

Abstract
Recognition of pathogen-associated molecular patterns by pattern recognition receptors of the innate immune system is crucial for the initiation of innate and adaptive responses and for immunological memory. We investigated the role of TLR7 in the induction of adaptive immunity and long-term memory following influenza virus infection and vaccination in C57BL/6 mice. During infection with influenza A/PR8/34 virus, the absence of either TLR7 or MyD88 leads to reduced virus-specific antibodies in the serum and antibody-secreting cells in their secondary lymphoid organs, particularly in bone marrow. In spite of this, the absence of TLR7/MyD88 signaling did not impair the production of protective antibodies. Following immunization with the 2009 pandemic inactivated split vaccine, TLR7−/− mice had significantly lower levels of germinal center formation, antibody-secreting cells, and circulating influenza virus-specific antibodies than control animals. Consequently, TLR7−/− mice failed to develop protective immunological memory upon challenge. Furthermore, the immunogenicity of the split vaccine was likely due to TLR7 recognition of virion RNA, as its removal from the split vaccine significantly reduced the levels of influenza virus-specific antibodies and compromised the vaccine protective efficacy in mice. Taken together, our data demonstrate that TLR7 plays an important role in vaccine-induced humoral immune responses to influenza virus through the interaction with viral RNA present in the split vaccine.
INTRODUCTION
Influenza viruses continue to be a considerable public health burden. Each year, influenza viruses infect 3 million to 5 million people worldwide, resulting in 250,000 to 500,000 deaths (61). In addition, influenza A viruses (IAVs) from animal reservoirs remain a pandemic threat which is highlighted by the 2009 H1N1 pandemic (12, 26, 56). Currently, vaccination remains the most cost-effective public health countermeasure to prevent seasonal and pandemic influenza. However, renewed efforts are needed to improve influenza vaccine efficacy in immunocompromised populations, older adults, and young children (3, 6, 9). Therefore, understanding the immune response to infection and vaccination with IAVs and especially how the interplay of host and viral components shapes the immune response is critical for designing influenza vaccines with improved immunogenicity and effectiveness.
The immune response to IAVs culminates in the production of protective neutralizing antibodies against the major surface protein, the hemagglutinin (HA) (14). Influenza virus infection can lead to production of neutralizing antibodies that provide life-long protection from infection with antigenically closely related viruses (2). This was exemplified by the recent spread of the 2009 pandemic influenza A/H1N1 virus [A(H1N1)pdm09], which caused an estimated 86 million cases and up to 17,620 deaths in the United States by April 2010 (49). Compared with seasonal influenza outbreaks, the overall impact of the 2009 H1N1 pandemic was lower in adults ≥65 years of age. This is possibly due to the presence of protective cross-reactive antibodies developed through childhood exposure to early 20th century H1N1 viruses which shared antigenic similarity with the A(H1N1)pdm09 virus (42). The immune response to influenza virus infection is initiated through the engagement of the innate immune system. The IAV genome consists of negative-sense, single-stranded RNA that is recognized by host pattern recognition receptors (PRRs). Many PRR ligands have previously been shown to improve the magnitude, duration, as well as breadth of neutralizing antibody responses (30, 51, 58).
Upon infection of host cells by IAV, viral RNAs (vRNAs) are sensed by PRRs, such as Toll-like receptor 7 (TLR7), retinoid acid inducible gene-I (RIG-I), and nucleotide-binding domain and leucine-rich-repeat-containing protein 3 (NLRP3), which form multimolecular complexes termed inflammasomes (45). Activation of these pathways leads to downstream signaling through myeloid differentiation primary response gene 88 (MyD88), TIR domain-containing adapter-inducing beta interferon (IFN-β) (TRIF), or caspase 1, respectively (55). The subsequent cascade signal induces type I interferons (IFN-α/β) and production of inflammatory cytokines (31). Of these PRRs, TLR7 is important not only for the activation of the innate antiviral response but also for the induction of adaptive immunity (7, 22, 25, 27, 34, 35). Heer and colleagues showed that TLR7 signaling is critical for antibody isotype class switching (22). This could be due to B-cell intrinsic TLR7 signaling or indirect B-cell stimulation by extrinsic TLR7-dependent production of IFN-α/β. Recently, we have shown that TLR7 signaling is involved in the recruitment of myeloid-derived suppressor cells (MDSCs) and for the shaping of humoral immunity in response to IAV infection (27). Boeglin and colleagues later showed that a combination of B-cell receptor, CD40, and TLR7 stimulation on B cells augments antibody-secreting cell (ASC) differentiation (7). Collectively, these data suggest that TLR7 signaling is important in adaptive immunity, particularly in the enhancement of B-cell responses.
In this study, we investigated the role of TLR7 in the long-term-memory responses to IAV infection and vaccination. In the case of infection, we found that serum IgM levels and the frequency of IgM-positive (IgM+) ASCs in secondary lymphoid organs were reduced in the absence of TLR7. However, TLR7 signaling played a minimal role in the production of HA-specific antibodies. Conversely, TLR7 was critical during immunization with an inactivated split influenza vaccine. The absence of TLR7 signaling led to reduced germinal center (GC) formation, expression of B7 on B cells, IAV-specific ASCs, and serum antibody responses. When challenged, TLR7−/− mice had significantly higher lung viral titers than wild-type mice. The immunogenicity of the split vaccine was likely due to the presence of vRNA and its potential interaction with TLR7. Overall, we demonstrate that TLR7 plays an important role in the induction of immune responses to an IAV monovalent split vaccine.
MATERIALS AND METHODS
Mice.
C57BL/6 (B6) mice were purchased from Charles River Laboratories (Wilmington, MA). TLR7−/− mice on the B6 background were a gift from Akiko Iwasaki (Yale University, New Haven, CT) (40) and Regeneron Pharmaceuticals, Inc. (Tarrytown, NY). MyD88−/− mice were also on the B6 background and a gift from Bruce Beutler (Scripps Research Institute, La Jolla, CA) (1). Both strains were bred at Charles River Laboratories. Animals were age matched and housed under pathogen-free conditions. Animal research was conducted under the guidance of the CDC's Institutional Animal Care and Use Committee in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited animal facility.
Influenza viruses and vaccines.
The mouse-adapted A/Puerto Rico/8/34 (PR8) virus and the A(H1N1)pdm09 A/Mexico/4108/2009 (Mex4108) and A/California/08/2009 (Cal/08) viruses were propagated by allantoic fluid inoculation of 10-day-old embryonated chicken eggs. The 50% egg infectious dose (EID50), 50% mouse infectious dose (MID50), and 50% mouse lethal dose (MLD50) were determined using methods described previously (54). For PR8, 1 MID50 was equivalent to 10 EID50s and 0.01 MLD50. For Mex4108, 1 PFU was equivalent to 28 EID50s. B6 or TLR7−/− mice were vaccinated intramuscularly (i.m.) in the hind leg with 10 μg of A/California/07/2009 X-179A (Cal/07) monovalent split vaccine (Sanofi Pasteur, Swiftwater, PA) in a volume of 100 μl. Viral genomic RNA components in the concentrated monovalent vaccine were inactivated by treatment with 20 μg/ml of RNase A (RNase; Qiagen, Valencia, CA) at 37°C for 30 min. RNase-treated vaccine (RTV) and heated untreated vaccine were run on a 1.2% ethidium bromide e-gel (Life Technologies, Grand Island, NY) to check for the presence of RNA. RTV was then diluted to the equivalent concentration used for the noninactivated vaccine inoculations and administered as described above.
Infections of mice and harvesting of tissues for flow cytometric analysis and viral titer quantification.
Infection experiments were carried out as described previously (27). Briefly, mice were infected intranasally (i.n.) with 25 MID50s of PR8 under anesthesia in a volume of 50 μl or mock infected with 50 μl of phosphate-buffered saline (PBS). On the indicated days postinfection (p.i.), animals were sacrificed to harvest lung, spleen, bone marrow (BM), and serum. Single-cell suspensions were stained with fluorochrome-conjugated antibodies for flow cytometric analysis, using an LSR-II flow cytometer (BD Biosciences, San Jose, CA). For the lethal challenge, the mice were infected with 100 MLD50s of PR8 or 9.3 × 104 EID50s of Mex4108.To determine viral titers, lungs were homogenized in 1 ml of cold PBS. Clarified homogenates were titrated in 10- to 11-day-old eggs to determine the EID50 viral titers in the lungs as previously described (48). Numbers were converted to a log10 scale to determine statistical significance.
IAV-specific antibody-secreting cell ELISpot assay.
To measure the frequency of virus-specific B cells, polyvinylidene difluoride multiscreen 96-well plates (Millipore, Billerica, MA) were first coated with 100 HA units (HAU) of UV-inactivated viruses for overnight at 4°C. For UV inactivation of virus, allantoic fluid-clarified PR8 or Cal/08 viruses were treated with 5,000 units of UV. To measure the B cells secreting total IgG or IgM, the plates were coated with either 5 μg/ml anti-mouse IgG or IgM antibodies (Southern Biotech, Birmingham, AL). On the next day, the plates were washed 3 times with PBS and blocked for 1 h at 37°C with 200 μl/well of RPMI 1640 supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin, l-glutamine, and 0.01 M HEPES buffer (cRPMI). Spleen, BM, and lung were harvested, and a single-cell suspension was prepared in cRPMI after red blood cell lysis. Red blood cell-cleared whole splenocytes or bone marrow cells (10 × 106 to 15 × 106 cells/ml) were added onto enzyme-linked immunosorbent spot (ELISpot) assay plates and incubated for overnight at 37°C in a humidified atmosphere with 5% CO2. The plates were washed 4 times with 0.5% Tween 20 in PBS (PBST) and incubated with biotinylated anti-mouse IgG or IgM antibodies (Southern Biotech, Birmingham, AL) for 1 h at room temperature. Plates were washed 4 times with PBST, and alkaline phosphatase-conjugated streptavidin (Vector, Burlingame, CA) in PBST was added for 1 h at room temperature. Plates were washed 4 times, and spot development was achieved by adding 100 μl of reagent from a Vector blue alkaline phosphatase substrate kit III (Vector, Burlingame, CA) to each well. Spot-forming units were counted using an ImmunoSpot analyzer (Cellular Technology Ltd., Cleveland, OH) and expressed as the number of spots per 106 cells.
HI.
Hemagglutination inhibition (HI) assay serum samples were treated with receptor-destroying enzyme (RDE; Denka Seiken Co., Tokyo, Japan) overnight at 37°C, followed by heat inactivation (56°C for 30 min). Serially diluted sera in V-bottom 96-well plates were tested in duplicate for their ability to inhibit the agglutination of 0.5% turkey red blood cells by 4 HAU of PR8 or Cal/08 virus in a standard HI assay as described previously (60). Numbers were converted to a log2 scale to determine statistical significance.
Antibody isotype determination by ELISA.
An antibody enzyme-linked immunosorbent assay (ELISA) was performed as described previously (27). Briefly, 96-well, flat-bottom plates were coated with 100 HAU/well of PR8, washed with PBST, and then blocked with 5% bovine serum albumin (BSA). Mouse sera were diluted 1:10. Absorbance was read at 450 nm. A standard curve of purified mouse IgM, IgG, IgG1, or IgG2c was used to convert absorbance readings to immunoglobulin concentration. Numbers were converted to a log10 scale to determine statistical significance.
Antibodies for surface staining for flow cytometric analysis.
Single-cell suspensions were stained with fluorochrome-conjugated antimouse antibodies to measure B-cell markers, as well as antigen-specific CD8+ T cells. The following antibodies were used for B cells: GL7 (fluorescein isothiocyanate) and CD19 (allophycocyanin [APC]-Cy7), purchased from BD Bioscience (San Jose, CA); CD80 (phycoerythrin [PE]), purchased from BioLegend (San Diego, CA); and CD38 (Alexa Fluor 700) and IgD (Pacific Blue), purchased from eBiosciences (San Diego, CA). The following antibodies were used for T cells: CD8 (Alexa Fluor 700) and CD3 (APC), purchased from BD Bioscience, and PR8 nucleoprotein (NP)-tetramer (H-2Db ASNENMETM) (PE), purchased from ProImmune (Sarasota, FL).
Intracellular cytokine staining.
Splenocytes were seeded at 1 × 106 in 100 μl of plain RPMI 1640 in 96-well round-bottom plates, and Cal/08 virus was added in a volume of 50 μl at a 0.1 multiplicity of infection (MOI) in each well. After 1 h infection, the remaining virus was neutralized by adding 50 μl RPMI 1640 containing 40% FBS, 400 U/ml penicillin, and 400 μg/ml streptomycin. Plates were then incubated for 5 days at 37°C in a humidified atmosphere with 5% CO2. After 5 days, cells were then collected and restimulated for 6 h with plate-bound anti-CD3 (10 μg/ml; clone 1452C11; eBioscience, San Diego, CA) and anti-CD28 (2 μg/ml, clone 37.51; eBioscience) in the presence of GolgiStop protein transport inhibitor (BD Bioscience, San Jose, CA) to enhance cytokine secretion and to aid flow cytometric detection (32, 59). Cells were surface stained with CD4 (PE-Cy7) and CD8 (Alexa Fluor 700), purchased from eBioscience, for 15 min at 4°C. Cells were then made permeable with Cytofix/Cytoperm solution (BD Biosciences), followed by intracellular staining with IFN-γ (peridinin chlorophyll protein-Cy5.5; purchased from BioLegend, San Diego, CA), tumor necrosis factor alpha (TNF-α; eFluor 450; purchased from eBioscience); and interleukin-17 (IL-17; Alexa 488), IL-4 (PE), and IL-10 (APC) (purchased from BD Bioscience) in Perm/Wash solution (BD Bioscience) for 30 min at 4°C. Cells were washed twice with Perm/Wash solution and resuspended in PBS–10% FBS. The samples were analyzed using an LSR II flow cytometer (BD Biosciences, Sam Jose, CA), and the cytometry data were analyzed using FlowJo software (TreeStar, Ashland, OR).
RNA purification/sequencing.
Viral RNA was purified from vaccine using an RNeasy Plus minikit (Qiagen, Valencia, CA). RNA was reverse transcribed and amplified with forward primer 5′-AGC AAA AAG CAG GGT GAC AAG ACA-3′ and reverse primer 5′-AGT AGA AAC AAG GGT GTT TTT TAT using a Superscript One Step reverse transcription-PCR system with Platinum Taq DNA polymerase (Life Technologies Corp., Carlsbad, CA). Prior to sequencing, the PCR product was cleaned using ExoSAP-IT reagent (Affymetrix, Santa Clara, CA). A sequencing reaction was performed using a BigDye Terminator (version 3.1) cycle sequencing kit, and sequences were run on a 3730xl sequencer (Life Technologies). Genetic analysis was performed using the BioEdit sequence alignment editor program (20).
Statistical analysis.
Statistical significance was determined by two-way analysis of variance with Bonferroni posttests to compare multiple groups of mice against control B6 mice. Significant P values are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Error bars represent the standard error of the mean (SEM), unless otherwise mentioned.
RESULTS
TLR7−/− and MyD88−/− mice had reduced GC reactions and IAV-specific IgM+ ASCs following IAV infection.
We and others have previously shown that TLR7 is important in the induction of the adaptive immune response to IAV infection (22, 27, 35). In this study, we investigated the long-term consequences of memory responses to IAV infection in the absence of signaling by TLR7. We also tested this in MyD88-deficient mice as an internal control because MyD88, a downstream adaptor protein for TLR7 and other TLRs, would share a signaling pathway with TLR7. B6, TLR7−/−, and MyD88−/− mice were infected with 25 MID50s PR8 or were mock infected with PBS. Primary B- and T-cell responses were then assessed at both 1 and 6 months p.i. Spleen, lung, and BM samples were harvested at each time point. For primary B-cell responses, we measured the frequency of GC B cells (CD19+ GL7+ CD38−) by flow cytometry and PR8-specific ASCs by ELISpot assay in spleen and lung. Both TLR7−/− and MyD88−/− mice had at least a 2-fold reduction in the frequency of GC B cells in response to IAV infection compared to control B6 mice, most strikingly in the spleen at 6 months p.i., with a greater than 3-fold reduction (Fig. 1A).
Fig 1.

TLR7−/− and MyD88−/− mice had fewer GC B cells and PR8-specific IgM+ ASCs following primary infection. Mice (n ≥ 4) were infected with 25 MID50s of PR8 virus, and spleen, lung, and BM were harvested at the time points indicated. (A) B cells entering GC reactions were identified as CD19+ IgD− GL7+ CD38− in both lung and spleen. Data are shown as the percentage of GC B cells out of the total number of CD19+ cells gated. (B and C) PR8-specific IgG+ ASCs (B) or IgM+ ASCs (C) from lung, spleen, and BM were measured by ELISpot assay against whole, UV-inactivated PR8 virus. Data are shown as the number of PR8-specific spots per million cells plated.
Despite a reduced frequency of GC B cells in these tissues, the numbers of class-switched IgG+ PR8-specific ASCs in the lung and spleen were not significantly decreased in either TLR7−/− or MyD88−/− mice (Fig. 1B). However, there was a general reduction in the frequency of IgM+, PR8-specific ASCs in these mice at the site of infection (the lung) as well as in memory lymphoid tissues (Fig. 1C). A lack of TLR7/MyD88 signaling did not impact the memory CD8+ T-cell response, as both TLR7−/− and MyD88−/− mice had comparable frequencies of NP-specific CD8+ T cells in both lungs and spleen following infection (see Fig. S1 in the supplemental material). Collectively, these data indicate that a deficiency in TLR7 signaling results in a minor reduction in primary B-cell responses but not in memory CD8+ T-cell responses following infection.
TLR7−/− and MyD88−/− mice had reduced GC reactions and lower levels of PR8-specific ASCs in the bone marrow following a lethal challenge.
Our data suggest that following influenza virus infection, loss of innate recognition of IAVs through TLR7 and its adaptor, MyD88, only modestly affects the development of B-cell adaptive immune memory. Nonetheless, it remains possible that these molecules regulate the quality of long-term protective memory responses, thereby impairing the ability of a host to respond to future challenge. To test this idea, we infected B6 or TLR7−/− mice with 25 MID50s PR8 virus first and then challenged with a lethal dose (100 MLD50s) of PR8 virus 1 or 6 months after the primary infection. We also set up a control group of mice that received PBS first and then were challenged. We then monitored the B- and T-cell recall responses 5 days following challenge. Lethal challenge induced a detectable level of GC reactions in both lung and spleen of B6 mice (Fig. 2A), with a higher response detected at 1 month p.i. in lung and 6 months p.i. in spleen. In contrast, such a recall response was drastically reduced in TLR7−/− or MyD88−/− mice at either time point (Fig. 2A; see Fig. S2 in the supplemental material). Nonetheless, the frequencies of PR8-specific IgG+ or IgM+ ASCs in these tissues (lung and spleen) from TLR7−/− mice were generally comparable to those in these tissues from B6 mice (Fig. 2B and C). However, in the bone marrow, the frequency of PR8-specific ASCs and particularly IgM+ ASCs was significantly reduced in the BM of both groups of knockout mice; by 6 months, TLR7−/− and MyD88−/− mice exhibited 7- and 11- fold reductions, respectively, compared to B6 mice (Fig. 2 And C). This result suggests a reduced recruitment of memory B cells to the BM from either the spleen or lung due to impaired GC reactions in the knockout animals (Fig. 2A). Consistent with the primary infection, the frequency of recall PR8-specific CD8+ T cells and the levels of cytokines secreted by CD4+ and CD8+ T cells were comparable in lung and spleen regardless of TLR7 or MyD88 signaling (see Fig. S3 in the supplemental material). Collectively, these findings suggest that the lack of TLR7 signaling compromises the recall B-cell response at late time points following IAV infection.
Fig 2.

TLR7−/− mice had reduced GC reactions and lower frequencies of PR8-specific ASCs in the bone marrow following a lethal IAV challenge. Mice (n ≥ 6) were infected with 25 MID50s of PR8 or mock infected with PBS (naïve) and then challenged at either 1 or 6 months following primary infection with 100 MLD50s of PR8. Lung, spleen, and BM were harvested on day 5 postchallenge. (A) GC B cells (CD19+ IgD− GL7+ CD38−) in either lung or spleen were measured by flow cytometry. Data are shown as the percentage of GC B cells out of the total number of CD19+ cells gated. (B and C) PR8-specific IgG+ ASCs (B) or IgM+ ASCs (C) from lung, spleen, and BM were measured by ELISpot assay against whole, UV-inactivated PR8 virus. Data are shown as the number of PR8-specific spots per million cells plated.
TLR7−/− and MyD88−/− mice had lower levels of serum PR8-specific antibodies, yet comparable HI titers, following IAV infection.
HA-specific serum antibodies are the major immune correlate of protection against IAV infection. Since both TLR7−/− and MyD88−/− mice had a generally reduced memory-B-cell response (Fig. 1 and 2), we wanted to determine if the virus-specific circulating antibodies were also reduced following infection in these mice. Sera were collected from the three groups of mice at 1, 3, and 6 months following primary infection and tested for the presence of PR8-specific antibodies by ELISA and HI assay (Fig. 3). Consistent with the observed differences in frequency of ASCs between control B6 and knockout mice (Fig. 1 And 2), the levels of PR8-specific IgM in TLR7−/− and MyD88−/− mice were generally reduced up to 3 months p.i. (Fig. 3A), while the IgG response was comparable to that of B6 mice (Fig. 3B). However, by 6 months p.i., both PR8-specific IgM and IgG antibodies were significantly reduced in knockout mice (Fig. 3A and B). When IgG subclasses were measured, both groups of knockout mice had elevated levels of PR8-specific IgG1 with reduced levels of IgG2c (Fig. 3C and D), indicating that the antibody responses in these mice was polarized toward a Th2 response, as demonstrated previously (22, 27, 35).
Fig 3.

TLR7−/− and MyD88−/− mice had comparable levels PR8-specific HI titers, despite reduced levels of IAV-specific IgM and IgG, by 6 months following IAV infection. Mice (n ≥ 15) were infected with 25 MID50s of PR8 virus, and sera were collected at 1, 3, and 6 months following infection. (A to D) The levels of circulating PR8-specfic IgM, IgG, IgG1, and IgG2c were measured by ELISA using whole, UV-inactivated PR8 virus. Each mouse represents one value, and the average is the geometric mean. (E) Sera were treated with RDE, and HI titers were measured by HI assay. Shown is the result for each individual mouse with the geometric mean.
Despite the reduction of PR8-specific antibody levels and evidence for Th2-skewed responses by 6 month p.i., HI titers in TLR7−/− and MyD88−/− mice were comparable to those from control B6 mice at all time points measured following infection (Fig. 3E). Not surprisingly, both groups of B6 and TLR7−/− mice were completely protected upon challenge, showing no lethality or weight loss, whereas PBS control mice succumbed to infection at between 6 and 9 days postchallenge (data not shown). This demonstrates that the signaling pathway through TLR7/MyD88 is dispensable for protective antibody responses following primary IAV infection, although the composition of this antibody response is different from that in control B6 mice. Collectively, these data suggest that although TLR7/MyD88 signaling regulates certain aspects of the memory-B-cell response, it plays a dispensable role in the protective humoral response against IAV infection.
The early-B-cell response to pandemic split vaccine was compromised in TLR7−/− mice.
Since TLR7 signaling was not critical for protective immunity following infection (Fig. 1 to 3), we next investigated whether TLR7 signaling affects the outcome of immunization with influenza vaccine. B6 and TLR7−/− mice were immunized i.m. with 10 μg of monovalent A(H1N1)pdm09 split vaccine. To assess the primary effector-B-cell response, we collected sera at day 15 following vaccination and then measured HI titers. B6 mice developed measurable HI titers (∼80) by day 15; however, TLR7−/− mice had undetectable levels of HI antibody (Fig. 4A). Also decreased were the levels of B cells entering GC reactions; TLR7−/− mice had fewer GC B cells than B6 mice on day 10 postimmunization (Fig. 4B and C). TLR7−/− B cells also exhibited a modest reduction in expression of the costimulatory molecule CD80 on their surface compared with B6 B cells, in both B cells that were actively part of a GC (GL7+ CD38−) and those B cells outside the GC (CD38+) (Fig. 4D). There was no significant difference in the frequencies of cytokine-producing CD4+ T cells and only a modest reduction of CD8+ TNF-α-producing T cells in the absence of TLR7 signaling (see Fig. S4A and B in the supplemental material). Therefore, these results suggest that TLR7 plays a key role in early-B-cell activation and antibody production in the context of immunization with split vaccine.
Fig 4.

The early-B-cell response to pandemic split vaccine was compromised in TLR7−/− mice. Mice (n = 5) were vaccinated i.m. with 10 μg pandemic A(H1N1)pdm09 split vaccine. (A) Sera were collected on day 15 (D15) following vaccination, RDE treated, and measured for HI titers against Cal/08 virus. Dashed line, lower limit of assay detection. Shown is the geometric mean with the 95% confidence interval. A representative dot plot of CD19+ IgD− GC B cells (GL7+ CD38−) in spleen (B) and the percentage of splenic GC B cells out of the total number of CD19+ B cells from 3 to 5 mice (C) are shown. (D) Representative histogram of CD80 expression pattern from GC B cells (GL7+ CD38−) or non-GC B cells (CD38+). Thin line, isotype control; gray filled line, TLR7−/− mice; thick black line, B6 mice.
The vRNA component of the split vaccine contributes to the vaccine's immunogenicity.
Our findings that the primary B-cell response was reduced in TLR7−/− mice indicate that TLR7 signaling is important for an optimal response to inactivated split influenza vaccine. They also suggest that the pandemic split vaccine may contain a TLR7 ligand(s) that potentiates the immunogenicity of this vaccine. To determine if the pandemic split vaccine contained RNA, the A(H1N1)pdm09 vaccine was treated with RNase or left untreated and then purified RNAs were run on an ethidium bromide gel. Indeed, untreated A(H1N1)pdm09 split vaccine contained detectable amounts of RNA, whereas RNase-treated vaccine was devoid of RNA (Fig. 5). Sequencing of the vRNA isolated from untreated vaccine confirmed the origin of the RNA to be PR8 virus, the parental high-growth donor used to produce influenza vaccine reassortant viruses.
Fig 5.
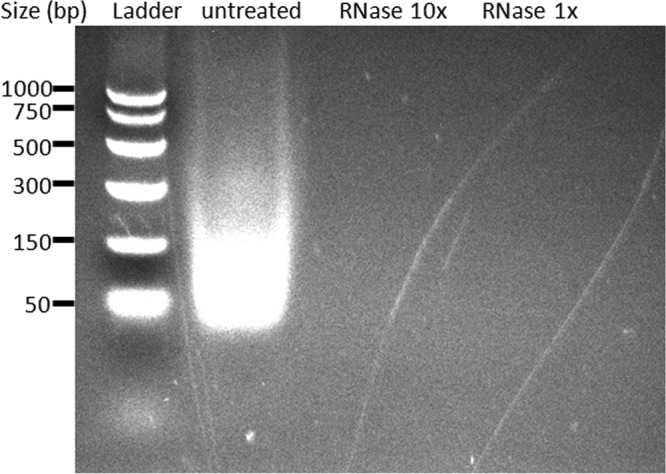
The A(H1N1)pdm09 split vaccine contains viral genomic RNA. Vaccine (150 μg HA) was treated with various concentrations of RNase (10× = 100 μg, 1× = 10 μg) at 37°C for 30 min or left untreated under the same conditions. RNA was then purified and concentrated from vaccine using a Qiagen kit. Samples were run on a 2% ethidium bromide nondenaturing agarose gel.
Next, we asked whether this RNA was responsible for the immunogenicity of the A(H1N1)pdm09 split vaccine. We immunized i.m. B6 and TLR7−/− mice with A(H1N1)pdm09 vaccine and compared the B-cell responses with those of B6 mice immunized with RNase-treated vaccine (RTV). We allowed these mice to proceed to memory stage (1 month following immunization) and measured the Cal/08-specific ASCs in spleen and BM (Fig. 6). TLR7−/− mice had reduced numbers of Cal/08-specific, IgG+ and IgM+ ASCs in both spleen and BM (Fig. 6A and B), with the most significant reduction being IgM+ ASCs in the BM. In the case of RTV-immunized mice, the BM but not the spleen exhibited reduced numbers of ACSs, similar to those detected in TLR7−/− mice immunized with untreated vaccine (Fig. 6A and B). These results suggest that the RNA component of the split vaccine is responsible for the immunogenicity of the vaccine and the host memory-B-cell response to vaccine.
Fig 6.

TLR7 recognition contributed to the B-cell response to split influenza vaccine. B6 and TLR7−/− mice (n = 10) were immunized i.m. with 10 μg A(H1N1)pdm09 split vaccine, and a control group of B6 mice was immunized with RNase-treated A(H1N1)pdm09 vaccine (RTV) (n = 5). Spleen (A) and BM (B) were harvested at 1 month postvaccination, and Cal/08-specific IgG+ or IgM+ ASCs were measured by ELISpot assay against whole, UV-inactivated Cal/08 virus. Data are shown as the number of spots from Cal/08-specific ASCs per million cells plated.
TLR7−/− mice failed to develop protective immunity following immunization.
Since an optimal B-cell response to split vaccine was dependent on TLR7 signaling, we next tested whether TLR7 was critical for the development of protective humoral immunity. We immunized i.m. B6 and TLR7−/− mice with the A(H1N1)pdm09 split vaccine and allowed the mice to proceed to the memory stage (1 month). We also included B6 mice immunized with RTV. We collected sera at 1 month following vaccination and measured HI titers (Fig. 7A). Consistent with the reduced frequency of Cal/08-specific ASCs (Fig. 6), the HI titers were significantly reduced in TLR7−/− mice immunized with untreated A(H1N1)pdm09 vaccine and undetectable in B6 mice immunized with RTV (Fig. 7A). Consequently, upon challenge with 2 × 105 MID50s of Mex4108 A(H1N1)pdm09, TLR7−/− mice had lung viral titers 100-fold higher than those in B6 mice at day 4 following challenge (Fig. 7B). RTV-immunized mice were equally unprotected from challenge. These data indicate that TLR7 signaling regulates the development of humoral immunity following split vaccine immunization. Additionally, the presence of a TLR7 ligand in the vaccine is critical for protective vaccine efficacy.
Fig 7.

TLR7−/− mice failed to develop protective immunity following immunization. B6 and TLR7−/− mice were immunized i.m. with 10 μg Cal/07 split vaccine, and a control group of B6 mice was immunized with RNase-treated Cal/07 vaccine (RTV). (A) Sera were collected at 1 month postvaccination for each group tested (n = 25), RDE treated, and measured for HI titers against Cal/08 virus. Shown are the geometric means of the titers collected with a 95% confidence interval. The HI titers of B6-RTV were under the lower limit of assay detection. (B) At 1 month following immunization, mice (n = 5) from all vaccinated groups were challenged with 2 × 105 MID50s Mex4108. A control group of B6 mice received PBS only and was then challenged. Lungs were harvested at day 4 postchallenge, and lung viral titers were measured in eggs. Shown is each individual titer with the geometric means of the titers collected.
DISCUSSION
Engaging innate PRRs is an important step in eliciting an effective adaptive response against IAV. Since TLR7 recognizes IAV genomic RNA, delineating the impact of TLR7 signaling on the innate and adaptive response is key to our understanding of the overall immune response to influenza virus infection and vaccination. Recently, we reported that deficient TLR7 signaling increased MDSC recruitment to the lungs and polarized the acute adaptive response toward Th2 following IAV infection (27). In the present study, we hypothesized that TLR7 signaling would enhance the long-term-memory response to IAV. We tested this hypothesis in the case of both infection and immunization. In our infection model, we found that a deficiency in TLR7 or MyD88 signaling reduced certain aspects of B-cell memory (Fig. 1 to 3). However, HI titers following primary infection were comparable and mice were equally well protected from a lethal challenge (Fig. 3E). These data indicate that TLR7 signaling is dispensable for the production of protective antibodies following IAV infection. In contrast, TLR7 signaling during immunization contributed to the development of a robust HI antibody response to the A(H1N1)pdm09 split vaccine and optimal clearance of challenge virus, which appeared to be dependent on the presence of RNA in the vaccine (Fig. 7). Together, our data demonstrate that TLR7 signaling is dispensable for the establishment of memory to IAV infection but is important for humoral responses following vaccination with split vaccine.
The differential role of TLR7 during infection versus immunization indicates that the context in which vRNA is recognized by host PRRs may be different in the two circumstances. In the context of infection, upon entry to host cell through endocytosis, vRNA is released from the late endosome following fusion of viral and host membranes (36). Throughout IAV's life cycle, vRNA can be recognized by at least 3 different PRRs: TLR7 in late endosomes, as well as RIG-I and NOD-like receptors (NLRs) in the cytosol (25). These innate immune receptors initiate an antigen-specific adaptive immune response by the induction of IFN-α/β and proinflammatory cytokines. Thus, it is possible that in the absence of TLR7, the collective signaling through other PRRs is sufficient to facilitate the induction of adaptive immunity during virus infection (4, 10, 35, 38, 52). In the context of immunization with split inactivated influenza vaccine, the vaccine components are confined to the major histocompatibility complex class II endocytic pathway. Therefore, endosomal TLR7 is the only PRR that can activate innate signals during vaccination.
There are three types of influenza vaccines licensed in the United States today: the live attenuated influenza vaccine (LAIV) and the split and subunit trivalent inactivated vaccines (TIVs) (15, 23, 44). IV remains the most widely administered influenza vaccine, but its immunogenicity is reduced in young children and older adults (3, 6, 58). Formulation of TIV with adjuvants is one strategy to improve vaccine immunogenicity, but adjuvanted influenza vaccines are not licensed in the United States. Since vRNA is a natural ligand for TLR7, having these innate signals in TIVs may improve their immunogenicity. Indeed, the antibody responses of B6 mice immunized with the A(H1N1)pdm09 split vaccine were dependent on RNA (Fig. 5), as RNase treatment abrogated the immunogenicity of this vaccine (Fig. 6 to 7), suggesting that the presence of vRNA is important for the immunogenicity of the split vaccine.
Our findings highlight the important role of TLR7 signaling in vaccine design. In support of this idea, other studies have shown that TLR7 is important for the induction of a protective memory response against influenza whole inactivated vaccine (18, 34, 35), while split vaccines lacking intact RNAs are defective in inducing a protective antibody response (34). In addition, Co and colleagues showed that a seasonal split vaccine activated human T cells more efficiently than a subunit vaccine, which contains only the highly purified surface antigens HA and neuraminidase (NA) (11). In a separate study, Kasturi and colleagues showed that purified HA alone did not elicit virus-neutralizing antibody titers, whereas the addition of a TLR7 ligand to HA increased antibody production in mice (30). The additions of synthetic TLR7 agonist to subunit-, purified protein-, or mRNA-based vaccines were shown to enhance the immunogenicity of vaccines against a variety of targets (17, 29, 39, 53, 62). Collectively, these studies show that the presence of vRNA that engages TLR7 signaling contributes to the immunogenicity of commercially available TIVs.
Induction of protective, durable antibody responses is a desirable outcome of vaccination. Protective antibodies are generally considered to be the products of class-switched, highly specific IgG-producing ASCs and memory B cells generated from GCs. However, growing evidence suggests that IgM-producing memory B cells and ASCs also actively participate in adaptive immune responses (33, 46). After infection with IAV, we found that TLR7−/− mice had reduced levels of circulating serum IgM (Fig. 3A) and IgM+ ASCs, particularly in the long-term-memory compartment of the BM (Fig. 1C and 2C). This IgM+ ASC deficiency was even more pronounced following immunization with split influenza vaccine (Fig. 6B). In addition, the average spot size of TLR7−/− IgM+-secreting ASCs was significantly reduced (data not shown). Other groups have similarly reported that IgM production is reduced in the absence of TLR7/MyD88 signaling (16, 41, 47, 57). Fink and colleagues showed that TLR7−/− mice produced lower levels of IgM but not IgG neutralizing antibodies against vesicular stomatitis virus (VSV) (16). They also found that TLR7−/− VSV-specific B cells did not expand or differentiate as well as the controls. Kang and colleagues showed that MyD88−/− mice produced lower levels of circulating IgM and fewer IgM+ ASCs in the spleen and BM following influenza virus-like particle vaccination (28). The association between TLR7 signaling and IgM production is also apparent in human B-cell responses (5, 19, 21). The precise long-term consequence of reduced IgM production as a result of TLR7 deficiency remains unclear. A recent study by Pape and colleagues showed that IgM+ memory B cells survive longer than class-switched memory B cells (46). Thus, it is possible that TLR7-mediated IgM memory cell formation and maintenance are critical after the humoral memory provided by IgG wanes. Alternatively, since IgM antibodies are considered an early adaptive defense due to their polyreactivity, TLR7 signaling in IgM production may be important in limiting viral spread at an early phase of infection (13, 16, 43, 63).
It remains unclear if the impaired B-cell response of TLR7−/− mice in response to vaccination is due to B-cell intrinsic or extrinsic effects. However, growing evidences suggest that TLR7 signaling directly on B cells contributes to enhanced antibody response. Kasturi and colleagues showed that TLR7 expression by B cells is required for an effective antibody response against an adjuvanted ovalbumin antigen (30). In addition, Hou and colleagues showed that intrinsic TLR7 signaling on B cells and less so in dendritic cells (DCs) is important for antiviral antibody responses (24). Later, Browne, using conditional-knockout mice, demonstrated that intrinsic MyD88 expression by B cells controlled GC formation as well as an animal's ability to control a retroviral infection (8). MyD88 expression by DCs was also important for viral control, but not to the extent of its expression by B cells. Thus, intrinsic TLR signaling on B cells may augment the signaling through B-cell receptor and T-cell help to stimulate B-cell proliferation and differentiation, as previously suggested (37, 50).
Our data demonstrate that TLR7 signaling is particularly important for the immunogenicity of some currently available TIVs. We propose that engaging TLR7 in the context of vaccination is one strategy to improve the immunogenicity of influenza vaccines and that this could be especially beneficial in populations for which the immunogenicity of influenza vaccine is reduced.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kortney Gustin and Steve Lindstrom for the use of their reagents for this study. We also greatly appreciate Sanofi Pasteur for allowing us the use of their A/California/07/2009 X-179A (H1N1) monovalent split vaccine. V.J.-S. received a postgraduate stipend from the Oak Ridge Institute for Science and Education.
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the funding agency or the Centers for Disease Control and Prevention.
Footnotes
Published ahead of print 25 July 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Adachi O, et al. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143–150 [DOI] [PubMed] [Google Scholar]
- 2. Ahmed R, Oldstone MB, Palese P. 2007. Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic. Nat. Immunol. 8:1188–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ambrose CS, Levin MJ, Belshe RB. 2011. The relative efficacy of trivalent live attenuated and inactivated influenza vaccines in children and adults. Influenza Other Respi. Viruses 5:67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barchet W, et al. 2005. Dendritic cells respond to influenza virus through TLR7- and PKR-independent pathways. Eur. J. Immunol. 35:236–242 [DOI] [PubMed] [Google Scholar]
- 5. Bekeredjian-Ding I, Jego G. 2009. Toll-like receptors; sentries in the B-cell response. Immunology 128:311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belshe RB, et al. 2007. Live attenuated versus inactivated influenza vaccine in infants and young children. N. Engl. J. Med. 356:685–696 [DOI] [PubMed] [Google Scholar]
- 7. Boeglin E, et al. 2011. Toll-like receptor agonists synergize with CD40L to induce either proliferation or plasma cell differentiation of mouse B cells. PLoS One 6:e25542 doi:10.1371/journal.pone.0025542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Browne EP. 2011. Toll-like receptor 7 controls the anti-retroviral germinal center response. PLoS Pathog. 7:e1002293 doi:10.1371/journal.ppat.1002293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cao W, et al. 2011. Improving immunogenicity and effectiveness of influenza vaccine in older adults. Expert Rev. Vaccines 10:1529–1537 [DOI] [PubMed] [Google Scholar]
- 10. Clingan JM, Ostrow K, Hosiawa KA, Chen ZJ, Matloubian M. 2012. Differential roles for RIG-I-like receptors and nucleic acid-sensing TLR pathways in controlling a chronic viral infection. J. Immunol. 188:4432–4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Co MD, et al. 2009. In vitro evidence that commercial influenza vaccines are not similar in their ability to activate human T cell responses. Vaccine 27:319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Danishuddin M, Khan SN, Khan AU. 2009. Phylogenetic analysis of surface proteins of novel H1N1 virus isolated from 2009 pandemic. Bioinformation 4:94–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diamond MS, et al. 2003. A critical role for induced IgM in the protection against West Nile virus infection. J. Exp. Med. 198:1853–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doherty PC, Turner SJ, Webby RG, Thomas PG. 2006. Influenza and the challenge for immunology. Nat. Immunol. 7:449–455 [DOI] [PubMed] [Google Scholar]
- 15. Fichera E, Felnerova D, Mischler R, Viret JF, Glueck R. 2009. New strategies to overcome the drawbacks of currently available flu vaccines. Adv. Exp. Med. Biol. 655:243–252 [DOI] [PubMed] [Google Scholar]
- 16. Fink K, et al. 2006. Early type I interferon-mediated signals on B cells specifically enhance antiviral humoral responses. Eur. J. Immunol. 36:2094–2105 [DOI] [PubMed] [Google Scholar]
- 17. Fotin-Mleczek M, et al. 2011. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 34:1–15 [DOI] [PubMed] [Google Scholar]
- 18. Geeraedts F, et al. 2008. Superior immunogenicity of inactivated whole virus H5N1 influenza vaccine is primarily controlled by Toll-like receptor signalling. PLoS Pathog. 4:e1000138 doi:10.1371/journal.ppat.1000138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Glaum MC, et al. 2009. Toll-like receptor 7-induced naive human B-cell differentiation and immunoglobulin production. J. Allergy Clin. Immunol. 123:224–230.e4 [DOI] [PubMed] [Google Scholar]
- 20. Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98 [Google Scholar]
- 21. Hanten JA, et al. 2008. Comparison of human B cell activation by TLR7 and TLR9 agonists. BMC Immunol. 9:39 doi:10.1186/1471-2172-9-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heer AK, et al. 2007. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J. Immunol. 178:2182–2191 [DOI] [PubMed] [Google Scholar]
- 23. Hickling J, D'Hondt E. 2006. A review of production technologies for influenza virus vaccines, and their suitability for deployment in developing countries for influenza pandemic preparedness. World Health Organization, Geneva, Switzerland [Google Scholar]
- 24. Hou B, et al. 2011. Selective utilization of Toll-like receptor and MyD88 signaling in B cells for enhancement of the antiviral germinal center response. Immunity 34:375–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. 2009. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206:79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Itoh Y, et al. 2009. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460:1021–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jeisy-Scott V, et al. 2011. Increased MDSC accumulation and Th2 biased response to influenza A virus infection in the absence of TLR7 in mice. PLoS One 6:e25242 doi:10.1371/journal.pone.0025242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kang SM, et al. 2011. MyD88 plays an essential role in inducing B cells capable of differentiating into antibody-secreting cells after vaccination. J. Virol. 85:11391–11400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kastenmuller K, et al. 2011. Protective T cell immunity in mice following protein-TLR7/8 agonist-conjugate immunization requires aggregation, type I IFN, and multiple DC subsets. J. Clin. Invest. 121:1782–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kasturi SP, et al. 2011. Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470:543–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kawai T, Akira S. 2006. TLR signaling. Cell Death Differ. 13:816–825 [DOI] [PubMed] [Google Scholar]
- 32. Kilpinen S, Hurme M. 1998. Low CD3+CD28-induced interleukin-2 production correlates with decreased reactive oxygen intermediate formation in neonatal T cells. Immunology 94:167–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kopf M, Brombacher F, Bachmann MF. 2002. Role of IgM antibodies versus B cells in influenza virus-specific immunity. Eur. J. Immunol. 32:2229–2236 [DOI] [PubMed] [Google Scholar]
- 34. Koyama S, et al. 2010. Plasmacytoid dendritic cells delineate immunogenicity of influenza vaccine subtypes. Sci. Transl. Med. 2:25ra24 doi10.1126/scitranslmed.3000759 [DOI] [PubMed] [Google Scholar]
- 35. Koyama S, et al. 2007. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J. Immunol. 179:4711–4720 [DOI] [PubMed] [Google Scholar]
- 36. Lakadamyali M, Rust MJ, Zhuang X. 2004. Endocytosis of influenza viruses. Microbes Infect. 6:929–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lanzavecchia A, Sallusto F. 2007. Toll-like receptors and innate immunity in B-cell activation and antibody responses. Curr. Opin. Immunol. 19:268–274 [DOI] [PubMed] [Google Scholar]
- 38. Lopez CB, et al. 2004. TLR-independent induction of dendritic cell maturation and adaptive immunity by negative-strand RNA viruses. J. Immunol. 173:6882–6889 [DOI] [PubMed] [Google Scholar]
- 39. Lorenzi JC, et al. 2010. Intranasal vaccination with messenger RNA as a new approach in gene therapy: use against tuberculosis. BMC Biotechnol. 10:77 doi:10.1186/1472-6750-10-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lund JM, et al. 2004. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U. S. A. 101:5598–5603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meyer-Bahlburg A, Khim S, Rawlings DJ. 2007. B cell intrinsic TLR signals amplify but are not required for humoral immunity. J. Exp. Med. 204:3095–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Neumann G, Kawaoka Y. 2011. The first influenza pandemic of the new millennium. Influenza Other Respi. Viruses 5:157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ochsenbein AF, et al. 1999. Control of early viral and bacterial distribution and disease by natural antibodies. Science 286:2156–2159 [DOI] [PubMed] [Google Scholar]
- 44. Palese P. 2006. Making better influenza virus vaccines? Emerg. Infect. Dis. 12:61–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pang IK, Iwasaki A. 2011. Inflammasomes as mediators of immunity against influenza virus. Trends Immunol. 32:34–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. 2011. Different B cell populations mediate early and late memory during an endogenous immune response. Science 331:1203–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pasare C, Medzhitov R. 2005. Control of B-cell responses by Toll-like receptors. Nature 438:364–368 [DOI] [PubMed] [Google Scholar]
- 48. Reed L. J., Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 49. Reed C, et al. 2009. Estimates of the prevalence of pandemic (H1N1) 2009, United States, April-July 2009. Emerg. Infect. Dis. 15:2004–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ruprecht CR, Lanzavecchia A. 2006. Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur. J. Immunol. 36:810–816 [DOI] [PubMed] [Google Scholar]
- 51. Sambhara S, et al. 2001. Heterosubtypic immunity against human influenza A viruses, including recently emerged avian H5 and H9 viruses, induced by FLU-ISCOM vaccine in mice requires both cytotoxic T-lymphocyte and macrophage function. Cell. Immunol. 211:143–153 [DOI] [PubMed] [Google Scholar]
- 52. Seo S-U, et al. 2010. MyD88 signaling is indispensable for primary influenza A virus infection but dispensable for secondary infection. J. Virol. 84:12713–12722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shukla NM, et al. 2011. Toward self-adjuvanting subunit vaccines: model peptide and protein antigens incorporating covalently bound Toll-like receptor-7 agonistic imidazoquinolines. Bioorg. Med. Chem. Lett. 21:3232–3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Szretter KJ, Balish AL, Katz JM. 2006. Influenza: propagation, quantification, and storage. Curr. Protoc. Microbiol. Chapter 15:Unit 15G.1 [DOI] [PubMed] [Google Scholar]
- 55. Takeda K, Akira S. 2004. TLR signaling pathways. Semin. Immunol. 16:3–9 [DOI] [PubMed] [Google Scholar]
- 56. Taubenberger JK. 2006. The origin and virulence of the 1918 “Spanish” influenza virus. Proc. Am. Philos. Soc. 150:86–112 [PMC free article] [PubMed] [Google Scholar]
- 57. Tsukamoto Y, et al. 2009. Toll-like receptor 7 cooperates with IL-4 in activated B cells through antigen receptor or CD38 and induces class switch recombination and IgG1 production. Mol. Immunol. 46:1278–1288 [DOI] [PubMed] [Google Scholar]
- 58. Vesikari T, et al. 2011. Oil-in-water emulsion adjuvant with influenza vaccine in young children. N. Engl. J. Med. 365:1406–1416 [DOI] [PubMed] [Google Scholar]
- 59. Vukmanovic-Stejic M, Vyas B, Gorak-Stolinska P, Noble A, Kemeny DM. 2000. Human Tc1 and Tc2/Tc0 CD8 T-cell clones display distinct cell surface and functional phenotypes. Blood 95:231–240 [PubMed] [Google Scholar]
- 60. Wei J, et al. 2010. Influenza A infection enhances cross-priming of CD8+ T cells to cell-associated antigens in a TLR7- and type I IFN-dependent fashion. J. Immunol. 185:6013–6022 [DOI] [PubMed] [Google Scholar]
- 61. WHO 2009. 2009 Influenza (seasonal) fact sheet. WHO, Geneva, Switzerland [Google Scholar]
- 62. Wille-Reece U, Wu CY, Flynn BJ, Kedl RM, Seder RA. 2005. Immunization with HIV-1 Gag protein conjugated to a TLR7/8 agonist results in the generation of HIV-1 Gag-specific Th1 and CD8+ T cell responses. J. Immunol. 174:7676–7683 [DOI] [PubMed] [Google Scholar]
- 63. Zhou ZH, Tzioufas AG, Notkins AL. 2007. Properties and function of polyreactive antibodies and polyreactive antigen-binding B cells. J. Autoimmun. 29:219–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.