

Abstract
Following the consumption of contaminated food or water by a human host, the Vibrio cholerae bacterium produces virulence factors, including cholera toxin (CT), which directly causes voluminous diarrhea, producing cholera. A complex regulatory network controls virulence gene expression and responds to various environmental signals and transcription factors. Ultimately, ToxT, a member of the AraC/XylS transcription regulator family, is responsible for activating the transcription of the virulence genes. ToxT-regulated promoters all contain one or more copies of the toxbox, a 13-bp DNA sequence which ToxT recognizes. Nucleotides 2 through 7 of the toxbox sequence are well conserved and contain an invariant tract of four consecutive T nucleotides, whereas the remainder of the toxbox sequence is not highly conserved other than being A/T rich. The binding of ToxT to toxboxes is required to activate the transcription of virulence genes, and toxboxes in several virulence gene promoters have been characterized. However, the toxboxes required for the activation of transcription from the cholera toxin promoter PctxAB have not been identified. PctxAB contains a series of heptad repeats (GATTTTT), each of which matches the 5′ end of the toxbox consensus sequence and is a potential binding site for ToxT. Using site-directed mutagenesis and high-resolution copper-phenanthroline footprinting, we have identified the functional toxboxes required for the ToxT activation of PctxAB. Our findings suggest that ToxT binds to only two toxboxes within PctxAB, despite the presence of several other potential ToxT binding sites within the promoter. Both toxboxes are essential for DNA binding and the full activation of ctxAB transcription.
INTRODUCTION
Vibrio cholerae is a curved, Gram-negative, noninvasive bacterium responsible for the severe diarrheal disease cholera. V. cholerae is found predominately in coastal regions and is transmitted to humans by the ingestion of contaminated water (12). The resulting infection is characterized by voluminous fluid loss, leading to extreme dehydration if left untreated. Of the more than 200 V. cholerae serogroups present in the environment, only the O1 and O139 serogroups are associated with epidemic disease (34, 35). The O1 serogroup is further divided into the classical and El Tor biotypes based upon phenotypic differences (11, 35). The current cholera pandemic, caused by El Tor V. cholerae strains, has persisted since 1961 and is estimated to affect approximately 5 million people annually (11, 35).
The diarrhea characteristic of cholera is caused directly by the secretion of cholera toxin (CT) in the upper small intestine. CT is a classic AB5 toxin, containing one active A subunit and 5 binding B subunits, which form a pentameric ring structure (14, 38). CT binds to the GM1 ganglioside of epithelial cells in the upper small intestine, allowing the active subunit to be translocated into the cells, where it is activated by proteolysis (7, 23). The resulting active A1 subunit ADP ribosylates the regulatory G protein Gαs, which results in the constitutive activity of adenylate cyclase, increasing cyclic AMP (cAMP) levels within the cells and resulting in the secretion of sodium, chloride, and water into the lumen (38).
V. cholerae virulence gene expression is controlled by a complex network of transcription regulators that is historically referred to as the ToxR regulon because that protein was the first to be discovered (27, 32). However, the direct activator of virulence gene expression is ToxT, whose expression depends upon ToxR (17, 18, 21). A pair of integral membrane proteins, ToxR and TcpP, in association with their respective cofactors, ToxS and TcpH, binds to the promoter region upstream of toxT and activates its transcription (9, 16–18, 22). Once produced, ToxT activates the transcription of the virulence genes necessary for pathogenesis. These virulence genes include the ctxAB genes encoding CT, which are located within the genome of a lysogenic bacteriophage, CTXΦ (19, 22, 41–45).
ToxT is a 32-kDa member of the AraC/XylS family of proteins, having a 100-amino-acid family domain in the C terminus that contains two helix-turn-helix domains for DNA binding (13, 17, 40). The N-terminal 176 amino acids of ToxT form another domain, the ToxT N-terminal domain (NTD), which does not have significant sequence similarity to any other proteins. However, the ToxT NTD was proposed previously to be important for effector binding and dimerization (6, 37, 42, 43). The ToxT crystal structure revealed some structural similarity between the ToxT NTD and the AraC NTD, which is responsible for the binding of AraC to its effector, arabinose, and for AraC dimerization (26). ToxT was monomeric in the crystals used for structural studies, and there is significant evidence that ToxT binds DNA as a monomer (42–44). However, bacterial two-hybrid studies and LexA fusion experiments revealed that the ToxT NTD is capable of dimerization when separated from the C-terminal domain (CTD), and ToxT dimerization after DNA binding may be important for the transcription activation of some virulence genes (6, 33, 37). The ToxT crystal structure also contained a buried unsaturated fatty acid, cis-palmitoleic acid, which was proposed previously to be a negative ToxT effector (26). The addition of unsaturated fatty acids or bile to V. cholerae growth medium causes reductions in virulence gene expression levels (5, 15).
ToxT binds to 13-bp sequences called toxboxes, which are located upstream of all the genes whose transcription is activated by ToxT (43). Toxboxes are characterized by a well-conserved 5′ portion containing a poly(T) tract and a degenerate 3′ portion that is generally A/T rich. In addition to having somewhat degenerate sequences, toxboxes also vary in configuration and location relative to the transcriptional start site (2, 42–44). However, toxboxes are invariably located upstream of the −35 sequence recognized by the RNA polymerase (RNAP) σ70, suggesting that ToxT interacts with RNA polymerase α-subunit CTDs (α-CTDs) to activate transcription (3). The different configurations of toxboxes likely dictate various interactions with the two RNA polymerase α-CTDs (43).
The ToxT-responsive toxboxes at V. cholerae virulence genes have been identified at every virulence promoter except for arguably the most important one, ctxAB (42–44). Previous DNase I footprinting studies localized the ToxT binding region within Pctx to between positions −111 and −41 relative to the transcription start site (45). Within this region, there is a series of heptad repeats of GATTTTT, which fits the highly conserved 5′ segment of the toxbox consensus sequence (43). The numbers of these repeats vary among the O1 biotypes: classical strain O395 has 6 perfect direct repeats, whereas most El Tor strains have 3 direct repeats. A nested deletion analysis of the ctxAB promoter (PctxAB) determined that the region extending from the transcription start site upstream to position −76 was sufficient for full transcriptional activation by ToxT, correlating with the three heptad repeats proximal to the promoter being involved in ToxT binding (45). However, the exact binding sites remain unidentified, as there are several potential toxboxes within this sequence.
In this study, we used a combination of site-directed mutagenesis and high-resolution copper-phenanthroline (CP) footprinting to characterize the ToxT binding sites that control ctxAB transcription. The location of the toxboxes was further confirmed by electrophoretic mobility shift assays (EMSAs) that assessed the effects of toxbox mutations on DNA binding by ToxT. Our results suggest that there are two functional toxboxes located upstream of ctxAB that are required for the control of the promoter.
MATERIALS AND METHODS
V. cholerae strains and plasmids.
The strains used in this study were Vibrio cholerae classical biotype strain O395 and its ΔtoxT derivative (JW150) (4). PctxAB::lacZ fusions for β-galactosidase assays were constructed on plasmid pTL61T (25) in strain O395 and the ΔtoxT strain. The strains were grown at 37°C in Luria broth (LB) medium for cultures grown overnight or in LB adjusted to start at pH 6.5 at 30°C under inducing conditions. Promoter constructs of ctxAB were constructed by using wild-type (WT) O395 colonies as a template for PCR. All promoter constructs were cloned between the HindIII and XbaI sites of pTL61T (25). The antibiotic concentrations used were 100 μg/ml ampicillin and 100 μg/ml streptomycin. Plasmid sequences were confirmed by the University of Michigan DNA sequencing core and Genewiz. V. cholerae was transformed with plasmid DNA by electroporation using a Bio-Rad MicroPulser.
DNA manipulation.
Plasmids were purified by using Promega Wizard Plus miniprep kits. PCR was performed by using Taq DNA polymerase (Denville Scientific), as specified by the manufacturer, in an Eppendorf Mastercycler gradient thermocycler. Restriction enzymes were purchased from New England BioLabs and used as specified by the manufacturer.
β-Galactosidase assays.
Vibrio cholerae strains were grown overnight at 37°C in LB and then subcultured at a 1:40 dilution into fresh inducing medium and grown for 3 h at 30°C with vigorous aeration. Bacteria were then placed on ice with the addition of 0.5 mg/ml chloramphenicol. Assays were performed according to a procedure described previously by Miller (29).
Protein purification.
Maltose binding protein (MBP)-ToxT was purified from Escherichia coli strain JM109 with plasmid pMALC2e containing the ToxT-MBP construct. E. coli cells were grown overnight at 37°C and then subcultured 1:40 into fresh LB and grown at 37°C until the optical density at 600 nm (OD600) reached 0.5. The culture was induced for 3 h by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to 0.25 mM. Bacterial cells were collected by centrifugation and then resuspended in buffer containing 20 mM Tris (pH 8.0). The cells were French pressed, and the lysate was centrifuged at 12,000 rpm for 10 min. The supernatant was passed over an amylose column (New England BioLabs) by using a peristaltic pump. The column was washed with 20 mM Tris (pH 8.0) buffer three times before the protein was eluted with 20 1-ml fractions of a solution containing 20 mM Tris (pH 8.0) and 10 mM maltose. Samples were analyzed by SDS-PAGE; eluates containing MBP-ToxT were dialyzed against a solution containing 50 mM Na2HPO4 (pH 8.0), 10 mM Tris (pH 8.0), and 100 mM NaCl and then dialyzed again against the same solution with 20% glycerol; and aliquots were frozen at −70°C. The protein concentration was determined by using ThermoScientific protein assay reagent according to the manufacturer's directions.
Electrophoretic mobility shift assays.
DNA probes were produced by PCR using plasmid templates containing the appropriate promoter fragments with one unlabeled primer and one primer end labeled with γ-32P (Perkin-Elmer) by T4 polynucleotide kinase (New England BioLabs). The assay mixtures were set up in a final volume of 30 μl with various concentrations of ToxT-MBP; 10 μg/ml salmon sperm DNA; 100 ng of labeled DNA probe; and binding buffer with a final concentration of 10 mM Tris (pH 7.4), 1 mM EDTA, 100 mM KCl, 1 mM dithiothreitol (DTT), 0.3 mg/ml bovine serum albumin (BSA), and 10% glycerol. The binding reaction mixtures were incubated at 30°C for 30 min prior to loading into a 6% acrylamide gel at 4°C. Gels were dried and then analyzed by autoradiography.
CP footprinting.
CP footprinting was performed as previously described (42–44). Chemical cleavage was done in gel after the separation of free DNA and the bound ToxT-DNA complex by EMSAs. Polyhistidine-tagged ToxT was purified as previously described (45). The ratio of ToxT to DNA used was adjusted empirically such that approximately 50% of the labeled DNA formed a bound complex with ToxT. The sequence ladder was created by using a SequiTherm Excel II DNA sequencing kit (Epicentre) with the same 32P-end-labeled primer used make the PCR products for EMSAs to minimize offset reactions, according to the manufacturer's instructions.
RESULTS
General map of ToxT binding.
We began our investigation of the requirements for ToxT binding to PctxAB in V. cholerae by analyzing the DNA sequence. The most notable feature of PctxAB is the presence of heptad repeat sequences of GATTTTT (Fig. 1), which were proposed previously to be binding sites for transcriptional activators such as ToxR and ToxT (4, 24, 28, 30, 39, 45). The numbers of perfect heptad repeats differ among V. cholerae strains: V. cholerae strains of the classical biotype typically have six repeats, whereas V. cholerae strains of the El Tor biotype typically have only three. The GATTTTT repeat sequence is consistent with the 5′ end of the toxbox consensus sequence illustrated in Fig. 2 by a sequence logo (8, 36, 43). In classical strain O395, used in all of the experiments described here, there are six perfect heptad repeats, followed by one repeat having two substitutions proximal to the promoter (Fig. 1). Therefore, several potential toxboxes could be identified by sequence analysis, but experimentation was required to determine which toxboxes are functional at PctxAB.
Fig 1.

Map of the ctxAB promoter region. The black bar over the sequence indicates DNase I footprinting protection by ToxT (45). The ToxT binding sites toxbox 1 and toxbox 2, determined in this study by mutagenesis and copper-1,10-phenanthroline footprinting, are illustrated by arrows. The base pair at position −76 is boxed to indicate the endpoint of the minimal ctxAB construct that is activated by ToxT. Heptad repeats are numbered underneath the sequence and are indicated by dotted arrows; imperfect repeat 7 is indicated by a dashed arrow. The transcriptional start site is indicated by a bent arrow, and the putative −10 and −35 promoter elements are boxed.
Fig 2.
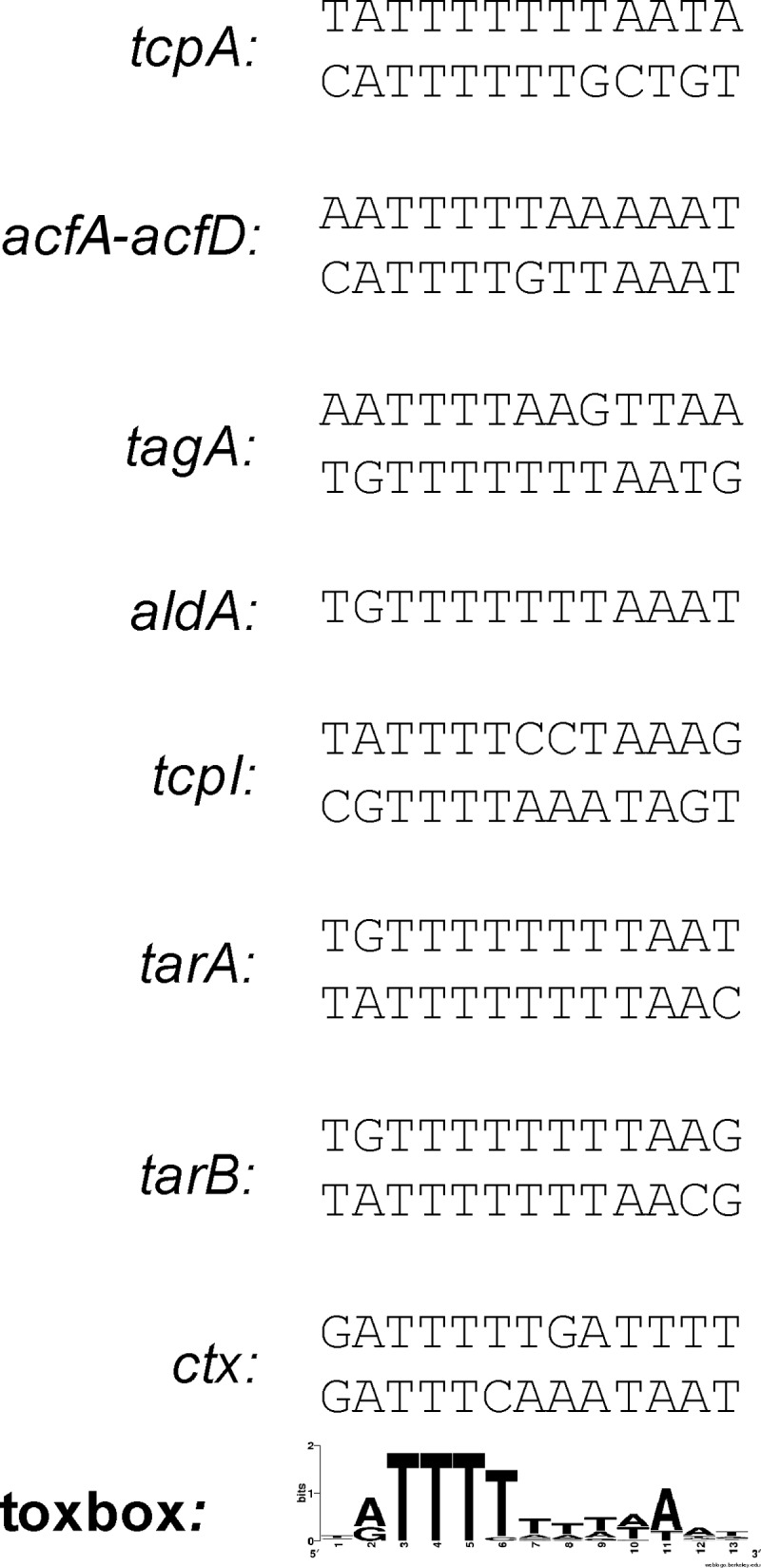
Alignment of ToxT binding sites. The toxbox sequences of ToxT-regulated genes are indicated. The gene names are identified on the left side, and the consensus sequence is shown at the bottom.
To pinpoint the location of the functional toxboxes within PctxAB, we performed site-directed mutagenesis of the heptad repeat sequences. In our initial analysis, we created two point mutations within each individual heptad repeat to produce a large effect on ToxT activity and clarify the most important repeat sequences. The poly(T) tract within a heptad repeat, corresponding to the highly conserved T tract present in toxboxes, was interrupted by mutating the fourth- and fifth-position nucleotides from thymidines to cytosines. The mutant promoter constructs were cloned into pTL61T (25), a vector containing a multicloning site upstream of a promoterless lacZ gene, allowing us to measure PctxAB activity in Miller units by β-galactosidase activity (29). These constructs were transformed into wild-type and ΔtoxT strains of classical V. cholerae strain O395, assessed for ToxT-dependent activity, and compared to the wild-type PctxAB::lacZ construct. In addition, we included a truncated mutant promoter, pJW211, which extends to position −76 relative to the transcription start site and thus includes truncated heptad repeat 4 and the promoter-proximal three perfect heptad repeats 5 to 7 (Fig. 1). Previous studies found that constructs extending to position −76 were fully activated by ToxT but that shorter constructs were not activated by ToxT (45), indicating that the DNA sequences required for ToxT activity are located between position −76 and the −35 box. As shown in Fig. 3, not only was the truncated promoter that extends only to position −76 activated by ToxT, the fold difference in transcription induced by ToxT was twice that of full-length PctxAB, indicating that this truncated promoter is fully functional.
Fig 3.

Effects of double point mutations of heptad repeats on ctxAB transcription. β-Galactosidase results from ctxAB::lacZ double point mutations are shown. Results from the WT full-length promoter strain are shown at the far left. The truncated promoter extending only to position −76 is marked as pJW211. The double point mutations are indicated in italics within the sequence. Heptad repeats are numbered and shown as dotted arrows; imperfect repeat 7 is indicated by a dashed arrow. The black bars indicate O395 WT toxT strains, and the gray bars indicate O395 ΔtoxT strains. The fold difference in β-galactosidase activities between the WT and ΔtoxT strains is labeled above each promoter. The asterisks indicate statistically significant fold differences in β-galactosidase results between O395 WT toxT PctxAB and O395 ΔtoxT PctxAB strains according to Student's t test (P < 0.03). Each experiment was repeated a minimum of three times, and the data shown are mean values, with the standard deviations indicated by error bars.
The results of our double-mutagenesis experiments provided the first evidence for the location of functional toxboxes at PctxAB (Fig. 3). The mutagenesis of repeats 1 and 2, at positions −97 and −96 (−97/−96) and positions −90/−89 relative to the transcriptional start site, caused no defects in ToxT-dependent transcription, which is consistent with previous work showing that only sequences downstream of position −76 are required for ToxT activity (45). The mutagenesis of repeat 3 at positions −83/−82 caused decreased transcription levels with or without ToxT; however, the fold difference between the wild-type and ΔtoxT strains was consistent with WT PctxAB constructs, indicating that ToxT could still function. In contrast to these results, the mutagenesis of repeat 4 at positions −76/−75 caused a complete loss of ToxT activity, suggesting that this sequence may be necessary for ToxT-dependent transcription activation. The mutagenesis of repeat 5 at positions −69/−68 also caused a complete loss of ToxT activity. Furthermore, the ToxT-independent transcriptional activity in this mutant strain was twice as high as that in the WT PctxAB strain, suggesting that this sequence may comprise part of a repressor binding site or may play a structural role that is favorable for ToxT-independent transcription when mutated. The mutagenesis of repeat 6 at positions −62/−61 reduced overall transcription levels but did not cause a significant defect in ToxT-dependent transcription, suggesting that it probably does not have a crucial role in ToxT binding. The mutagenesis of imperfect heptad repeat 7 at positions −55/−54, which has substitutions at the two 3′ nucleotides, resulted in significantly reduced ToxT activity, suggesting that it does have an important role. Finally, a region downstream of the heptad repeats, at positions −52/−51, was included in the mutagenesis analysis because of its rich A/T content, similar to the somewhat degenerate 3′ portion of the consensus toxbox sequence. Mutations at positions −52/−51 caused a complete loss of activation by ToxT, suggesting that this sequence is also necessary for ToxT activity, and therefore, this region was further analyzed in subsequent experiments.
The above-described results indicate that the ToxT activation of PctxAB transcription requires the region downstream of position −76 and are consistent with previous studies of PctxAB by Yu and DiRita (45). However, we now see that specific mutations at positions −76/−75, −69/−68, −55/−54, and −52/−51 abrogate ToxT activity. This suggests that the corresponding heptad repeats 4, 5, and 7 as well as the A/T-rich region immediately downstream of the repeats are necessary for ToxT activity.
Comprehensive site-directed mutagenesis of PctxAB.
To characterize more precisely which nucleotides are necessary for ToxT activity within PctxAB, single point mutations were created at each position ranging from positions −79 to −39, coinciding with the 5′ end of repeat 4 and extending through the A/T-rich region downstream of the heptad repeats (Fig. 1). Each A or T nucleotide was changed to a G or C, respectively, and each G or C was changed to an A. These mutant promoter constructs were cloned into pTL61T and transformed into wild-type strain O395 and its ΔtoxT derivative. Promoter activity was measured by β-galactosidase activity, and results are shown in Fig. 4.
Fig 4.

Effects of single point mutations on ctxAB transcription. β-Galactosidase results from ctxAB::lacZ single point mutations are shown. Results for full-length WT and truncated WT (pJW211) promoter constructs are shown at the far left. Individual mutation results correlate with the nucleotide underneath the x axis. Heptad repeats included for mutagenesis are numbered and indicated by dotted arrows; imperfect repeat 7 is illustrated by a dashed arrow. Black bars are O395 WT toxT strains, and white bars are O395 ΔtoxT strains. The asterisks indicate statistically significant differences in β-galactosidase results between O395 WT toxT PctxAB and O395 ΔtoxT PctxAB strains according to Student's t test (P < 0.03).
This analysis identified numerous individual base pairs that are important for ToxT activity. Any single point mutation within heptad repeat 5 abrogated the ToxT activation of PctxAB transcription, indicating that this sequence is essential for ToxT activity. Similarly, the mutagenesis of the T tract within heptad repeat 6 also abrogated ToxT activity. Surprisingly, the single-point mutagenesis of heptad repeat 4 had little effect on ToxT activity, with the exception of positions −75 and −76, which caused decreased ToxT activation when mutated. These results are consistent with the double-point mutagenesis studies described above, in which mutations at positions −76/−75 abrogated ToxT activity. However, these results also suggest that the remainder of repeat 4 is not important for ToxT activity.
The single-point mutagenesis of imperfect heptad repeat 7 did not cause significant defects in ToxT activity. However, the A/T-rich region downstream of heptad repeat 7 revealed that some of these base pairs are important for ToxT activity. Individual mutations between positions −52 and −45 caused significant defects in the ToxT-dependent transcription of PctxAB, suggesting that this region is important for ToxT function. Mutations between positions −43 and −41 caused significant defects in both ToxT-dependent and -independent transcription, indicating that these nucleotides may be more important for RNAP than ToxT.
Previous work with other ToxT-activated promoters characterized the toxbox as a somewhat degenerate 13-bp sequence with a conserved poly(T) tract near the 5′ end (Fig. 2) (42–44). The PctxAB mutagenesis experiments described here revealed a clear region required for ToxT activity between positions −72 and −59, comprising repeats 5 and 6, which we designate toxbox 1 (Fig. 1 [arrows], 3, and 4). Additionally, single-point mutagenesis of the A/T-rich region downstream of the heptad repeats caused some defects in activation, suggesting a second ToxT binding site, toxbox 2, between positions −58 and −46 (Fig. 1, 3, and 4), that apparently has less sequence specificity. Interestingly, the mutation at position −53, which changes a C nucleotide to the consensus toxbox T nucleotide at this position, resulted in elevated transcription levels (Fig. 4).
Copper-phenanthroline footprinting of ToxT at PctxAB.
To complement our genetic analysis and confirm the locations of ToxT binding, we performed in vitro DNA footprinting experiments. Previous studies using DNase I footprinting identified a region of ToxT protection from positions −111 to −41 upstream of PctxAB (45). However, DNase I footprinting is problematic at PctxAB due to the presence of numerous A tracts, which interfere with DNase I cleavage even in the absence of bound proteins. To achieve higher-resolution footprinting of ToxT on PctxAB, we performed a copper-1,10-phenanthroline (CP) footprinting analysis, which generates a higher-resolution image of the base pairs protected by ToxT. CP cleaves within the DNA minor groove, as does DNase I, but because CP is a much smaller molecule, it is much less sensitive to the sequence-specific narrowing of the minor groove and thus cleaves at every individual base pair. The CP footprinting technique was previously used to characterize ToxT binding at the tcpA, aldD, acfA, acfD, and tagA promoters (42–44).
The CP footprint of ToxT at PctxAB revealed two distinct regions of protection. The upstream region encompassing toxbox 1, spanning positions −72 to −60 and including heptad repeats 5 and 6, is very strongly protected by ToxT (Fig. 5). These data correlate well with the results of the mutagenesis experiments described above, suggesting that this sequence is an authentic toxbox that is required for DNA binding and PctxAB activation by ToxT. The second region of protection, within toxbox 2, ranges from positions −58 to −49 (ATTTCAAAT). This region includes imperfect heptad repeat 7 and the A/T-rich region directly downstream of the heptad repeats indicated to be important for ToxT activity by mutagenesis studies (Fig. 3 and 4). In general, the protection of this region was much weaker than the protection observed at toxbox 1. In particular, positions −51 and −52 were found to be important for ToxT activity and are somewhat protected in the CP footprint (Fig. 5, dots). Similarly, position −48, which is important for ToxT activity, is weakly protected in the footprint. However, in general, the footprinting of toxbox 2 is significantly weaker than that of toxbox 1. Although it is possible that ToxT binding to the major groove of toxbox 2 does not protect the minor groove, the weaker footprint is more likely due to weaker ToxT binding. These CP footprinting experiments were performed with both full-length PctxAB, which includes all seven heptad repeats, and the truncated promoter pJW211, and the results were essentially identical for both constructs (data not shown). Figure 5 shows the results for pJW211, which includes the PctxAB sequence to position −76 from the transcriptional start site and is comprised of the T tract of repeat 4 as well as repeats 5 to 7.
Fig 5.

CP footprinting of ToxT on PctxAB. Toxbox 1 and toxbox 2 are indicated by solid arrows with the correlating numerical position from the transcriptional start site. “C,” “T,” “A,” and “G” at the top left refer to the nucleotide lanes of the sequencing ladder. The dotted arrows and numbers to the left show the locations of the GATTTTT repeats, and the solid arrow indicates the position of the vector. The solid lines and black dots to the right of the toxboxes indicate the locations of ToxT-dependent transcriptional defects identified in the single-point-mutagenesis experiments. This footprint was created by using the pJW211 construct, which extends to position −76.
These results suggest that PctxAB contains two toxboxes, both of which are generally consistent with the previously described toxbox consensus sequence. The PctxAB toxboxes are also consistent with other ToxT-activated virulence genes in both number and relative distance from the transcriptional start site (42–44).
ToxT binding to wild-type and mutant PctxAB constructs.
The genetic and biochemical analyses described above narrowed down the region of ToxT binding to two specific binding sites that are consistent with the toxbox consensus sequence. However, the footprinting experiments showed a relatively weak protection of toxbox 2, calling into question whether it is truly a ToxT binding site or instead a region possibly important for contact between RNA polymerase and ToxT. To confirm that mutations of these designated toxboxes cause defects in DNA binding by ToxT, we performed EMSAs using DNA probes that contain the double point mutations created for the general mapping of ToxT binding (Fig. 3). ToxT binding to DNA was compared between wild-type PctxAB and the mutant promoter sequences (Fig. 6). In these experiments, the first lane of each gel contained a DNA probe only, and the subsequent lanes from left to right had increasing amounts of ToxT. As the ToxT concentration was increased in combination with the wild-type probe, two different shifted species were observed. This observation is consistent with one ToxT monomer occupying one toxbox at a lower ToxT concentration ([ToxT]) and then a second ToxT monomer occupying the second toxbox at a higher [ToxT], producing the slower-migrating species. In contrast, the mutant promoter sequences shown in Fig. 6A to D did not produce the slower-migrating band, even at the highest [ToxT], suggesting that only the nonmutated toxbox could be occupied. This result is evident with all the mutants that alter one of the two toxboxes that we identified by mutational analysis, verifying their importance for ToxT binding.
Fig 6.

ToxT binding to wild-type and mutant PctxAB constructs. Electrophoretic mobility shift assays (EMSAs) were carried out with each mutant PctxAB construct, as indicated below the right panel of each EMSA. Lane 1 of each gel is the free probe with no ToxT present. ToxT-MBP concentrations increase across the gel from left to right, as indicated by the black triangle. The ToxT-MBP concentrations used for each EMSA are 2.3 nM, 4.6 nM, 6.9 nM, 9.2 nM, 11.5 nM, and 13.8 nM.
To confirm that the abrogation of ToxT binding to toxboxes is specific to mutations within the identified toxboxes, we analyzed ToxT binding to a probe with mutations at positions −76/−75. These mutations are located within heptad repeat 4 upstream of toxbox 1 and caused a defect in ToxT-dependent transcription activation in β-galactosidase assays (Fig. 3). However, when results of EMSAs of the −76/−75 mutant probe were compared to the results of EMSAs of the wild-type probe, no difference was evident, suggesting that the defects in transcription activation caused by the −76/−75 mutations are perhaps due to other factors, such as a reduced RNA polymerase interaction with DNA, and not the result of decreased ToxT binding.
The above-described results are consistent with our designation of two toxboxes within PctxAB being correct. To test our hypothesis that the disruption of both toxboxes would eliminate ToxT binding, we performed EMSAs using probes with both toxboxes mutated, at positions −69/−68 and −55/−54 (Fig. 6F). A very weak shifted band was observed in these experiments, which did not significantly increase in intensity as the [ToxT] was increased. These results suggest that ToxT is unable to bind specifically to a probe having mutations in both toxboxes even at higher ToxT concentrations. This in vitro result is supported by in vivo β-galactosidase assays with the PctxAB::lacZ construct containing the double toxbox mutations in the WT and ΔtoxT O395 backgrounds, which produced 1,776 ± 22.79 and 1,755 ± 89.35 Miller units of activity, respectively.
DISCUSSION
The experiments described here were designed to characterize the DNA sequence requirements for ToxT to activate the transcription of ctxAB, resulting in the production of CT and, subsequently, diarrhea in cholera patients. Previous studies characterized the ToxT binding sites, or toxboxes, at several other known ToxT-activated promoters, but detailed information about the functional toxboxes at ctxAB, arguably the most important virulence locus in V. cholerae, remained lacking (Fig. 2) (42–44). The presence of GATTTTT heptad repeat sequences, each of which resembles the conserved 5′ portion of a toxbox (43), made the identification of the functional ToxT binding sites impossible without further experimentation. Double and single point mutations were made within the GATTTTT heptad repeats to identify which of the seven repeats within the classical V. cholerae ctxAB promoter are vital for transcription activation, and these results were verified by CP footprinting experiments and EMSAs using purified DNA and ToxT.
Double point mutations of heptad repeats 4, 5, 6, and 7, as well as the A/T-rich region downstream of the repeats, caused severe defects in ToxT-dependent transcriptional activity, strongly suggesting that these sequences are important for ToxT binding. In addition to abrogating the ToxT activation of ctxAB, the mutations of repeat 5 also increased ToxT-independent transcription levels. This result could be due to the disruption of an H-NS binding site previously identified by Stonehouse et al. (39), which would prevent the repression of ctxAB expression by H-NS. H-NS preferentially binds to A/T-rich regions such as this one, which cause DNA to be intrinsically curved, and the interruption of this stretch of nucleotides with a G or C may prevent H-NS from binding at nucleation sites and oligomerizing along the DNA (10, 31). Another possible explanation is that altering the DNA curvature may enhance the interaction of RNA polymerase with the promoter region, diminishing the requirement for ToxT to activate transcription. The difference in DNA curvature may also explain the decreased expression levels observed when heptad repeat 3 was mutated. In this case, the overall magnitude of transcription decreased, but the fold difference in expression levels between the wild-type and ΔtoxT strains was similar to that of wild-type PctxAB constructs, indicating that ToxT activity was not affected by the mutations.
The DNA sequence requirements for ToxT activity at ctxAB were determined at a higher resolution by using ctxAB::lacZ constructs with single point mutations in the region between positions −79 and −39. Individual point mutations within a region spanning positions −72 to −59 caused severe defects in ToxT-dependent activity, with the exception of positions −65 and −64. This 13-bp sequence, which we designated toxbox 1, is consistent with previously characterized ToxT binding sites in both sequence and proximity to the transcriptional start site (2, 42–44). Interestingly, there are no single point mutations within toxbox 1 that significantly increased ToxT-independent activity, unlike the double point mutation within heptad repeat 5. This finding suggests that a single nucleotide change from a thymidine to a cytosine is not enough to enhance ToxT-independent transcription by whatever mechanism is responsible for this effect. However, this does not rule out the possibility that mutations of nucleotides other than cytosine may be sufficient to enhance ToxT-independent transcription.
Unlike the mutations that led us to identify toxbox 1, the single point mutations that led us to identify toxbox 2 did not reveal such an obvious contiguous region important for ToxT binding. Only four mutations, at positions −53, −52, −51, and −48, caused statistically significant decreases in levels of ToxT-dependent transcription. This difference between the two toxboxes and ToxT sequence requirements can be visualized with the CP footprinting experiments. These results indicate two separate regions of DNA protection by ToxT: positions −72 to −60 (toxbox 1) and positions −58 to −49 (toxbox 2). These regions strongly correlate with the results from the mutagenesis experiments. Based on the strong correlation between the footprinting and mutagenesis results, we specify toxbox 2 as spanning the region between positions −58 and −46. This is consistent with some other ToxT-activated promoters in which toxboxes most proximal to the −35 promoter element are less specific in their sequence requirements than toxboxes located distally (42–44). Furthermore, the orientation and position of this toxbox most closely resembles that of the single toxbox at the aldA promoter, which produces relatively weak activation (41).
The sequences of each of the toxboxes identified within PctxAB fit the consensus sequence, although toxbox 2 has a variation at position 6, which is part of the conserved T tract in every other toxbox (43). This change from T to C in toxbox 2 could explain the weaker protection in footprinting experiments, and it is notable that the mutation of that position to the consensus T resulted in higher transcription levels.
The designation of the functional toxboxes was confirmed by EMSAs that compared ToxT binding to wild-type or double point mutant PctxAB DNA probes. Mutations that are within the identified toxboxes visibly altered ToxT binding compared to that of WT PctxAB. The absence of the second, slower-migrating ToxT-bound species suggests that ToxT could occupy only the nonmutated toxbox and is unable to bind to the mutated toxbox, supporting the in vivo transcriptional activation experiments with the PctxAB double point mutants. Additionally, double point mutations within both toxboxes resulted in the complete abrogation of ToxT binding and transcription activation in vivo, as expected. The EMSA results also support the hypothesis that mutations within heptad repeat 4 do not disrupt the ToxT binding region, as suggested by the results shown in Fig. 3. Instead, this region of PctxAB may be important for the interaction of ToxT bound to toxbox 1 with the α-CTDs of RNA polymerase, and the mutations negatively affected this interaction, resulting in lower levels of transcription activation in the presence of ToxT. One other observation from the EMSAs is that binding to a single DNA fragment by a second ToxT monomer, producing the higher complex, occurs at a relatively low [ToxT], with substantial amounts of free DNA remaining in the reaction mixture. This finding suggests that ToxT binding to PctxAB may be cooperative. The role of ToxT dimerization in DNA binding remains unclear, as monomers are clearly able to bind to individual toxboxes (42, 43), and most ToxT is monomeric in solution (M. Bellair and J. H. Withey, unpublished data). However, these results suggest that after the initial binding to one toxbox by a ToxT monomer, binding to the second toxbox may be enhanced, potentially by ToxT dimerization.
The PctxAB toxboxes are located upstream of the −35 promoter element, classifying it as a class I promoter (3). This is also the case for the toxboxes identified in every other ToxT-activated promoter that has been characterized (42–44). Class I promoters require an interaction between the activator protein and the α-CTD of RNAP for transcriptional activation (3). Because two toxboxes were identified within PctxAB, we hypothesize that there is a specific interaction between two ToxT monomers and two α-CTDs. Our previously described ToxT and α-CTD interaction models proposed that when two toxboxes are present, there are two distinct points of interaction between individual ToxT monomers and the α-CTD (43). An alternative hypothesis is that one ToxT monomer contacts RNA polymerase and that the other ToxT monomer stabilizes this interaction, possibly by ToxT dimerization. The mutagenesis experiments illustrated that the mutation of one toxbox, particularly toxbox 1, is sufficient to decrease the overall level of transcription, suggesting that ToxT must occupy both toxboxes for full activation. The weak protection conferred by ToxT to toxbox 2 raises the possibility that the interaction with the α-CTD may be important for enhanced binding to this sequence by ToxT. Another possible explanation for the weak footprint observed at toxbox 2 is that a positive ToxT effector, such as bicarbonate, is required to increase the binding specificity (1); future experiments will determine if either of these possibilities is indeed the case.
In this study, we focused on V. cholerae classical biotype strain O395, which contains six perfect GATTTTT repeats and one imperfect repeat. However, other strains possess various numbers of repeats. El Tor biotype strains generally contain only three of the heptad repeats but otherwise retain the same DNA sequence as that of classical strain O395 at PctxAB. The absence of the distal heptad repeats does not negatively impact ToxT-activated transcription, as the toxboxes that we identified in O395 encompass the heptad repeats that are most proximal to the transcriptional start site and would be included in the El Tor promoter region. ToxT was also not observed to bind to the distal heptad repeats in the footprinting experiments (data not shown). The significance of the distal heptad repeats in V. cholerae strains of the classical biotype is still unclear, but they may play a role in H-NS binding, may contribute to the curvature of the DNA, or could be important for the ToxR-mediated activation of PctxAB in the presence of bile, which was observed only for strains of the classical biotype (20).
In summary, we have characterized the specific sequence requirements for binding to PctxAB and transcription activation by ToxT. The DNA sequences of the identified toxboxes are consistent with the consensus toxbox in that they are degenerate but contain the 5′ poly(T) tracts common among all known ToxT DNA binding sites (42–44). The toxboxes in PctxAB are also consistent with other ToxT-activated promoters in their positioning relative to the transcriptional start site (2, 42–44). Although ToxT is a flexible transcription activator in regard to sequence requirements, configuration, and the number of binding sites, it has specific requirements for the activation of PctxAB, and a single mutation within one the two toxboxes is enough to severely decrease transcription activation by ToxT.
ACKNOWLEDGMENTS
We thank the members of the Withey and Neely laboratories for helpful discussions.
This work was supported by NIH grant 1K22AI071011 from the NIAID and by Wayne State University laboratory start-up funds.
Footnotes
Published ahead of print 20 July 2012
REFERENCES
- 1. Abuaita BH, Withey JH. 2009. Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect. Immun. 77:4111–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bellair M, Withey JH. 2008. Flexibility of Vibrio cholerae ToxT in transcription activation of genes having altered promoter spacing. J. Bacteriol. 190:7925–7931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Busby S, Ebright RH. 1994. Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell 79:743–746 [DOI] [PubMed] [Google Scholar]
- 4. Champion GA, Neely MN, Brennan MA, DiRita VJ. 1997. A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Mol. Microbiol. 23:323–331 [DOI] [PubMed] [Google Scholar]
- 5. Chatterjee A, Dutta PK, Chowdhury R. 2007. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect. Immun. 75:1946–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Childers BM, et al. 2007. Identification of residues critical for the function of the Vibrio cholerae virulence regulator ToxT by scanning alanine mutagenesis. J. Mol. Biol. 367:1413–1430 [DOI] [PubMed] [Google Scholar]
- 7. Chinnapen DJ, Chinnapen H, Saslowsky D, Lencer WI. 2007. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 266:129–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DiRita VJ, Parsot C, Jander G, Mekalanos JJ. 1991. Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 88:5403–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dorman CJ. 2004. H-NS: a universal regulator for a dynamic genome. Nat. Rev. Microbiol. 2:391–400 [DOI] [PubMed] [Google Scholar]
- 11. Dziejman M, et al. 2002. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc. Natl. Acad. Sci. U. S. A. 99:1556–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Faruque SM, Albert MJ, Mekalanos JJ. 1998. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 62:1301–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL. 1997. Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61:393–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gill DM. 1976. The arrangement of subunits in cholera toxin. Biochemistry 15:1242–1248 [DOI] [PubMed] [Google Scholar]
- 15. Gupta S, Chowdhury R. 1997. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect. Immun. 65:1131–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hase CC, Mekalanos JJ. 1998. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A. 95:730–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Higgins DE, DiRita VJ. 1994. Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae. Mol. Microbiol. 14:17–29 [DOI] [PubMed] [Google Scholar]
- 18. Higgins DE, Nazareno E, DiRita VJ. 1992. The virulence gene activator ToxT from Vibrio cholerae is a member of the AraC family of transcriptional activators. J. Bacteriol. 174:6974–6980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hulbert RR, Taylor RK. 2002. Mechanism of ToxT-dependent transcriptional activation at the Vibrio cholerae tcpA promoter. J. Bacteriol. 184:5533–5544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hung DT, Mekalanos JJ. 2005. Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT-independent manner. Proc. Natl. Acad. Sci. U. S. A. 102:3028–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krukonis ES, DiRita VJ. 2003. DNA binding and ToxR responsiveness by the wing domain of TcpP, an activator of virulence gene expression in Vibrio cholerae. Mol. Cell 12:157–165 [DOI] [PubMed] [Google Scholar]
- 22. Krukonis ES, Yu RR, DiRita VJ. 2000. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol. Microbiol. 38:67–84 [DOI] [PubMed] [Google Scholar]
- 23. Lencer WI, Saslowsky D. 2005. Raft trafficking of AB5 subunit bacterial toxins. Biochim. Biophys. Acta 1746:314–321 [DOI] [PubMed] [Google Scholar]
- 24. Li CC, Crawford JA, DiRita VJ, Kaper JB. 2000. Molecular cloning and transcriptional regulation of ompT, a ToxR-repressed gene in Vibrio cholerae. Mol. Microbiol. 35:189–203 [DOI] [PubMed] [Google Scholar]
- 25. Linn T, St Pierre R. 1990. Improved vector system for constructing transcriptional fusions that ensures independent translation of lacZ. J. Bacteriol. 172:1077–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lowden MJ, et al. 2010. Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc. Natl. Acad. Sci. U. S. A. 107:2860–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matson JS, Withey JH, DiRita VJ. 2007. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect. Immun. 75:5542–5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mekalanos JJ, et al. 1983. Cholera toxin genes: nucleotide sequence, deletion analysis and vaccine development. Nature 306:551–557 [DOI] [PubMed] [Google Scholar]
- 29. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 30. Miller VL, Mekalanos JJ. 1985. Genetic analysis of the cholera toxin-positive regulatory gene toxR. J. Bacteriol. 163:580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Navarre WW, et al. 2006. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 313:236–238 [DOI] [PubMed] [Google Scholar]
- 32. Peterson KM, Mekalanos JJ. 1988. Characterization of the Vibrio cholerae ToxR regulon: identification of novel genes involved in intestinal colonization. Infect. Immun. 56:2822–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prouty MG, Osorio CR, Klose KE. 2005. Characterization of functional domains of the Vibrio cholerae virulence regulator ToxT. Mol. Microbiol. 58:1143–1156 [DOI] [PubMed] [Google Scholar]
- 34. Reidl J, Klose KE. 2002. Vibrio cholerae and cholera: out of the water and into the host. FEMS Microbiol. Rev. 26:125–139 [DOI] [PubMed] [Google Scholar]
- 35. Sack DA, Sack RB, Nair GB, Siddique AK. 2004. Cholera. Lancet 363:223–233 [DOI] [PubMed] [Google Scholar]
- 36. Schneider TD, Stephens RM. 1990. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18:6097–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shakhnovich EA, Hung DT, Pierson E, Lee K, Mekalanos JJ. 2007. Virstatin inhibits dimerization of the transcriptional activator ToxT. Proc. Natl. Acad. Sci. U. S. A. 104:2372–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spangler BD. 1992. Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol. Rev. 56:622–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stonehouse EA, Hulbert RR, Nye MB, Skorupski K, Taylor RK. 2011. H-NS binding and repression of the ctx promoter in Vibrio cholerae. J. Bacteriol. 193:979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tobes R, Ramos JL. 2002. AraC-XylS database: a family of positive transcriptional regulators in bacteria. Nucleic Acids Res. 30:318–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Waldor MK, Mekalanos JJ. 1996. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272:1910–1914 [DOI] [PubMed] [Google Scholar]
- 42. Withey JH, DiRita VJ. 2005. Activation of both acfA and acfD transcription by Vibrio cholerae ToxT requires binding to two centrally located DNA sites in an inverted repeat conformation. Mol. Microbiol. 56:1062–1077 [DOI] [PubMed] [Google Scholar]
- 43. Withey JH, DiRita VJ. 2006. The toxbox: specific DNA sequence requirements for activation of Vibrio cholerae virulence genes by ToxT. Mol. Microbiol. 59:1779–1789 [DOI] [PubMed] [Google Scholar]
- 44. Withey JH, DiRita VJ. 2005. Vibrio cholerae ToxT independently activates the divergently transcribed aldA and tagA genes. J. Bacteriol. 187:7890–7900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu RR, DiRita VJ. 2002. Regulation of gene expression in Vibrio cholerae by ToxT involves both antirepression and RNA polymerase stimulation. Mol. Microbiol. 43:119–134 [DOI] [PubMed] [Google Scholar]