

Abstract
We examined whether tachyzoite proliferation in the brains of immunocompetent hosts during the chronic stage of infection with Toxoplasma gondii induces production of IgG antibodies that recognize parasite antigens different from those recognized by the antibodies of infected hosts that do not have tachyzoite growth. For this purpose, two groups of CBA/J mice, which display continuous tachyzoite growth in their brains during the later stage of infection, were infected, and one group received treatment with sulfadiazine to prevent tachyzoite proliferation during the chronic stage of infection. T. gondii antigens recognized by the IgG antibodies from these two groups of mice were compared using immunoblotting following separation of tachyzoite antigens by two-dimensional gel electrophoresis. Several antigens, including the microneme protein MIC2, the cyst matrix protein MAG1, and the dense granule proteins GRA4 and GRA7, were commonly recognized by IgG antibodies from both groups of mice. There were multiple antigens recognized mostly by IgG antibodies of only one group of mice, either with or without cerebral tachyzoite growth. The antigens recognized only by or mostly by the antibodies of mice with cerebral tachyzoite growth include MIC6, the rhoptry protein ROP1, GRA2, one heat shock protein HSP90, one (putative) HSP70, and the myosin heavy chain. These results indicate that levels of IgG antibody to only selected T. gondii antigens increase in association with cerebral tachyzoite proliferation (reactivation of infection) in immunocompetent hosts with chronic infection.
INTRODUCTION
Toxoplasma gondii is an obligate intracellular protozoan parasite that can infect many warm-blooded mammals, including humans. Acute infection is characterized by proliferation of tachyzoites in a variety of cells throughout the body and can cause various diseases, such as lymphadenitis and congenital infection of fetuses (24). Gamma interferon (IFN-γ)-mediated immune responses limit proliferation of tachyzoites, but the parasite establishes a chronic infection by forming cysts which can contain hundreds to thousands of bradyzoites, primarily in the brain. Chronic infection with T. gondii is one of the most common parasitic infections in humans. It is estimated that 500 million to 2 billion people worldwide are chronically infected with this parasite (5, 10). Despite the fact that such a large number of people are infected and most likely harbor T. gondii cysts in their brains (29), the clinical importance of this chronic infection remains largely unexplored and therefore unappreciated.
Recent studies demonstrated increased Toxoplasma IgG, but not IgM, antibody levels in sera of patients with recent onset of schizophrenia (22, 39). These schizophrenia patients do not appear to be in the acute stage of acquired infection with T. gondii, since both IgM and IgG antibody titers increase during this stage of infection (6). Therefore, the patients appear to be in a specific condition of chronic infection that causes an increase only in IgG antibody titers. A correlation between chronic T. gondii infection and cryptogenic epilepsy has also been reported (34), and Toxoplasma IgG antibody levels were greater in the patients than controls in this case as well (34).
We recently addressed what condition causes an increase in only the IgG antibody levels during chronic T. gondii infection using immunocompetent mice. Our studies revealed that an occurrence of tachyzoite proliferation in the brain during the chronic stage of infection causes an increase in Toxoplasma IgG antibody titers but not in IgM antibody titers in the sera (33). Of interest, the IgG antibody titers significantly correlated with amounts of mRNA for tachyzoite-specific SAG1 in the brains of chronically infected mice (33). Therefore, it appears that cerebral proliferation of tachyzoites during the chronic stage of infection provides sufficient stimulation to enhance production of IgG antibodies to the parasite in immunocompetent hosts. Tachyzoites and bradyzoites express antigens specific to each stage of the parasite in addition to those expressed in both stages (21), and infected hosts produce antibodies that recognize different T. gondii antigens in the acute and chronic stages of infection (11, 19, 37). Since T. gondii exists mostly as bradyzoites within tissue cysts during the chronic stage of infection, it is possible that an occurrence of cerebral tachyzoite growth in this stage induces production of IgG antibodies that recognize T. gondii antigens different from those recognized by the antibodies of infected hosts that do not have such tachyzoite proliferation. To address this possibility, in the present study, we examined T. gondii antigens recognized by IgG antibodies of mice with and without active proliferation of tachyzoites in their brains during the chronic stage of infection.
MATERIALS AND METHODS
Mice and infection with T. gondii.
T. gondii has three predominant clonal genotypes (types I, II, and III) (9, 16, 18), and type II constitutes a majority of clinical cases of toxoplasmosis and asymptomatic infections in humans in North America and Europe (2, 9, 18). Therefore, the ME49 strain, a type II strain, was used for this study. CBA/J mice were chosen for a murine model, since they are one of the strains susceptible to chronic infection with the type II parasite and display active proliferation of tachyzoites in their brains during the later stage of infection (7, 35). Acute inflammatory changes are noted in the brain but not in the lungs, livers, spleens, or kidneys of susceptible mice during the chronic stage of infection (2 months after infection) (36). Female CBA/J mice (Jackson Laboratories, Bar Harbor, ME) were orally infected by gavage with 10 cysts of the ME49 strain (20). Oral infection is the natural route of infection of this parasite. Cysts were obtained from the brains of chronically infected female Swiss-Webster mice (40). Swiss-Webster mice were from Taconics (Germantown, NY). One group of CBA/J mice (n = 8) received sulfadiazine (400 mg/liter) in drinking water beginning at 3 weeks after infection for the entire period of the experiment to prevent proliferation of tachyzoites during the chronic stage of infection. Another group of mice (n = 12) did not receive the treatment. Sera were obtained from the sulfadiazine-treated and untreated mice at 2 months (8 to 9 weeks) after infection. As a control, sera were also obtained from uninfected mice (n = 5). Mice were housed in a specific-pathogen-free condition and were 2 to 3 months old when used. Uninfected mice were older than 2 to 3 months to be approximately age matched with chronically infected animals.
Quantification of mRNA for tachyzoite-specific SAG1, bradyzoite-specific BAG1, and other T. gondii antigens in the brain.
At 2 months after infection, RNA was isolated from brains of CBA/J mice with and without sulfadiazine treatment using RNA STAT-60 (TEL-TEST “B,” Inc., Friendswood, TX), and cDNA was synthesized using 1 μg of RNA (20). To quantify tachyzoites in their brains, cDNA was applied for real-time PCR to measure the amounts of mRNA for tachyzoite-specific SAG1 using a StepOnePlus thermal cycler (Applied Biosystems, Foster City, CA) with the following primers and probe: CACAGAGTTGTATGGTCACAGTGA (forward), GCACCGTAGGAGCACCTT (reverse), and 6-carboxyfluorescein (FAM)-TCGGTCGTCAATAATG-minor groove binder (MGB)-nonfluorescent quencher (NFQ) (probe) (33). Bradyzoite-specific BAG1 mRNA levels were measured as described previously (38). mRNA levels for T. gondii ROP1, GRA2, GRA7, and MAG1 were also measured with the following primers and probes: CCGCCGGTGGCTCTAG (forward), TTCGGAGGCGGATTCATTAGTG (reverse), and CCCCTCCCCTTCTGCA (probe) for ROP1; GCCAAAGAAGCAGCTGGAA (forward), CTCCACATTCGCGAGTTTCTTG (reverse), and ACGGTCACCATGCCCC (probe) for GRA2; CACGAGACGAAAGGGTGGTT (forward), GCCGCTGTTCTCGACAAAGA (reverse), and CCTGGCAGCATCACGT (probe) for GRA7; and CTGACACGCGCACTGAAG (forward), CCTCAATCGGTGCCATATCCT (reverse), and CAGACGGGCATCCAT (probe) for MAG1. All probes are labeled with FAM at the 5′ ends and MGB-NFQ at the 3′ ends. All primers and probes were from Applied Biosystems. The quantification of mRNA was normalized to the β-actin level, which was measured using a commercial kit (Applied Biosystems).
Preparation of tachyzoite antigens.
Tachyzoites of the ME49 strain were maintained by serial passages in human foreskin fibroblasts (HFF) (HFF-1; American Type Culture Collection, Manassas, VA) maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen, Camarillo, CA) supplemented with 10% fetal bovine serum (Invitrogen), 1% l-glutamine (Invitrogen), 20 μg/ml gentamicin (Gemini Bioproducts, West Sacramento, CA), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). To prepare tachyzoite antigens, infected monolayers were scraped at 2 days after a passage, which is the time that the majority of HFF were holding large numbers of intracellular tachyzoites. The harvested infected cells were ruptured by being passed through a 27-gauge needle, and tachyzoites were then purified from the suspension by being filtered through a 3-μm-pore membrane (Whatman, Piscataway, NJ). The purified tachyzoites were washed in DMEM by centrifugation at 1,400 rpm for 10 min three times. Thereafter, the tachyzoites were solubilized by suspending them in sample buffer containing 8 M urea, 50 mM dithiothreitol, 2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 0.2% Bio-Lyte 3/10 ampholyte solution, and 0.001% bromophenol blue (Bio-Rad, Hercules, CA) at a concentration of 6.67 × 108 parasites/ml and stored at −80°C. Multiple tachyzoite antigen preparations were then pooled, divided into 150-μl aliquots, and stored at −80°C until used.
2-DE.
All electrophoresis equipment and supplies for two-dimensional gel electrophoresis (2-DE) were from Bio-Rad Inc. (Hercules, CA) unless otherwise described. Solubilized tachyzoite antigen preparations in sample buffer were thawed and each diluted with 150 μl of sample buffer to bring the final concentration of each sample to 1 × 108 parasite equivalents per 300 μl (6). To separate the proteins in the first dimension by isoelectric focusing (IEF), each sample was applied to a ReadyStrip pH 3 to 10 immobilized pH gradient (IPG) strip and allowed to incubate at room temperature overnight. Strips were then placed in the PROTEAN IEF cell apparatus and covered with mineral oil to prevent drying. Current was applied to IPG strips until it reached 50,000 V · h. Strips were then drained of excess mineral oil and stored at −80°C overnight. Before subjecting the focused IPG strips to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the strips were thawed and soaked in two equilibration buffers, each for 10 min while shaking. Equilibration buffer I contained 375 mM Tris-HCl (pH 8.8), 6 M urea, 2% SDS, and 2% dithiothreitol. Equilibration buffer II contained 375 mM Tris-HCl (pH 8.8), 6 M urea, 2% SDS, and 2.5% iodoacetamide. SDS-PAGE was conducted using the PROTEAN II Xi cell and 10% acrylamide gels (Jule Biotechnologies, Milford, CT). IPG strips were attached to acrylamide gels using agarose containing bromophenol blue. Both gels were then placed in the PROTEAN II Xi cell. Upper and lower buffer reservoirs were filled with SDS running buffer containing 0.1% SDS (Fisher, Waltham, MA), 192 mM glycine (Fisher), and 25 mM Tris (Invitrogen, Carlsbad, CA). Electrophoresis was carried out at 16 mA/gel for 30 min, followed by 24 mA/gel until proteins migrated to the bottom of the gel. In some experiments, gels were stained with a Coomassie brilliant blue solution containing 10% acetic acid (Sigma, St. Louis, MO), 40% methanol (Fisher), and 0.1% Coomassie brilliant blue R250 (Bio-Rad). Thereafter, the gels were washed several times with destaining solution containing 10% acetic acid and 40% methanol. The gels were then destained overnight in 150 ml of 5% acetic acid. The gels were scanned with an HP Scanjet G4050 (Hewlett-Packard, Palo Alto, CA) and then subjected to immunoblotting or stored in 5% acetic acid.
Immunoblotting.
Transfer of proteins separated by 2-DE to a nitrocellulose membrane was carried out as described previously (8). Prior to transfer, Coomassie-stained or unstained gels were soaked in a buffer containing 50 mM Tris (pH 7.5) and 1% SDS (Fisher) for 1 h. The gel and nitrocellulose membrane were then soaked in transfer buffer containing 25 mM Tris, 192 mM glycine, and 20% (vol/vol) methanol for 15 min. Transfer of separated proteins from the 2-DE gels to nitrocellulose membranes was carried out using a Trans-Blot electrophoretic transfer cell (Bio-Rad) at 10 V at 4°C for 12 h. After transfer, membranes were washed with Tris-buffered saline containing 0.1% Tween 20 (Fisher) (TBS-T) and incubated with 20 ml of blocking buffer containing 0.5% enhanced chemiluminescence (ECL) blocking agent (GE Healthcare, Piscataway, NJ) in TBS-T for 1 h at room temperature on an orbital shaker. Thereafter, membranes were washed in TBS-T and incubated with 20 ml of pooled sera from uninfected or infected mice at a 1:4,000 dilution in blocking buffer at room temperature for 1 h. Membranes were then washed in TBS-T and incubated in a 1:2,000 dilution of peroxidase-conjugated goat anti-mouse IgG (γ chain-specific) antibodies (Invitrogen) at room temperature for 1 h. After incubation, membranes were thoroughly washed in TBS-T and incubated in Supersignal West Pico chemiluminescent substrate (Thermo, Waltham, MA) for 5 min. The volume of substrate was 0.1 ml/cm2 of membrane. After developing in substrate, the membranes were imaged using a LAS-4000 image analyzer (Fuji Film, Tokyo, Japan).
Determination of the identity of T. gondii antigens using mass spectrometry.
After identification of protein spots corresponding to the antigens recognized by IgG antibodies on immunoblots, those protein spots were excised from a Coomassie-stained gel and the samples were submitted to the University of Kentucky Mass Spectrometry Facility to determine the identities of these antigens. The gel pieces were digested with trypsin, and liquid chromatography-electrospray ionization-tandem mass spectrometry was performed using a ThermoFinnigan LTQ. Resulting tandem mass spectrometry spectra were searched against proteins in the ToxoDB database (http://toxodb.org/toxo/) using the Mascot search engine (Matrix Science).
Dot blotting with recombinant dense granule protein GRA2.
The construction of the pUET-GRA2 plasmid expressing amino acids 21 to 185 of GRA2 (protein with a deletion of its signal peptide) of type II T. gondii (the ME49 strain) and the purification of the corresponding recombinant protein from Escherichia coli were performed by the Proteogenix company (Oberhausbergen, France), similarly to what was described previously by Golkar et al. (15). One microliter of either a solution (15.6 μg/ml) of recombinant pUET-GRA2 fusion protein (rGRA2) or pUET peptide was deposited onto 5-mm-diameter discs of a nitrocellulose membrane. The samples were allowed to dry for 2 to 3 min. The membranes were then blocked and incubated with 2-fold serial dilutions (1:1,000 to 1:16,000) of pooled sera of an experimental group or individual mouse serum at a 1:20,000 dilution for immunoblotting as described in “Immunoblotting” above.
Statistical analysis.
Levels of significance for amounts of mRNA for T. gondii antigens in the brain were determined by a Student t test, Mann-Whitney test, or one-way analysis of variance (ANOVA) with Newman-Keuls posttest. Differences which provided a P value of <0.05 were considered significant.
RESULTS
T. gondii antigens recognized by IgG antibodies of mice treated with sulfadiazine to prevent tachyzoite growth during the chronic stage of infection.
Antigen preparation of tachyzoites of the ME49 strain was applied for 2-DE and the gels were stained with Coomassie brilliant blue. Numerous protein spots were observed in the range of pI 3 to 10 with various molecular masses (Fig. 1B). We subjected these proteins separated by 2-DE to immunoblotting to determine T. gondii antigens recognized by IgG antibodies of chronically infected CBA/J mice treated with sulfadiazine to prevent tachyzoite proliferation during the chronic stage of infection. Tachyzoite-specific SAG1 mRNA was undetectable or detected at very low levels in the brains of these animals (Fig. 2), confirming that tachyzoite growth in their brains was efficiently inhibited by sulfadiazine treatment. In contrast, bradyzoite-specific BAG1 mRNA was detected in their brains, indicating that these mice were in the chronic stage of infection (Fig. 2). The immunoblot with pooled sera of these animals revealed six spots within a range of molecular masses, 150 to 25 kDa, clearly recognized by their IgG antibodies (indicated as C1 to C6 in Fig. 1A). There were also several spots below 25 kDa recognized by the antibodies (Fig. 1A). As a control, pooled sera from uninfected mice were also subjected to the immunoblotting. There were no spots detected in reaction with the normal mouse sera (data not shown).
Fig 1.

2-DE immunoblot showing T. gondii antigens recognized by IgG antibodies in sera of infected CBA/J mice treated with sulfadiazine to inhibit proliferation of tachyzoites during the chronic stage of infection (A) and a Coomassie-stained 2-DE gel indicating protein spots corresponding to each of the antigens recognized by the IgG antibodies (B). Tachyzoites (1 × 108 parasite) were solubilized in sample buffer and subjected to 2-DE. The gel was stained with Coomassie brilliant blue and scanned with an HP Scanjet G4050. The tachyzoite antigens separated by 2-DE were transferred to a nitrocellulose membrane, and the membrane was subjected to immunoblotting with the pooled sera from the infected and sulfadiazine-treated mice (see Materials and Methods). The identity of each antigen indicated (C1 to C6) on the immunoblot is described in Table 1.
Fig 2.
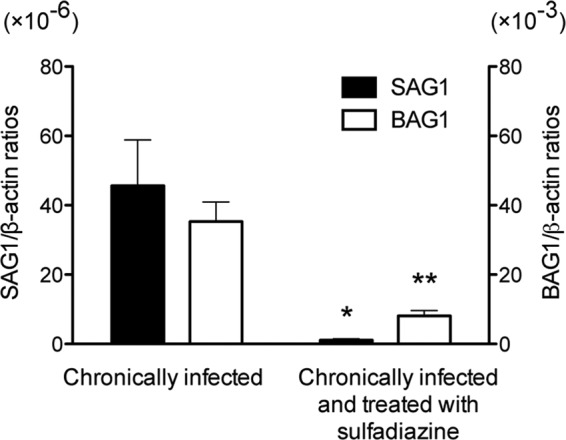
Quantification of mRNA for tachyzoite-specific SAG1 and bradyzoite-specific BAG1 in the brains of T. gondii-infected CBA/J mice treated or not with sulfadiazine. Two groups of CBA/J mice were infected with 10 cysts of the ME49 strain of T. gondii, and one group (n = 8) was treated with sulfadiazine beginning at 3 weeks after infection for the entire period of the experiment to prevent proliferation of tachyzoites during the chronic stage of infection. Another group of mice (n = 12) did not receive the treatment. Brain samples were obtained from both groups of animals at 2 months after infection. cDNAs were prepared from the brains of these animals, and amounts of mRNA for SAG1 and BAG1 were measured by real-time reverse transcription PCR (see Materials and Methods). Results are shown as the mean ± SE of each group. *, P < 0.01; **, P < 0.001.
We first performed the immunoblotting using unstained gels. However, identifying protein spots in a Coomassie-stained gel that corresponds to each of the antigens recognized by the IgG antibodies of the chronically infected mice was difficult because of the presence of multiple protein spots in the areas where the immunogenic antigens were located. Thus, we next performed immunoblotting consecutively to Coomassie staining of the 2-DE gels. In this way, we were able to see both the blue protein spots and those recognized by the antibodies on an identical nitrocellulose membrane. Staining with Coomassie brilliant blue did not affect the antigenicity of T. gondii proteins, and we observed the identical spots in immunoblotting using gels with and without Coomassie staining (data not shown). By using the stained gel for immunoblotting, we determined the protein spots in a Coomassie-stained gel (indicated as C1 to C6 in Fig. 1B), which correspond to each of the C1 to C6 spots in the immunoblot (Fig. 1A). There were no visible protein spots corresponding to T. gondii antigens below 25 kDa recognized by the antibodies.
To determine the identity of the T. gondii antigens C1 to C6, the protein spot corresponding to each of these antigens was excised from a Coomassie-stained gel and applied for mass spectrometry. The identity of each of these six antigens is shown in Table 1. These antigens were the microneme protein MIC2, one putative heat shock protein HSP70, the cyst matrix protein MAG1, and the dense granule proteins GRA4 and GRA7. In regard to GRA4, two spots (C4 and C5) with similar molecular masses but different pI values were identified as this molecule. A possible reason for their different pI values is posttranslational modification, such as sialidization. Indeed, GRA4 has been shown to be glycosylated (1), although the nature of the glycane is unknown.
Table 1.
Identities of T. gondii antigens recognized by the IgG antibodies of mice treated with sulfadiazine to inhibit tachyzoite growth during the chronic stage of infection (indicated in Fig. 1)
| Sample ID in Fig. 1 | Protein ID | Gene ID | Protein scorea |
|---|---|---|---|
| C1 | Microneme protein 2 (MIC2) | TGME49_001780 | 515 |
| C2 | Heat shock protein 70 (HSP70), putative | TGME49_073760 | 1,660 |
| C3 | Cyst matrix protein (MAG1) | TGME49_070240 | 922 |
| C4 | Dense granule protein 4 (GRA4) | TGME49_110780 | 163 |
| C5 | Dense granule protein 4 (GRA4) | TGME49_110780 | 178 |
| C6 | Dense granule protein 7 (GRA7) | TGME49_003310 | 715 |
Protein scores of >50 indicate identity or extensive homology in mass spectrometry analyses.
T. gondii antigens recognized by IgG antibodies of mice with active proliferation of tachyzoites in their brains during the chronic stage of infection.
We next performed immunoblotting to examine whether IgG antibodies of mice with cerebral proliferation of tachyzoites during the chronic stage of infection recognize T. gondii antigens different from those recognized by the antibodies of chronically infected animals in which tachyzoite growth was inhibited. For this purpose, we used sera obtained from CBA/J mice at 2 months after infection without sulfadiazine treatment. As expected, large amounts of SAG1 mRNA were detectable in their brains (Fig. 2), indicating the presence of active tachyzoite growth in their brains. In addition, the amounts of BAG1 mRNA in their brains were 4 times greater than those of animals treated with sulfadiazine (Fig. 2). Since sulfadiazine is not effective on bradyzoites, active proliferation of tachyzoites in the brains of untreated mice most likely resulted in formation of new cysts. Figure 3A shows the results of immunoblotting with sera of mice with cerebral tachyzoite growth. This immunoblot was performed using the same tachyzoite antigen preparation in parallel with the immunoblot shown in Fig. 1A with sera of mice treated with sulfadiazine to prevent tachyzoite growth. Therefore, the intensity of reaction of each spot recognized by IgG antibodies of the two groups of mice shown in Fig. 1A and 3A can be compared. The antibodies of the mice with active cerebral tachyzoite growth recognized each of the antigens, C1 to C6, recognized by the antibodies of mice without the tachyzoite proliferation shown in Fig. 1A (Fig. 3A). In addition to these antigens, there were multiple antigens recognized only by or mostly by the IgG antibodies of mice with cerebral tachyzoite proliferation (S1 to S10 [Fig. 3A]). The selective recognition of these 10 antigens (S1 to S10) by these antibodies is clearly visible in Fig. 3C, in which the blots shown in Fig. 1A and 3A are overlaid using two different colors: red for the spots shown in Fig. 1A and green for the spots shown in Fig. 3A. Whereas the spots corresponding to C1 to C6 are yellow, the spots corresponding to S1 to S10 are exclusively green (Fig. 3C). Densitometry analysis of the strength of reaction of each of these 10 spots on the immunoblots shown in Fig. 1A and 3A clearly indicates recognition of these antigens only by or markedly more strongly (at least 6.1 times greater) by IgG antibodies of the mice with active cerebral tachyzoite growth compared to the antibodies of animals without such tachyzoite growth (Table 2). The strength of reaction of S6 to S8 was relatively less than that of the other spots, and therefore, the results of densitometry analysis on these spots may not be as accurate as that on the others. However, the S6 to S8 spots were not recognized at all by the antibodies of mice without cerebral tachyzoite growth (Table 2).
Fig 3.

2-DE immunoblot showing T. gondii antigens recognized by IgG antibodies in sera of CBA/J mice with cerebral proliferation of tachyzoites during the chronic stage of infection (A) and a Coomassie-stained 2-DE gel indicating protein spots corresponding to each of the antigens recognized by the IgG antibodies (B). Tachyzoites (1 × 108 parasite) were solubilized in sample buffer and subjected to 2-DE. The gel was stained with Coomassie brilliant blue and scanned with an HP Scanjet G4050. The tachyzoite antigens separated by 2-DE were transferred to a nitrocellulose membrane, and the membrane was subjected to immunoblotting with the pooled sera from the infected mice (see Materials and Methods). The spots indicated (S1 to S10) are those recognized only by or mostly by the IgG antibodies of mice with cerebral proliferation of tachyzoites compared to the immunoblot with the IgG antibodies of mice treated with sulfadiazine to inhibit tachyzoite growth during the chronic stage of infection shown in Fig. 1A. The identity of each antigen indicated on the immunoblot is described in Table 3. Panel C is an overlay of the blots shown in Fig. 1A and 3A using two different colors: red for the spots shown in Fig. 1A and green for the spots shown in Fig. 3A. Whereas the spots corresponding to C1 to C6 are yellow, the spots corresponding to S1 to S10 are exclusively green. There are multiple spots shown exclusively red (D1 to D4), indicating that these antigens are recognized only by or mostly by IgG antibodies from mice that did not have active tachyzoite proliferation.
Table 2.
Densitometry analysis of the strength of the reactions in immunoblots with IgG antibodies of mice with (Fig. 3A) and without (Fig. 1A) active cerebral tachyzoite growth to T. gondii antigens indicated as S1 to S10 in Fig. 3A and those indicated as C1 to C6 in Fig. 1A
| Sample ID | Densitometry readings of the spot with sera of mice (×103) |
Fold increase (R1/R2) | |
|---|---|---|---|
| With cerebral tachyzoite growth (R1) | Without tachyzoite growth (R2) | ||
| S1 | 24.0 | 0 | ∞ |
| S2 | 11.4 | 0.045 | 256 |
| S3 | 4.15 | 0 | ∞ |
| S4 | 10.4 | 0.29 | 36.0 |
| S5 | 5.76 | 0.15 | 38.5 |
| S6 | 1.97 | 0 | ∞ |
| S7 | 2.05 | 0 | ∞ |
| S8 | 3.21 | 0 | ∞ |
| S9 | 20.5 | 3.38 | 6.06 |
| S10 | 88.3 | 0 | ∞ |
| C1 | 175 | 34.8 | 5.06 |
| C2 | 24.3 | 10.6 | 2.30 |
| C3 | 103 | 25.3 | 4.08 |
| C4 | 97.6 | 61.4 | 1.59 |
| C5 | 76.3 | 41.2 | 1.85 |
| C6 | 240 | 134 | 1.79 |
The strength of reaction on the C1 to C6 antigens also increased approximately 2 to 5 times in the blot shown in Fig. 3A compared to that in Fig. 1A (Table 3). However, of importance, increases in the strength of reaction with T. gondii antigens by IgG antibodies from mice with cerebral tachyzoite proliferation did not occur on all antigens recognized by IgG antibodies of mice treated with sulfadiazine to inhibit tachyzoite proliferation. For example, antigens indicated as D1 to D4 in Fig. 3C are seen exclusively in red, indicating that these antigens were recognized only by or mostly by the antibodies from mice without active cerebral tachyzoite proliferation (mass spectrometry to determine the identities of D1 to D4 was not performed in the present study). These results indicate that increases in IgG antibody levels in association with cerebral tachyzoite growth occur only to selected antigens of T. gondii.
Table 3.
Identities of T. gondii antigens recognized only by or mostly by the IgG antibodies of mice with cerebral proliferation of tachyzoites during the chronic stage of infection (indicated in Fig. 3) compared to the antigens recognized by the antibodies of mice treated with sulfadiazine to inhibit tachyzoite growth (shown in Fig. 1)
| Sample ID in Fig. 3 | Protein ID | Gene ID | Protein scorea |
|---|---|---|---|
| S1 | Myosin heavy chain, putative (most likely) | TGME49_008370 | 600 |
| S2 | Myosin heavy chain, putative (most likely) | TGME49_008370 | 132 |
| S3 | Heat shock protein 90 (HSP90) | TGME49_088380 | 224 |
| S4 | Heat shock protein 70 (HSP70), putative | TGME49_111720 | 1,996 |
| S5 | Hypothetical protein, conserved (coronin) | TGME49_016970 | 1,896 |
| S6 | Rhoptry protein 1 (ROP1) | TGME49_109590 | 384 |
| S7 | Rhoptry protein 1 (ROP1) | TGME49_109590 | 317 |
| S8 | Rhoptry protein 1 (ROP1) | TGME49_109590 | 487 |
| S9 | Microneme protein 6 (MIC6) | TGME49_018520 | 177 |
| S10 | Dense granule protein 2 (GRA2) | TGME49_027620 | 285 |
Protein scores of >50 indicate identity or extensive homology in mass spectrometry analyses.
We were able to determine the protein spots corresponding to each of the 10 antigens, S1 to S10, recognized only by or mostly by IgG antibodies of mice with active tachyzoite proliferation in their brains, in a Coomassie-stained gel. Those protein spots are indicated as S1 to S10 in Fig. 3B. The identity of each of these 10 antigens is shown in Table 3. They include one HSP90, one putative HSP70, three isoforms of the rhoptry protein ROP1, MIC6, and GRA2. Although the location of GRA2 in Fig. 3A is similar to that of one antigen recognized by IgG antibodies of mice without active growth of tachyzoites shown in Fig. 1A, the difference in the locations of these two spots is clearly visible in green (GRA2) and red (the spot seen in Fig. 1A) in the overlay shown in Fig. 3C. The HSP70 identified here (gene identity [ID]: ToxoDB database no. TGME49_111720) is different from the HSP70 (gene ID: TGME49_073760) recognized by IgG antibodies of mice without active proliferation of tachyzoites. Another spot (S5) was identified as a hypothetical protein. This protein was recently identified as coronin in the other two genotypes, GT1 (type I) and VEG (type III) strains, of T. gondii (Table 3). Two additional spots (S1 and S2) were determined as myosin heavy chain (putative) (Table 3), although Coomassie-stained spots corresponding to these two spots were not visible on nitrocellulose membranes but only on a stained gel. Therefore, the identity of these two spots is listed as “most likely” in Table 3.
Differences in amounts of mRNA for ROP1, GRA2, MAG1, and GRA7 in the brains of mice with and without active cerebral proliferation of tachyzoites during the chronic stage of infection.
It might be possible that levels of expression of S1 to S10 antigens in the brain increase in correlation with cerebral proliferation of tachyzoites, and this might be a reason for increases in levels of IgG antibody to these antigens in mice with cerebral tachyzoite growth. To address this possibility, we selected two representatives, ROP1 (S6 to S8) and GRA2 (S10), from this group of antigens and measured their mRNA levels in the brains of mice with and without active cerebral tachyzoite growth. For a comparison, we also measured mRNA levels of two antigens, MAG1 and GRA7, selected from the group of antigens, C1 to C6, commonly recognized by IgG antibodies of these two groups of animals. mRNA levels for all of these four antigens were 4.0 to 6.7 times greater in the brains of mice with active cerebral tachyzoite growth than in those treated with sulfadiazine to inhibit the tachyzoite growth (P < 0.001 [Fig. 4]). Unexpectedly, their mRNA levels did not directly correlate with strength of reactions by IgG antibodies to these antigens in the immunoblots shown in Table 2. For example, mRNA expression levels of GRA2, GRA7, and MAG1 did not differ in the brains of mice treated with sulfadiazine (Fig. 4), whereas only MAG1 and GRA7 were recognized by IgG antibodies of these animals (Fig. 1A). These results suggest that serum levels of IgG antibody to T. gondii antigens during the chronic stage of infection do not simply reflect amounts of the antigens expressed in the brains of infected hosts. Bradyzoites may express GRA2 but may not secrete it, whereas tachyzoites actively secrete GRA2 during their invasion and proliferation in host cells. Quantity may be another factor involved. Bradyzoites may secrete much larger amounts of GRA7 than GRA2, and this may be a reason why IgG antibodies to GRA7 but not those to GRA2 were detected in sera from chronically infected mice that did not have active proliferation of tachyzoites. Other factors, such as localization, antigenicity, and/or stability of the antigens, could also be involved in determining the strength of IgG antibody responses to these molecules.
Fig 4.
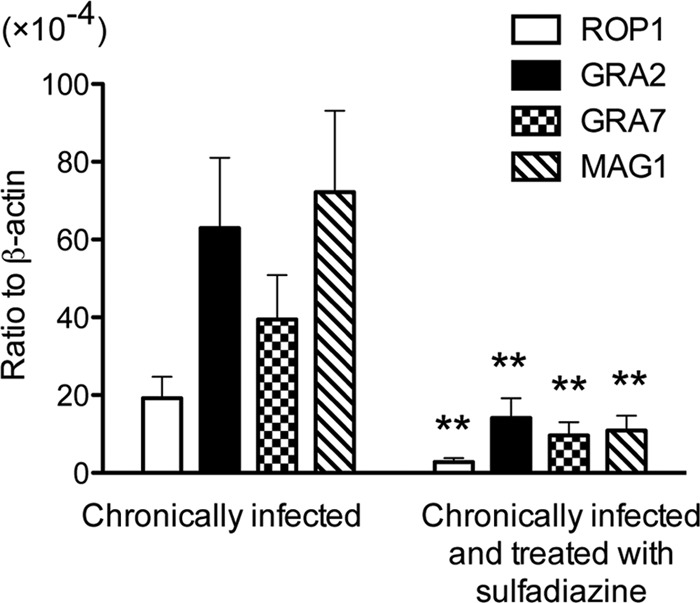
Quantification of mRNA for ROP1, GRA2, GRA7, and MAG1 in the brains of T. gondii-infected CBA/J mice treated or not with sulfadiazine. Two groups of CBA/J mice were infected with 10 cysts of the ME49 strain of T. gondii, and one group (n = 8) was treated with sulfadiazine beginning at 3 weeks after infection for the entire period of the experiment to prevent proliferation of tachyzoites during the chronic stage of infection. Another group of mice (n = 12) did not receive the treatment. Brain samples were obtained from both groups of animals at 2 months after infection. cDNA was prepared from the brains of these animals, and amounts of mRNA for ROP1, GRA2, GRA7, and MAG1 were measured by real-time reverse transcription-PCR (see Materials and Methods). Results are shown as the mean ± SE of each group. **, P < 0.001.
Difference in reactivity of IgG antibodies of mice with and without active cerebral proliferation of tachyzoites to rGRA2.
Since GRA2 was revealed to be one of the T. gondii antigens recognized by IgG antibodies associated with active tachyzoite proliferation in the brain during the chronic stage of infection, we compared the reactivities of the antibodies from chronically infected mice with and without sulfadiazine treatment using dot blotting with recombinant GRA2 (rGRA2; fusion protein with plasmid pUET-GRA2 expressing amino acids 21 to 185 of GRA2). Twofold serial dilutions (1:1,000 to 1:16,000) of sera from these two groups of animals were incubated with discs of a nitrocellulose membrane on which either rGRA2 or pUET alone as a control was spotted. rGRA2 was strongly recognized by IgG antibodies of mice with cerebral tachyzoite proliferation at dilutions of 1:1,000 to 1:4,000 (Fig. 5A). Some positive reactions were detected with their sera even at higher dilutions, 1:8,000 and 1:16,000 (Fig. 5A). At a 1:1,000 dilution of the sera, some reactions were seen on the disc onto which the pUET control peptide had been spotted. However, this reaction was not observed in repeated experiments (data not shown). In contrast, no reaction was observed with each dilution of sera from mice treated with sulfadiazine to prevent tachyzoite growth in their brains (Fig. 5A). No reaction was observed with sera from uninfected animals either (Fig. 5A).
Fig 5.
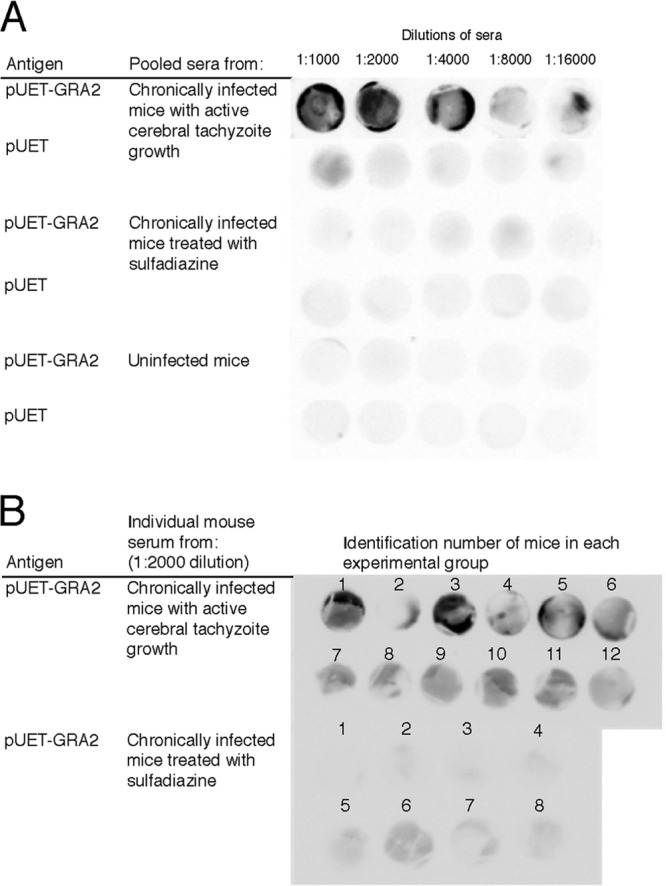
Recognition of rGRA2 of T. gondii by IgG antibodies of infected CBA/J mice with and without active cerebral proliferation of tachyzoites during the chronic stage of infection. One microliter of either rGRA2 (a fusion protein with pUET; 15.6 μg/ml) or pUET solution (15.6 μg/ml) was absorbed onto a nitrocellulose membrane. (A) The membranes were incubated with serial 2-fold dilutions (1:1,000 to 1:16,000) of pooled sera from chronically infected CBA/J mice with cerebral tachyzoite proliferation and those treated with sulfadiazine to prevent the tachyzoite growth (see Materials and Methods). Sera from uninfected mice were also used as a control. (B) The membranes were incubated with a 1:2,000 dilution of sera from individual chronically infected CBA/J mice with cerebral tachyzoite proliferation (n = 12) and those treated with sulfadiazine to prevent the tachyzoite growth (n = 8).
To further confirm the differences in reactivity of IgG antibodies to rGRA2 in mice with and without active cerebral tachyzoite growth during the chronic stage of infection, we performed dot blotting using rGRA2 with sera obtained from individual animals of each group at a 1:2,000 dilution. Sera from 11 of 12 mice (all but mouse no. 2 [92%]) with active tachyzoite proliferation in their brains showed positive reactions, whereas only 1 (mouse no. 6) of 8 animals (12.5%) treated with sulfadiazine to inhibit tachyzoite growth showed a positive reaction (Fig. 5B). These results indicate that the presence of elevated IgG antibodies to GRA2 is a good indicator to distinguish these two groups of mice. Of interest, in the mice with tachyzoite proliferation, the mouse that did not show a clear positive reaction had the lowest SAG1 mRNA level in the brain in this group (data not shown). The mouse that showed a positive reaction in the sulfadiazine-treated group is one of two mice with the highest levels of SAG1 mRNA in their brains in this group (data not shown). These results further support the possibility that the presence of elevated levels of anti-GRA2 IgG antibodies is associated with cerebral tachyzoite growth during the chronic stage of T. gondii infection.
DISCUSSION
The present study demonstrated that IgG antibodies of mice with active tachyzoite proliferation in their brains during the chronic stage of infection recognize T. gondii antigens different from those recognized by the antibodies of chronically infected animals treated with sulfadiazine to prevent tachyzoite growth. ROP1, GRA2, MIC6, HSP90 (TGME49_088380), and HSP70 (TGME49_111720) were among the antigens recognized only by or mostly by the IgG antibodies of mice with cerebral tachyzoite growth. To our knowledge, this is the first evidence indicating differences in recognition of T. gondii antigens by IgG antibodies of immunocompetent hosts with and without active tachyzoite growth in the brain during the chronic stage of infection. In addition, the identity of T. gondii antigens recognized by IgG antibodies associated with cerebral growth of tachyzoites in chronically infected hosts has not been reported before.
Microneme and rhoptry proteins are secreted from tachyzoites and play important roles in their attachment to and invasion of host cells (30). Dense granule proteins are also secreted from the parasite into parasitophorous vacuoles after their invasion of host cells (25, 26). Among the seven antigens recognized only by or mostly by IgG antibodies of mice with active tachyzoite proliferation in their brains, three antigens (ROP1, GRA2, and MIC6) belong to these secretory proteins. Therefore, these proteins secreted from tachyzoites during their proliferation in the brain appear to stimulate production of antibody to these molecules. In humans, IgG antibodies to ROP1 and GRA2 are detected significantly more frequently in individuals with acute infection than in those with chronic infection (3, 17). In addition, significantly greater levels of anti-GRA2 IgG antibodies were detected in sera from pregnant women with acute infection than in sera from those with chronic infection (14). Therefore, it appears that production of antibodies to GRA2 and ROP1 is associated with proliferation of tachyzoites not only in the brain but also in other organs.
Although increased serum levels of anti-GRA2 and anti-ROP1 IgG antibodies most likely occur not only during reactivation of T. gondii infection but also during acute acquired infection, the latter can be diagnosed by existing serological tests detecting elevated IgM antibody levels and low avidity of IgG antibodies. Therefore, increased amounts of IgG antibodies to GRA2 and ROP1 in the absence of anti-T. gondii IgM antibodies and the presence of high-avidity IgG antibodies might be good potential indicators of an occurrence of tachyzoite proliferation in the brain during the chronic stage of infection. This possibility is supported by the evidence that GRA2 and ROP1 are among the antigens that showed the largest differences in the reactivities of IgG antibodies between mice with and without cerebral tachyzoite growth as shown in Table 2, although the strength of the reaction of IgG antibodies to ROP1 is weaker than that to GRA2 in the mice with the cerebral proliferation.
In regard to differences in antigen recognition by antibodies during acute acquired infection and reactivation of chronic infection, we performed a preliminary study using immunoblotting with sera obtained at 2 weeks after infection using the same dilution of sera from chronically infected mice as used in the blots shown in Fig. 1A and 3A. IgG antibodies at 2 weeks after infection showed spots corresponding to HSP70 (C2), MAG1 (C3), GRA4 (C4 and C5), GRA7 (C6), and ROP1 (S6, S7, and S8), but not other antigens described in this article (unpublished data). Furthermore, there are additional multiple spots strongly recognized by these acute-stage IgG antibodies, but these spots are not observed in either of the immunoblots shown in Fig. 1A and 3A (unpublished data). Therefore, recognition of T. gondii antigens by IgG antibodies of chronically infected hosts with cerebral tachyzoite proliferation appears to differ from that by IgG antibodies of acute acquired infection, at least in their relative quantitative levels. More detailed studies with sera obtained from different time points during acute acquired infection would be needed to address this point.
Dot blotting using rGRA2 confirmed the difference in levels of IgG antibodies in sera of mice with and without active cerebral proliferation of tachyzoites during the chronic stage of infection. Pooled sera from mice with active cerebral tachyzoite growth showed strong positive reactions at a 1:4,000 dilution. In contrast, no reaction was observed with a 1:1,000 dilution of sera from animals treated with sulfadiazine to prevent the tachyzoite proliferation. In reactions with sera from individual mice at a 1:2,000 dilution, 92% (11/12) of animals with cerebral tachyzoite growth showed positive reactions, whereas only 12.5% (1/8) of mice without the tachyzoite proliferation showed a positive reaction. These results from dot blotting with rGRA2 correlate well with the results of 2-DE gel immunoblotting performed with a 1:4,000 dilution of pooled sera shown in Fig. 1 and 3, as well as those in Table 2. These results all together support the possibility that elevated anti-GRA2 IgG antibody levels in sera can be a good indicator of an occurrence of reactivation (cerebral tachyzoite proliferation) of T. gondii infection during the chronic stage of infection.
In regard to MIC6, we were unable to find literature reporting production of antibodies to this protein in T. gondii-infected hosts. Microneme proteins are discharged onto the parasite surface and form tight interactions with host cell receptors. MIC6 forms a multimeric complex with MIC1 and MIC4 and contributes to invasion of host cells by tachyzoites in vitro and to virulence in vivo (4, 8, 31). Therefore, it appears that infected hosts recognize MIC6 and produce antibodies when active growth of tachyzoites occurs. In the present study, it is unclear why antibodies to MIC1 and MIC4 were not detected in association with cerebral tachyzoite proliferation. MIC6 might have stronger antigenicity to induce antibody production than the other two MIC proteins.
Two heat shock proteins, HSP90 (TGME49_088380) and putative HSP70 (TGME49_111720), were also selectively recognized by IgG antibodies associated with cerebral proliferation of tachyzoites in the present study. HSP90 has been shown to be induced during the stage conversion of the parasite from tachyzoite to bradyzoite (13). Therefore, it is possible that cerebral proliferation of tachyzoites is associated with formation of new cysts and that the HSP90 expressed during the formation of new cysts stimulates production of antibody to this molecule. This possibility is supported by the evidence that greater mRNA levels for bradyzoite-specific BAG1 were detected in the brains of mice with cerebral tachyzoite growth than in those without the tachyzoite growth. In regard to HSP70, it is known that T. gondii has multiple HSP70 molecules. Silva et al. (32) previously reported expression of high levels of HSP70 during a short period of conversion of bradyzoites to tachyzoites using polyclonal antibodies to HSP70 of Leishmania braziliensis. Although the function(s) of HSP70 (TGME49_111720) has not been determined, this molecule may also be expressed during the stage conversion from the bradyzoite to tachyzoite. Therefore, detecting increased IgG levels to these two heat shock proteins, HSP90 (TGME49_088380) and putative HSP70 (TGME49_111720), in addition to GRA2 and ROP1, which are discussed above, might be another option for detecting an occurrence of reactivation of chronic T. gondii infection in the brain.
In regard to six T. gondii antigens recognized by IgG antibodies of mice in which tachyzoite proliferation was inhibited by sulfadiazine treatment, MAG1 and GRA7 were among these antigens. In humans, IgG antibodies to these two molecules can be detected from the acute to chronic stages of infection (3, 12, 28). Therefore, infected hosts appear to produce the antibodies to these molecules throughout the course of infection with the parasite. Since reactivity to MAG1 and GRA7 increased in mice with cerebral tachyzoite proliferation during the chronic stage of infection, detecting increased levels of IgG antibodies to these antigens may be considered an alternative option to detect reactivation of infection during the chronic stage of infection. For example, MAG1 is expressed in cyst matrix and cyst wall of T. gondii (27), and cerebral tachyzoite growth is associated with increases in cyst formation as shown in Fig. 2. Therefore, detecting increased levels of IgG antibodies to MAG1 (and GRA7), in combination with detecting increased levels of anti-GRA2 and anti-ROP1 IgG antibodies, in the absence of Toxoplasma IgM antibodies to exclude acute acquired infection, might be a possible option to detect reactivation of T. gondii infection in the brain.
In the present study, tachyzoites harvested from their cultures with host HFF were used to prepare antigens for 2-DE followed by immunoblotting. However, two antigens, HSP90 (TGME49_088380) and MAG1, associated with encystment of the parasite were detected as a part of the antigens recognized by IgG antibodies of infected mice. Spontaneous differentiation of tachyzoites to bradyzoites appears to have occurred in a portion of the parasites in the cultures with HFF, and this would be the reason for the presence of both HSP90 and MAG1 in the antigen preparation. Spontaneous differentiation from tachyzoites to bradyzoites is known to occur during cultures of type II strains.
The molecular masses and the pI values of the T. gondii antigens identified in the present study are approximately consistent with their predicted values of these molecules, except for ROP1 and MIC2. The pI values of ROP1 (S6 to S8) detected on the immunoblot were lower than the predicted value, 5.16, of this molecule. A possible reason for this difference is posttranslational modification such as sialidization. A recent report indeed suggested a possible glycosylation of ROP1 (23). The spot identified as MIC2 (C1) was located in the area where the expected molecular mass is around 150 kDa, which is much greater than the predicted molecular mass of this molecule, 78 kDa. The reason for this difference is unclear, but ubiqutination may be one possible mechanism for its larger apparent molecular mass.
Our recent studies showed that mice with cerebral tachyzoite proliferation during the chronic stage of infection have greater Toxoplasma IgG, but not IgM, antibody levels in their sera than animals in which such tachyzoite proliferation was inhibited (33). Increased Toxoplasma IgG, but not IgM, antibody levels have been observed in sera of patients with recent onset of schizophrenia (22, 39). Increased IgG antibody levels were also observed in patients with cryptogenic epilepsy (6). These observations may suggest that reactivation of chronic T. gondii infection has occurred in these patients and that such reactivation of infection might be involved in the etiology of these diseases. It would be valuable to examine whether IgG antibodies of the patients with schizophrenia and cryptogenic epilepsy recognize T. gondii antigens different from those recognized by the antibodies of healthy individuals chronically infected with the parasite, and whether the patients' antibodies recognize the same antigens as those detected by the antibodies of mice with cerebral tachyzoite proliferation in the present study.
ACKNOWLEDGMENTS
We thank Bert Lynn and Jack Goodman at the University of Kentucky Mass Spectrometry Facility for helpful discussion and Marie Gehman for assistance in preparing the manuscript.
This work was supported by a grant (number 08R-2047 to Y.S.) from The Stanley Medical Research Institute and grants from the National Institutes of Health (AI078756, AI095032, and AI077887 to Y.S.). rGRA2 used in this work was produced using the GRAVIT grant (number 070304) to C.M.
Footnotes
Published ahead of print 30 July 2012
REFERENCES
- 1. Achbarou A, et al. 1991. Differential targeting of dense granule proteins in the parasitophorous vacuole of Toxoplasma gondii. Parasitology 103(Part 3):321–329 [DOI] [PubMed] [Google Scholar]
- 2. Ajzenberg D, et al. 2002. Genotype of 86 Toxoplasma gondii isolates associated with human congenital toxoplasmosis, and correlation with clinical findings. J. Infect. Dis. 186:684–689 [DOI] [PubMed] [Google Scholar]
- 3. Aubert D, et al. 2000. Recombinant antigens to detect Toxoplasma gondii-specific immunoglobulin G and immunoglobulin M in human sera by enzyme immunoassay. J. Clin. Microbiol. 38:1144–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blumenschein TM, et al. 2007. Atomic resolution insight into host cell recognition by Toxoplasma gondii. EMBO J. 26:2808–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boyer K, Marcinak J, McLeod R. 2007. Toxoplasma gondii (toxoplasmosis), p 1267–1288 In Long S, Pickering LK, Prober CG. (ed), Principles and practice of pediatric infectious diseases, 3rd ed Churchill Livingstone, New York, NY [Google Scholar]
- 6. Brooks RG, McCabe RE, Remington JS. 1987. Role of serology in the diagnosis of toxoplasmic lymphadenopathy. Rev. Infect. Dis. 9:1055–1062 [DOI] [PubMed] [Google Scholar]
- 7. Brown CR, et al. 1995. Definitive identification of a gene that confers resistance against Toxoplasma cyst burden and encephalitis. Immunology 85:419–428 [PMC free article] [PubMed] [Google Scholar]
- 8. Cerede O, et al. 2005. Synergistic role of micronemal proteins in Toxoplasma gondii virulence. J. Exp. Med. 201:453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Darde ML, Bouteille B, Pestre-Alexandre M. 1992. Isoenzyme analysis of 35 Toxoplasma gondii isolates and the biological and epidemiological implications. J. Parasitol. 78:786–794 [PubMed] [Google Scholar]
- 10. Denkers EY, Gazzinelli RT. 1998. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 11:569–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Derouin F, Rabian-Herzog C, Sulahian A. 1989. Longitudinal study of the specific humoral and cellular response to Toxoplasma gondii in a patient with acquired toxoplasmosis. J. Clin. Lab. Immunol. 30:97–102 [PubMed] [Google Scholar]
- 12. Di Cristina M, et al. 2004. The Toxoplasma gondii bradyzoite antigens BAG1 and MAG1 induce early humoral and cell-mediated immune responses upon human infection. Microbes Infect. 6:164–171 [DOI] [PubMed] [Google Scholar]
- 13. Echeverria PC, et al. 2005. Toxoplasma gondii Hsp90 is a potential drug target whose expression and subcellular localization are developmentally regulated. J. Mol. Biol. 350:723–734 [DOI] [PubMed] [Google Scholar]
- 14. Golkar M, et al. 2007. The dense granule protein GRA2, a new marker for the serodiagnosis of acute Toxoplasma infection: comparison of sera collected in both France and Iran from pregnant women. Diagn. Microbiol. Infect. Dis. 58:419–426 [DOI] [PubMed] [Google Scholar]
- 15. Golkar M, et al. 2007. Evaluation of protective effect of recombinant dense granule antigens GRA2 and GRA6 formulated in monophosphoryl lipid A (MPL) adjuvant against Toxoplasma chronic infection in mice. Vaccine 25:4301–4311 [DOI] [PubMed] [Google Scholar]
- 16. Grigg ME, Bonnefoy S, Hehl AB, Suzuki Y, Boothroyd JC. 2001. Success and virulence in Toxoplasma as the result of sexual recombination between two distinct ancestries. Science 294:161–165 [DOI] [PubMed] [Google Scholar]
- 17. Holec-Gasior L, Kur J, Hiszczynska-Sawicka E. 2009. GRA2 and ROP1 recombinant antigens as potential markers for detection of Toxoplasma gondii-specific immunoglobulin G in humans with acute toxoplasmosis. Clin. Vaccine Immunol. 16:510–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Howe DK, Sibley LD. 1995. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J. Infect. Dis. 172:1561–1566 [DOI] [PubMed] [Google Scholar]
- 19. Huskinson J, et al. 1989. Toxoplasma antigens recognized by immunoglobulin G subclasses during acute and chronic infection. J. Clin. Microbiol. 27:2031–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kang H, Suzuki Y. 2001. Requirement of non-T cells that produce gamma interferon for prevention of reactivation of Toxoplasma gondii infection in the brain. Infect. Immun. 69:2920–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kasper LH. 1989. Identification of stage-specific antigens of Toxoplasma gondii. Infect. Immun. 57:668–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leweke FM, et al. 2004. Antibodies to infectious agents in individuals with recent onset schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 254:4–8 [DOI] [PubMed] [Google Scholar]
- 23. Luo Q, et al. 2011. Analysis of the glycoproteome of Toxoplasma gondii using lectin affinity chromatography and tandem mass spectrometry. Microbes Infect. 13:1199–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCabe RE, Remington JS. 1990. Toxoplasma gondii, p 2090 In Mandell GL, Douglas RG, Bennett JE. (ed), Principles and practice of infectious diseases, 3rd ed Churchill Livingstone Inc., New York, NY [Google Scholar]
- 25. Mercier C, Adjogble KD, Daubener W, Delauw MF. 2005. Dense granules: are they key organelles to help understand the parasitophorous vacuole of all apicomplexa parasites? Int. J. Parasitol. 35:829–849 [DOI] [PubMed] [Google Scholar]
- 26. Mercier C, Travier L, Bittame A, Gendrin C, Cesbron-Delauw MF. 2010. The dense granule proteins of Toxoplasma gondii, p 1–31 In De Bruyn O, Peeters S. (ed), Parasitology research trends. Nova Science Publishers Inc., Hauppauge, NY [Google Scholar]
- 27. Parmley SF, et al. 1994. Molecular characterization of a 65-kilodalton Toxoplasma gondii antigen expressed abundantly in the matrix of tissue cysts. Mol. Biochem. Parasitol. 66:283–296 [DOI] [PubMed] [Google Scholar]
- 28. Pfrepper KI, et al. 2005. Seroreactivity to and avidity for recombinant antigens in toxoplasmosis. Clin. Diagn. Lab. Immunol. 12:977–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pusch L, Romeike B, Deckert M, Mawrin C. 2009. Persistent toxoplasma bradyzoite cysts in the brain: incidental finding in an immunocompetent patient without evidence of a toxoplasmosis. Clin. Neuropathol. 28:210–212 [DOI] [PubMed] [Google Scholar]
- 30. Santos JM, Soldati-Favre D. 2011. Invasion factors are coupled to key signalling events leading to the establishment of infection in apicomplexan parasites. Cell. Microbiol. 13:787–796 [DOI] [PubMed] [Google Scholar]
- 31. Sawmynaden K, et al. 2008. Structural insights into microneme protein assembly reveal a new mode of EGF domain recognition. EMBO Rep. 9:1149–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Silva NM, et al. 1998. Expression of Toxoplasma gondii-specific heat shock protein 70 during in vivo conversion of bradyzoites to tachyzoites. Infect. Immun. 66:3959–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh J, et al. 2010. Toxoplasma IgG and IgA, but not IgM, antibody titers increase in sera of immunocompetent mice in association with proliferation of tachyzoites in the brain during the chronic stage of infection. Microbes Infect. 12:1252–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stommel EW, et al. 2001. Cryptogenic epilepsy: an infectious etiology? Epilepsia 42:436–438 [DOI] [PubMed] [Google Scholar]
- 35. Suzuki Y, Joh K, Orellana MA, Conley FK, Remington JS. 1991. A gene(s) within the H-2D region determines the development of toxoplasmic encephalitis in mice. Immunology 74:732–739 [PMC free article] [PubMed] [Google Scholar]
- 36. Suzuki Y, Orellana MA, Wong SY, Conley FK, Remington JS. 1993. Susceptibility to chronic infection with Toxoplasma gondii does not correlate with susceptibility to acute infection in mice. Infect. Immun. 61:2284–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Suzuki Y, Thulliez P, Desmonts G, Remington JS. 1988. Antigen(s) responsible for immunoglobulin G responses specific for the acute stage of Toxoplasma infection in humans. J. Clin. Microbiol. 26:901–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suzuki Y, et al. 2010. Removal of Toxoplasma gondii cysts from the brain by perforin-mediated activity of CD8+ T cells. Am. J. Pathol. 176:1607–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Torrey EF, Yolken RH. 2003. Toxoplasma gondii and schizophrenia. Emerg. Infect. Dis. 9:1375–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang X, Michie SA, Xu B, Suzuki Y. 2007. Importance of IFN-gamma-mediated expression of endothelial VCAM-1 on recruitment of CD8+ T cells into the brain during chronic infection with Toxoplasma gondii. J. Interferon Cytokine Res. 27:329–338 [DOI] [PubMed] [Google Scholar]