late transcripts were found. Transcripts are mapped on the genome in Fig. 2. Our results indicated that early transcription progresses inward from the ends of the linear genome, with transcripts E1, E2, and E3 from the right end and transcript E4 from the left. Predicted genes on late transcripts were found on the positive strand downstream of E4 with the exception of orf21, transcript L3, which was found on the negative strand.
Putative promoters, resembling the consensus Escherichia coli σ70 promoters, were identified upstream of all transcript regions. Upstream of early transcripts E1 and E2 were also found promoter signals resembling recognition sites for the lactococcal two-component regulator LlrB (52). Moreover, putative promoters upstream of E1 were found in a tandem repeat region comprised of two repeat units, 1 (66 bp) and 2 (35 bp), repeated in the order 1-2-1-2-1-2-1. Tandemly repeated promoters have been shown to result in strong promoter activity in the Ralstonia solanacearum phage ϕRSB1 (34).
As shown in Fig. 2B, putative Rho-independent terminators were detected in seven of the eight intergenic regions found by PCR to exert transcription termination. A putative mechanism for transcription termination in the orf46 (E2)-orf45 (E3) intergenic region was not identified.
Several putative promoters and terminators identified outside the confirmed start-stop regions are probably involved in modulating transcription activity and, consequently, downstream gene expression.
Functional assignment of ORFs.
BLAST searches revealed that 18 of the 48 ORFs were similar to other known sequences, whereas 30 ORFs produced no significant hits in database searches. Putative function was assigned to 16 ORFs by sequence comparison or MS identification of structural proteins. Information on the BLAST alignments is given in Table 1. In some cases the functional assignment was based on genomic location, presence of conserved domains, or identification of conserved motifs by manual inspection, discussed below. Based on putative function, ORFs could be grouped in functional modules. Predicted module borders matched well with transcript borders identified by PCR (Fig. 2).
ORFs on early transcripts E1, E2, and E3.
Twenty open reading frames, orf28 to orf47, were predicted on early transcripts E1 to E3. BLAST searches revealed similarity to known sequences for five of these ORFs (Table 1). ORF41 showed weak resemblance to a Lactobacillus transcriptional regulator (YP_004286453). A putative function was assigned to the orf42 gene product by its significant similarity to proteins of the HNH family of homing endonucleases commonly found in phage genomes.
DNA replication.
Four open reading frames (orf 1 to orf 4) were predicted in the genomic region corresponding to early transcript E4. BLAST searches revealed that orf4 putatively encoded a DNA polymerase (DNAP) type B, similar to that of Streptococcus phage Cp-1 (48) and the ϕ29-like phages (49). We identified the highly conserved DNAP motifs ExoI, ExoII, ExoIII, CT, and motif-1, -2a, -2b, and -3. Also present were insertion sequences corresponding to terminal protein region 1 (TPR 1) and TPR 2 conserved among protein-primed DNA polymerases (33, 49). Sequence comparison of ORF4 and the 36 seed sequences of the Pfam DNAP family B (PF03175) showed that ORF4 is likely a distant relative of the phage-encoded protein-primed DNA polymerases (Fig. 3). In addition to the polymerase, three proteins are essential to DNA replication in the ϕ29-like phages (10). These are the terminal DNA-bound protein (TP; gp3 in ϕ29), a single-strand DNA binding protein (SSB; gp5 in ϕ29), and a double-strand DNA binding protein (DSB; gp6 in ϕ29), and they are encoded by genes immediately upstream of the DNAP-encoding gene. Based on their genomic localization, orf1, orf2, and orf3 possibly encode proteins with similar involvement in DNA replication. The high pI values of the predicted ORF1 to ORF3 proteins might indicate interactions with negatively charged molecules such as DNA, but BLAST results did not corroborate this. The presence of a covalently bound terminal protein was, however, indicated by the observation that proteinase K treatment was essential for purification of ϕYS61 DNA. An alignment of the deduced 261-amino-acid (aa) sequence of ϕYS61 ORF3 to the terminal proteins (aa 167 to 266) of Bacillus phages ϕ29, B103, GA-1, and Nf, Enterobacteria phages PRD1 and L17, Lactococcus phage asccϕ28, and Streptococcus phage Cp-1 revealed conservation of functional amino acids (33) and predicted secondary structures, despite the low overall sequence similarity (see Fig. S1 in the supplemental material). These data and the presence of inverted terminal repeats in the ϕYS61 genome strongly suggest that ϕYS61 DNA replication occurs by a protein-primed mechanism and that orf3 and orf4 encode the terminal protein and the protein-primed DNA polymerase, respectively.
Fig 3.

Maximum-likelihood (ML) tree based on amino acid sequence alignment of ϕYS61 ORF4, Lactococcus phage asccϕ28 ORF7, Bacillus phage ϕ29 gp2, and the 36 seed sequences of the Pfam type B DNA polymerase family (PF03175). The alignment was produced in CLC after alignment fix points had been introduced at the positions of conserved motifs ExoI, ExoII, ExoIII, CT, and motif-1, -2a, and -3 (49). Branch lengths are indicated in red.
DNA packaging.
A strong indication of homology was found between the predicted orf5-encoded protein and the DNA encapsidation protein of Bacillus phage B103 and other ϕ29-like phages. The ϕ29-like DNA encapsidation protein is a DNA packaging ATPase that, together with packaging RNA (pRNA) and the head-tail connector protein, forms a packaging motor (58). Strong similarity was also observed between the putative head-tail connector of ϕYS61 and the connectors of the ϕ29-like phages. Comparison of ϕ29 and Cp-1 pRNA structures (5, 47) to computer-predicted ϕYS61 RNA secondary structures revealed no significant pRNA prediction.
Structural proteins.
Structural proteins were analyzed by SDS-PAGE, and five protein bands were identified by mass spectrometry analysis (Fig. 4). Three distinct SDS-PAGE bands were identified as variants of ORF12. Of these, the lower two bands were identified as ORF12A and ORF12B. The predicted masses of ORF12A and ORF12B, 45.4 and 53.9 kDa, respectively, were consistent with the observed protein migration during SDS-PAGE. The third ORF12 variant with an observed mass of about 56 kDa could possibly result from an additional frameshift event or from posttranslational modification. We were unable to identify additional peptides that could cause the observed slower migration of this band. BLAST searches revealed that ORF12A is homologous to the major capsid protein of Enterococcus phage EF62ϕ. ORF12B contains a bacterial group 2 immunoglobulin-like domain (Big_2; Pfam accession number PF02368) and is more similar to the major capsid protein of the ϕ29-like phages. Immunoglobulin-like (Ig-like) domains are frequently found on the surface of tailed dsDNA phages and have been proposed to interact with carbohydrates on the cell surface and thereby facilitate phage adsorption (25). Moreover, Ig-like domains are commonly added to phage structural proteins by programmed ribosomal frameshifts (26). We identified two possible slippery sequences in the orf12A-orf12B overlapping region: CAGGGGTAA at position 9609 to 9618 and GACCCGTCC at position 9598 to 9606 (+1 frame codons are shown in boldface and the + 3 frame alternative codons are underlined). Frameshifting in the −1 direction could potentially be achieved at both positions but by different mechanisms: in the first position a tRNAGly (3′-CCC-5′) could slip back one base, and at the second a tRNAThr (3′-UGG-5′) could bind the proline CCG codon, thereby making the third codon base available for pairing with the following tRNAVal anticodon (7). Our MS data did not distinguish between the two frameshift positions.
Fig 4.
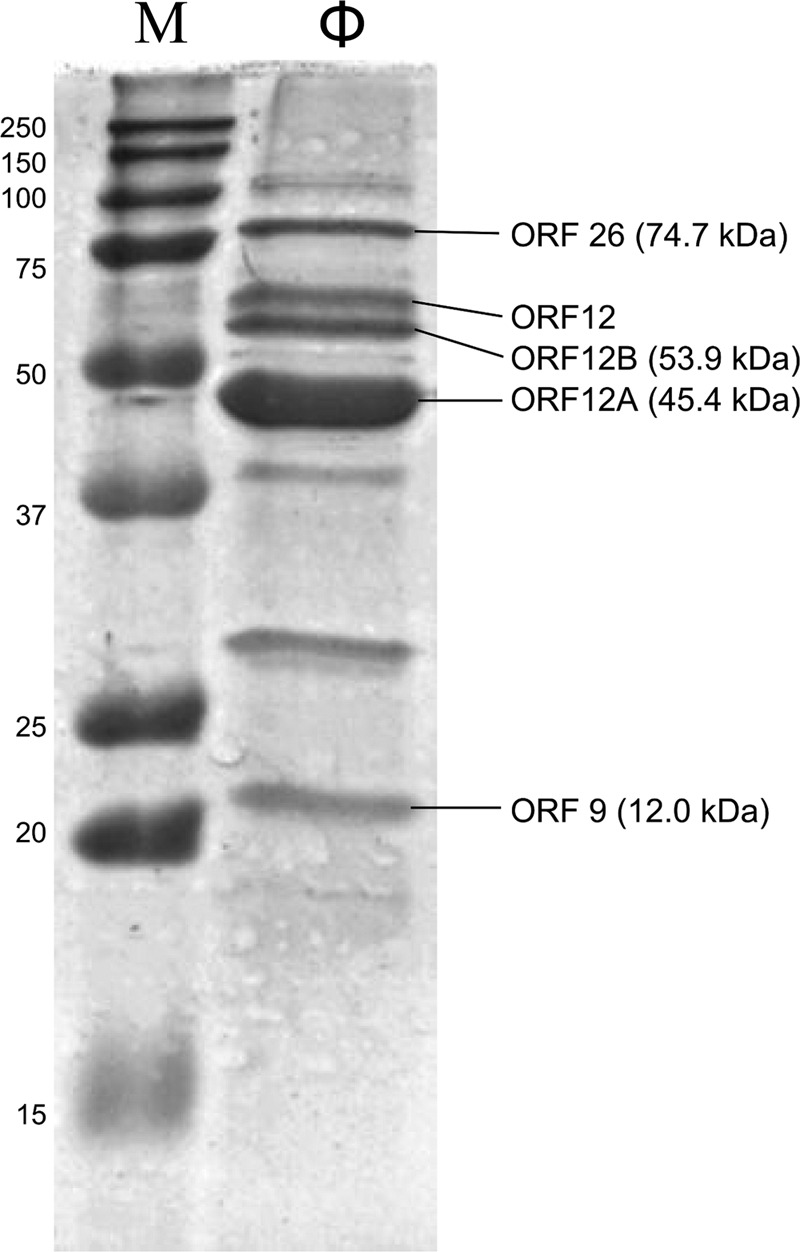
SDS-PAGE analysis of ϕYS61 structural proteins. Lane M, molecular mass marker, with masses in kilodaltons indicated on the left; lane ϕ, ϕYS61 proteins visualized with Coomassie stain. Protein identities determined by MS analysis are shown on the right side with deduced molecular masses shown in parentheses. No molecular mass could be predicted for the protein band marked ORF12 since we were unable to identify the mechanism by which a protein of this size is produced from the coding sequence of orf12A or orf12B.
The gene products of orf9 and orf26 were also identified by MS as components of the ϕYS61 virion. Based on its genomic location, we suspect that orf9 encodes a capsid protein; however, no sequence similarity was found to known proteins. ORF26 is probably a tail protein, based on its location on transcript L5 downstream of the genes encoding the putative tail, connector, lower collar, and tail fiber proteins (orf21, orf22, orf23, and orf24, respectively).
The predicted orf20-encoded protein shares homology and essential catalytic residues with the tail-associated cell wall-degrading enzyme, gp13, of Bacillus phage B103 and the ϕ29-like phages. gp13 of ϕ29 is most likely involved in the penetration of the phage tail through the peptidoglycan layer (61). gp13 has also been shown to be essential for phage tail assembly interacting with gp9 (tail protein) after gp11 (lower collar) has assembled onto the phage capsid (27, 29).
Host cell lysis.
We identified a putative muramidase (ORF18) with 50% amino acid identity to LytA of prophages integrated in Weissella paramesenteroides ATCC 33313 and several Leuconostoc spp. A putative holin (ORF27) was identified by its sequence similarity to the holin (gp24) of Leuconostoc phage 1-A4. Significant sequence similarity was also found to the Pfam holin_4 family (PF05105).
Comparison to other members of the Podoviridae.
Inverted terminal repeats, ORF similarities, and the probable replication by a protein-primed mechanism strongly suggest that the closest relatives of ϕYS61 are podoviruses of the subfamily Picovirinae. To investigate this, whole-genome comparisons were done using the Easyfig software that utilizes the tBLASTx algorithm. The genome of ϕYS61 was individually compared to the genomes of Bacillus phages ϕ29 (NC_011048) and GA-1 (NC_002649), Streptococcus phage Cp-1 (NC_001825), Lactococcus phage asccϕ28 (NC_010363), and enterobacteria phage PRD1. The two phages which are most similar, ϕ29 and Cp-1, are shown in Fig. 5. As can be seen in the figure, there are several small regions of sequence similarity and some likeness in genome organization.
DISCUSSION
In this study, we report the characterization of Weissella cibaria phage ϕYS61 and present the first complete genome sequence of a phage infecting the genus Weissella. The host strain was originally classified as Weissella kimchii (19); however, the two species W. kimchii and W. cibaria (9) are very closely related and have been proposed to be one species named W. cibaria (23). The G+C content of the 33.594-bp ϕYS61 genome is 43.9%, which is more similar to the reported 43.9 to 44.9% G+C of W. cibaria (9) than the 48.2% reported for the W. kimchii strain CHJ3 (19). Bacteria of the genus Weissella are commonly encountered in vegetable fermentations, and bacteriophages infecting them could be responsible for variability in fermentation progress and product quality. Better understanding of bacteriophages in industrial fermentations is of great importance for the use and improvement of phage countermeasures.
The bacteriophage ϕYS61 infecting W. cibaria YS61 was isolated from kimchi, a Korean fermented vegetable product. During serial propagation of ϕYS61, it was necessary to filter out the readily appearing resistant YS61 mutants. Twelve resistant isolates showed various degrees of bacteriophage resistance. Phage resistance is often associated with reduced fitness (11), and kimchi fermentations are highly competitive environments (32). Thus, the readily appearing ϕYS61-resistant mutants might not be able to prevail in actual kimchi fermentations.
Another aspect of kimchi fermentation where bacteriophage knowledge could be useful is in the prevention of overripening. Weissella spp. have been associated with excess acid formation and a reduced shelf life for kimchi. Bacteriophages or phage lysins could possibly be used to reduce the growth of Weissella and other unwanted bacteria during kimchi storage. This could also be implemented for other fermented products, for instance, in Korean rice wine, where W. cibaria is a likely cause of quality deterioration (S. K. Yum, Seoul Takju Manufacturers' Association, personal communication). The putative lysin identified in this study showed homology to LytA of a Weissella paramesenteroides prophage (Table 1). It is possible that the ϕYS61 lysin could be applied to increase shelf life of kimchi and other fermented vegetable products.
Morphologically, phage ϕYS61 resembled the recently reported W. cibaria phage ϕ22 isolated from a fermented pork sausage (54). The C2 morphotype of Podoviridae is relatively rare and had by the year 2000 been reported only for 39 bacteriophages, whereas the C1 Podoviridae morphotype had been reported 631 times (2). The observed capsid dimensions (85 by 36 nm) were smaller than the 92- by 50-nm capsid reported for Podoviridae ϕ22 infecting W. cibaria N22 (54). Genome size is often proportional to capsid dimensions; however, the genome of ϕYS61 (34.5 kb) is larger than the genome size (29 kb) reported for ϕ22 (54).
Most of the predicted ϕYS61 ORFs with significant similarity to known sequences showed homology to proteins of phages and bacteria commonly associated with soil, vegetables, and vegetable fermentation (Table 1). This is in agreement with the extensive horizontal gene transfer observed between bacteriophages in closely related environments (14, 22).
Putative structural proteins of the ϕYS61 genome resembled structural proteins of phages of the Podoviridae subfamily Picovirinae, as well as the predicted enterococcal phage EF62ϕ (13) (Table 1). EF62ϕ is a phage-like extrachromosomal linear genetic element identified during Enterococcus faecalis EF62 genome sequencing. Enterococcus species have been found along with Weissella in vegetable fermentation (32), and genetic exchange between these species is thus likely. The morphology of the EF62ϕ-like enterococcal phages is unknown. An overview of capsid dimensions and genome sizes of ϕYS61 and its closest relatives is given in Table S2 in the supplemental material. Even though ϕYS61 resembles the ϕ29-like phages, it is significantly larger both in capsid and genome size. Moreover, the baseplate structure of ϕYS61 differs from the structures of the ϕ29-like phages (49).
Bacteriophage ϕYS61 also resembles the members of the Picovirinae with respect to DNA replication and packaging. Although we were unable to purify TP-DNA from ϕYS61, the finding that proteinase treatment was essential for DNA purification and the presence of TPR-1 and TPR-2 insertion sequences in the DNA polymerase gene (orf4) strongly indicate that ϕYS61 replicates by a protein-primed mechanism. This hypothesis is supported by the presence of inverted terminal repeats in the ϕYS61 genome, a trait that is characteristic for phages with protein-primed replication within the order Caudovirales (43). To our knowledge, this is the largest Podoviridae genome to replicate by this mechanism.
Similarity to the phages with protein-primed DNA replication was further analyzed by whole-genome comparison (Fig. 5). Similarities in genome organization were observed, and we find it likely that ϕYS61 is a distant relative of Cp-1 and ϕ29-like phages.
The characterization and genome sequence analysis of ϕYS61 have revealed a novel member of the Podoviridae with a clear relationship to the Picovirinae. ϕYS61 differs, however, in typical Picovirinae traits such as genome and capsid size. We propose that the bacteriophage ϕYS61 should represent a new subfamily within the family Podoviridae.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Morten Skaugen for performing the mass spectrometry analysis.
This work was supported by Tine SA, The Norwegian Research Council, and the Korea Research Foundation (grant number KRF-2007-013-F00007).
Footnotes
Published ahead of print 10 August 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Ackermann HW. 1998. Tailed bacteriophages: the order Caudovirales. Adv. Virus Res. 51:135–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ackermann HW. 2001. Frequency of morphological phage descriptions in the year 2000. Brief review. Arch. Virol. 146:843–857 [DOI] [PubMed] [Google Scholar]
- 3. Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Altschul SF, et al. 2005. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 272:5101–5109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anderson D, Bodley JW. 1990. Role of RNA in bacteriophage phi 29 DNA packaging. J. Struct. Biol. 104:70–74 [DOI] [PubMed] [Google Scholar]
- 6. Bae JW, et al. 2005. Development and evaluation of genome-probing microarrays for monitoring lactic acid bacteria. Appl. Environ. Microbiol. 71:8825–8835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baranov PV, Gesteland RF, Atkins JF. 2004. P-site tRNA is a crucial initiator of ribosomal frameshifting. RNA 10:221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barrangou R, Yoon SS, Breidt F, Jr, Fleming HP, Klaenhammer TR. 2002. Characterization of six Leuconostoc fallax bacteriophages isolated from an industrial sauerkraut fermentation. Appl. Environ. Microbiol. 68:5452–5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bjorkroth KJ, et al. 2002. Taxonomic study of Weissella confusa and description of Weissella cibaria sp. nov., detected in food and clinical samples. Int. J. Syst. Evol. Microbiol. 52:141–148 [DOI] [PubMed] [Google Scholar]
- 10. Blanco L, Lazaro JM, de Vega M, Bonnin A, Salas M. 1994. Terminal protein-primed DNA amplification. Proc. Natl. Acad. Sci. U. S. A. 91:12198–12202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bohannan BJM, Lenski RE. 2000. Linking genetic change to community evolution: insights from studies of bacteria and bacteriophage. Ecol. Lett. 3:362–377 [Google Scholar]
- 12. Boulanger P. 2009. Purification of bacteriophages and SDS-PAGE analysis of phage structural proteins from ghost particles. Methods Mol. Biol. 502:227–238 [DOI] [PubMed] [Google Scholar]
- 13. Brede DA, Snipen LG, Ussery DW, Nederbragt AJ, Nes IF. 2011. Complete genome sequence of the commensal Enterococcus faecalis 62, isolated from a healthy Norwegian infant. J. Bacteriol. 193:2377–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brüssow H, Desiere F. 2001. Comparative phage genomics and the evolution of Siphoviridae: insights from dairy phages. Mol. Microbiol. 39:213–223 [DOI] [PubMed] [Google Scholar]
- 15. Chang JY, Chang HC. 2011. Growth inhibition of foodborne pathogens by kimchi prepared with bacteriocin-producing starter culture. J. Food Sci. 76:M72–M78 [DOI] [PubMed] [Google Scholar]
- 16. Chang JY, Chang HC. 2010. Improvements in the quality and shelf life of kimchi by fermentation with the induced bacteriocin-producing strain, Leuconostoc citreum GJ7 as a starter. J. Food Sci. 75:M103–M110 [DOI] [PubMed] [Google Scholar]
- 17. Chevenet F, Brun C, Banuls A-L, Jacq B, Christen R. 2006. TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics 7:439 doi:10.1186/1471-2105-7-439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cho J, et al. 2006. Microbial population dynamics of kimchi, a fermented cabbage product. FEMS Microbiol. Lett. 257:262–267 [DOI] [PubMed] [Google Scholar]
- 19. Choi HJ, et al. 2002. Weissella kimchii sp. nov., a novel lactic acid bacterium from kimchi. Int. J. Syst. Evol. Microbiol. 52:507–511 [DOI] [PubMed] [Google Scholar]
- 20. Choi IK, et al. 2003. Novel Leuconostoc citreum starter culture system for the fermentation of kimchi, a fermented cabbage product. Antonie Van Leeuwenhoek 84:247–253 [DOI] [PubMed] [Google Scholar]
- 21. Dereeper A, et al. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36:W465–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Desiere F, McShan WM, van Sinderen D, Ferretti JJ, Brüssow H. 2001. Comparative genomics reveals close genetic relationships between phages from dairy bacteria and pathogenic streptococci: evolutionary implications for prophage-host interactions. Virology 288:325–341 [DOI] [PubMed] [Google Scholar]
- 23. Ennahar S, Cai Y. 2002. 2004 Genetic evidence that Weissella kimchii Choi et al. 2002 is a later heterotypic synonym of Weissella cibaria Björkroth et al. Int. J. Syst. Evol. Microbiol. 54:463–465 [DOI] [PubMed] [Google Scholar]
- 24. Eom HJ, Park JM, Seo MJ, Kim MD, Han NS. 2008. Monitoring of Leuconostoc mesenteroides DRC starter in fermented vegetable by random integration of chloramphenicol acetyltransferase gene. J. Ind. Microbiol. Biotechnol. 35:953–959 [DOI] [PubMed] [Google Scholar]
- 25. Fraser JS, Maxwell KL, Davidson AR. 2007. Immunoglobulin-like domains on bacteriophage: weapons of modest damage? Curr. Opin. Microbiol. 10:382–387 [DOI] [PubMed] [Google Scholar]
- 26. Fraser JS, Yu Z, Maxwell KL, Davidson AR. 2006. Ig-like domains on bacteriophages: a tale of promiscuity and deceit. J. Mol. Biol. 359:496–507 [DOI] [PubMed] [Google Scholar]
- 27. Garcia JA, Carrascosa JL, Salas M. 1983. Assembly of the tail protein of the Bacillus subtilis phage phi 29. Virology 125:18–30 [DOI] [PubMed] [Google Scholar]
- 28. Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704 [DOI] [PubMed] [Google Scholar]
- 29. Hagen EW, Reilly BE, Tosi ME, Anderson DL. 1976. Analysis of gene function of bacteriophage ϕ29 of Bacillus subtilis: identification of cistrons essential for viral assembly. J. Virol. 19:501–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hyatt D, et al. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119 doi:10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hyman P, Abedon ST. 2009. Practical methods for determining phage growth parameters. Methods Mol. Biol. 501:175–202 [DOI] [PubMed] [Google Scholar]
- 32. Jung JY, et al. 2011. Metagenomic analysis of kimchi, a traditional Korean fermented food. Appl. Environ. Microbiol. 77:2264–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kamtekar S, et al. 2006. The phi29 DNA polymerase:protein-primer structure suggests a model for the initiation to elongation transition. EMBO J. 25:1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kawasaki T, et al. 2009. Genomic characterization of Ralstonia solanacearum phage ϕRSB1, a T7-like wide-host-range phage. J. Bacteriol. 191:422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim J, Chun J, Han HU. 2000. Leuconostoc kimchii sp. nov., a new species from kimchi. Int. J. Syst. Evol. Microbiol. 50:1915–1919 [DOI] [PubMed] [Google Scholar]
- 36. Kim M, Chun J. 2005. Bacterial community structure in kimchi, a Korean fermented vegetable food, as revealed by 16S rRNA gene analysis. Int. J. Food Microbiol. 103:91–96 [DOI] [PubMed] [Google Scholar]
- 37. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 38. Lee D, Kim S, Cho J, Kim J. 2008. Microbial population dynamics and temperature changes during fermentation of kimjang kimchi. J. Microbiol. 46:590–593 [DOI] [PubMed] [Google Scholar]
- 39. Lee JS, et al. 1997. Classification of isolates originating from kimchi using carbon-source utilization patterns. J. Microbiol. Biotechnol. 7:68–74 [Google Scholar]
- 40. Lee JS, et al. 2005. Analysis of kimchi microflora using denaturing gradient gel electrophoresis. Int. J. Food Microbiol. 102:143–150 [DOI] [PubMed] [Google Scholar]
- 41. Lee JS, et al. 2002. Weissella koreensis sp. nov., isolated from kimchi. Int. J. Syst. Evol. Microbiol. 52:1257–1261 [DOI] [PubMed] [Google Scholar]
- 42. Lu Z, Breidt F, Plengvidhya V, Fleming HP. 2003. Bacteriophage ecology in commercial sauerkraut fermentations. Appl. Environ. Microbiol. 69:3192–3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maniloff J, Ackermann HW. 1998. Taxonomy of bacterial viruses: establishment of tailed virus genera and the order Caudovirales. Arch. Virol. 143:2051–2063 [DOI] [PubMed] [Google Scholar]
- 44. Marchler-Bauer A, et al. 2009. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 37:D205–D210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marchler-Bauer A, Bryant SH. 2004. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 32:W327–W331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marchler-Bauer A, et al. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martin AC, Lopez R, Garcia P. 1996. Analysis of the complete nucleotide sequence and functional organization of the genome of Streptococcus pneumoniae bacteriophage Cp-1. J. Virol. 70:3678–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martin AC, Lopez R, Garcia P. 1995. Nucleotide sequence and transcription of the left early region of Streptococcus pneumoniae bacteriophage Cp-1 coding for the terminal protein and the DNA polymerase. Virology 211:21–32 [DOI] [PubMed] [Google Scholar]
- 49. Meijer WJ, Horcajadas JA, Salas M. 2001. ϕ29 family of phages. Microbiol. Mol. Biol. Rev. 65:261–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moineau S, Lévesque C. 2005. Control of bacteriophages in industrial fermentations, p 286–296 In Kutter E, Sulakvelidze A. (ed), Bacteriophages: biology and applications. CRC Press, Boca Raton, FL [Google Scholar]
- 51. Nam YD, Chang HW, Kim KH, Roh SW, Bae JW. 2009. Metatranscriptome analysis of lactic acid bacteria during kimchi fermentation with genome-probing microarrays. Int. J. Food Microbiol. 130:140–146 [DOI] [PubMed] [Google Scholar]
- 52. O'Connell-Motherway M, et al. 2000. Six putative two-component regulatory systems isolated from Lactococcus lactis subsp. cremoris MG1363. Microbiology 146:935–947 [DOI] [PubMed] [Google Scholar]
- 53. Park JM, et al. 2010. Identification of the lactic acid bacteria in kimchi according to initial and over-ripened fermentation using PCR and 16S rRNA gene sequence analysis. Food Sci. Biotechnol. 19:541–546 [Google Scholar]
- 54. Pringsulaka O, Patarasinpaiboon N, Suwannasai N, Atthakor W, Rangsiruji A. 2011. Isolation and characterisation of a novel Podoviridae-phage infecting Weissella cibaria N 22 from Nham, a Thai fermented pork sausage. Food Microbiol. 28:518–525 [DOI] [PubMed] [Google Scholar]
- 55. Rappsilber J, Ishihama Y, Mann M. 2003. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75:663–670 [DOI] [PubMed] [Google Scholar]
- 56. Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1:2856–2860 [DOI] [PubMed] [Google Scholar]
- 57. Shin MS, Han SK, Ryu JS, Kim KS, Lee WK. 2008. Isolation and partial characterization of a bacteriocin produced by Pediococcus pentosaceus K23-2 isolated from kimchi. J. Appl. Microbiol. 105:331–339 [DOI] [PubMed] [Google Scholar]
- 58. Simpson AA, et al. 2000. Structure of the bacteriophage phi29 DNA packaging motor. Nature 408:745–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Solheim M, Aakra A, Vebo H, Snipen L, Nes IF. 2007. Transcriptional responses of Enterococcus faecalis V583 to bovine bile and sodium dodecyl sulfate. Appl. Environ. Microbiol. 73:5767–5774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sullivan MJ, Petty NK, Beatson SA. 2011. Easyfig: a genome comparison visualizer. Bioinformatics 27:1009–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xiang Y, et al. 2008. Crystal and cryoEM structural studies of a cell wall degrading enzyme in the bacteriophage φ29 tail. Proc. Natl. Acad. Sci. U. S. A. 105:9552–9557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119 [DOI] [PubMed] [Google Scholar]
- 63. Zomer AL, Buist G, Larsen R, Kok J, Kuipers OP. 2007. Time-resolved determination of the CcpA regulon of Lactococcus lactis subsp. cremoris MG1363. J. Bacteriol. 189:1366–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.