

Abstract
In lung cancer, frequent loss of one allele of chromosome 3p is seen in both small cell lung cancer and non-small cell lung cancer (NSCLC), providing evidence of tumor suppressor genes (TSGs) in this chromosomal region. The mechanism of Fus1 tumor suppressor activity is unknown. We have found that a Fus1 peptide inhibits the Abl tyrosine kinase in vitro (IC50 35 μM). The inhibitory Fus1 sequence was derived from a region that was deleted in a mutant FUS1 gene (FUS1 (1–80)) detected in some lung cancer cell lines. Importantly, a stearic acid-modified form of this peptide was required for the inhibition, but stearic acid alone was not inhibitory. Two NSCLC cell lines, which lack expression of wild-type Fus1, contain activated c-Abl. Forced expression of an inducible FUS1 cDNA in H1299 NSCLC cells decreased levels of activated c-Abl and inhibited its tyrosine kinase activity. Similarly, treatment of c-Abl immune complexes with the inhibitory Fus1 peptide also reduced the level of c-Abl in these immune complexes. The size and number of colonies of the NSCLC cell line, H1299, in soft agar was strongly inhibited by the Abl kinase inhibitor imatinib mesylate. Co-expression of FUS1 and c-ABL in COS1 cells blocked activation of c-Abl tyrosine kinase. In contrast, co-expression of mutant FUS1 (1–80) with c-ABL had little inhibitory activity against c-Abl. These findings provide strong evidence that c-Abl is a possible target in NSCLC patients that have reduced expression of Fus1 in their tumor cells.
Keywords: c-Abl, FUS1, NSCLC, lung cancer, imatinib
Introduction
Lung cancer, the leading cause of cancer related mortalities in the United States (US Cancer Statistics Working Group, 2004), is divided into two major sub-groups, small cell lung carcinomas (SCLC) and non-small cell lung carcinomas (NSCLC). Depending on the type and stage of the disease, treatment for lung cancer includes tumor resection, chemotherapy, and radiation therapy. Recently, new approaches in the treatment of NSCLC have arisen from the discovery that the epithelial growth factor receptor (EGFR) is frequently overexpressed and activated in NSCLC (Dowell and Minna, 2005). In lung cancer, loss of sequences within chromosome 3p is frequently seen in both SCLC and NSCLC, providing evidence of tumor suppressor genes (TSGs) in this chromosomal region (Girard et al., 2000; Zabarovsky et al., 2002). The FUS1 gene has been identified in the 3p21.3 critical chromosomal region and is considered a novel TSG (Zabarovsky et al., 2002; Ji et al., 2002). The mechanism of Fus1 tumor suppressor activity is unknown. A deletion mutant of FUS1 has been characterized (FUS1 (1–80)); it lacks the last 30 amino acids encoded by the FUS1 gene (Kondo et al., 2001; Ji et al., 2002). Myristoylation of Fus1 at the N-terminus is required for the tumor suppressing function of FUS1 as myristoylation of Fus1 has been shown to increase in the half-life of the Fus1 protein (Uno et al., 2004). The lack of Fus1 myristoylation has been found in NSCLC primary tumors (Uno et al., 2004). Of interest, the myristic fatty acid, which is bound to the N-terminal segment of c-Abl isoform 1b, is thought to be involved in autoinhibition of c-Abl (Hantschel et al., 2003).
The c-Abl protein is a tightly regulated non-receptor tyrosine kinase that is involved in the regulation of cell proliferation, cell survival, cell adhesion, cell migration and apoptosis. The c-Abl protein is located in both the nucleus and the cytoplasm. In the nucleus, c-Abl protein is known to be activated in response to DNA damage and contributes to apoptosis (Wang, 2000). Cytoplasmic c-Abl is associated with growth factor receptor signaling that can affect cell mobility and cell adhesion (Taagepera et al., 1998). Several studies have show that c-Abl contains intramolecular interactions that provide autoinhibitory mechanisms (Pendergast, 2002; Pluk et al., 2002; Hantschel et al., 2003). Studies have also found that c-Abl kinase activity is regulated by intermolecular interactions with negative regulators, which include Bcr (Liu et al., 1996; Ling et al., 2003), PAG (Wen and Van Etten, 1997) and F-actin (Woodring et al., 2003).
The c-Abl protein is widely expressed in all tissues and, until recently, oncogenically activated forms of Abl were believed to be restricted to hematopoietic malignancies (for example, Philadelphia chromosome+ chronic myeloid leukemia involving BCR–ABL fusions). A recent report has shown that c-Abl tyrosine kinase is activated in aggressive human breast cancer cell lines (Srinivasan and Plattner, 2006). Activation of c-Abl was linked to EGFR expression and activation in the breast cancer cell lines (Srinivasan and Plattner, 2006).
We have widened the scope of Abl kinase’s role in solid tumors with the discovery there is an activated c-Abl kinase in human NSCLC cell lines, A549 and H1299 (Lin J, Sun T, Ji L, Minna J, Roth J and Arlinghaus R. Activated c-Abl in FUS1 haploinsufficient NSCLC. 96th Annual American Association for Cancer Research, April 2005). Of note, both the A549 and H1299 NSCLC cell lines are known have the 3p21.3 deletion and are deficient in the Fus1 protein (Kondo et al., 2001; Ito et al., 2004).
Results
A stearate-Fus1 peptide inhibits the tyrosine kinase activity of c-Abl
Fus1 is a TSG associated with NSCLC (Kondo et al., 2001; Zabarovsky et al., 2002). A C-terminal deletion mutant of FUS1 was isolated from a lung cancer tumor cell line, which encodes the first 80 amino acids of the 110-amino-acid wild-type Fus1 protein (Kondo et al., 2001). We synthesized a stearate-Fus1 peptide (KLRRVHKNLIPQGIVKLDHR) originating from sequences that are deleted in the C-terminal truncated mutant FUS1. This peptide strongly inhibited a bacterially purified Abl in a cell-free kinase assay using Crk, a known Abl substrate (Feller et al., 1994), as a target (Figure 1a). Surprisingly, the Fus1 peptide lacking the stearate N-terminal moiety was inactive as an Abl kinase inhibitor (Figure 1b). Stearate alone was also not inhibitory. A stearate-modified Bcr peptide, GQSSRVEPSPTTYRMFDIC, which has about the same number of amino acids as the stearate Fus1 peptide, did not have any inhibitory effect on the bacterially purified 45 kDa recombinant Abl kinase (Figure 1c). Next, we assayed the effects of the stearate Fus1 peptide against a full-length purified human c-Abl. An insect purified human full-length active c-Abl was used in a cell-free kinase assay using a known peptide target of c-Abl, EAIYAAPFAKKK. Only the stearate Fus1 peptide was able to decrease the phosphorylation of the target peptide when compared to the stearate Bcr peptide and non-stearate Fus1 peptide (Figure 1d). Together, these results indicate that both the stearate moiety and the amino-acid sequences within the Fus1 peptide were necessary for the inhibition of the c-Abl kinase.
Figure 1. Stearic acid-modified Fus1 peptide inhibits constitutively activated commercial Abl tyrosine kinase.
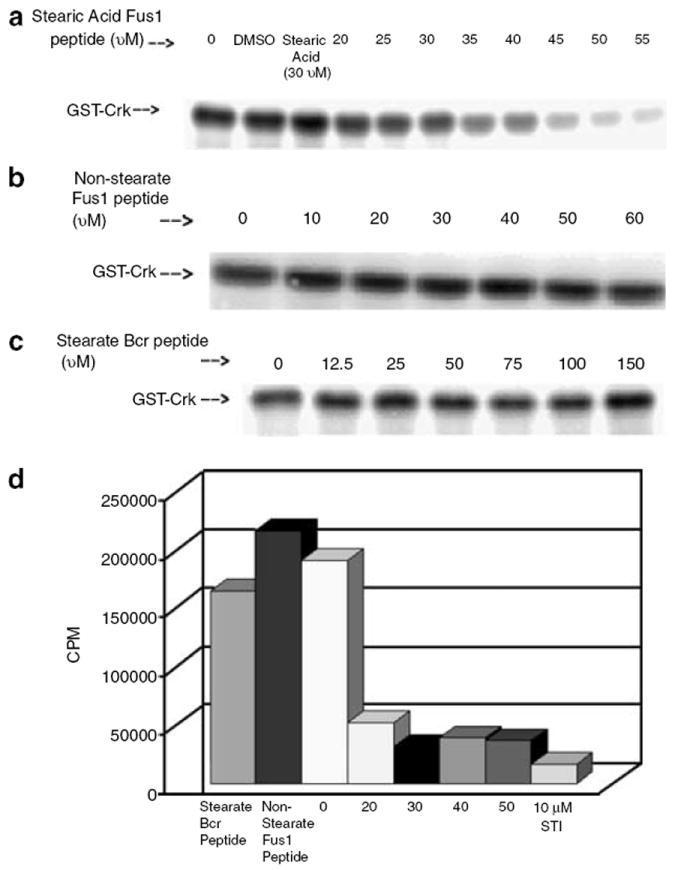
(a) Stearic acid-modified Fus1 peptide inhibits GST–Crk phosphorylation by a bacterially purified, constitutively active, Abl kinase (IC50 35uM). (b) Without the stearic acid modification, the Fus1 peptide does not inhibit GST–Crk phosphorylation by the bacterially purified, constitutively active, Abl kinase. (c) A stearic acid-modified Bcr peptide does not inhibit GST–Crk phosphorylation by the bacterially purified, constitutively active, Abl kinase. (d) Stearate Fus1 peptide inhibits phosphorylation of a known Abl kinase peptide target, EAIYAAPFAKKK, by full-length human Abl.
The c-Abl tyrosine kinase is activated in NSCLC cell lines
We examined several NSCLC cell lines for the presence of activated c-Abl (Figure 2). The NSCLC cell lines A549 and H1299 contained a tyrosine kinase-active c-Abl protein as determined immune complex kinase assays (Figure 2a). In contrast, normal lung fibroblast cell line CCD16 did not express an activated c-Abl tyrosine kinase. Lysates of the NSCLC cell lines also contained a tyrosine-phosphorylated c-Abl protein, as shown by immune complex western blot (Figure 2b). Figure 2b also indicates that there is an elevated level of c-Abl protein in the NSCLC cell lines A549 and H1299 when compared to the normal lung fibroblast cell line CCD16. There is a general increase in the phosphortyrosine protein in NSCLC cell lines A549 and H1299 compared to CCD16 (Figure 2c), although all phosphotyrosine proteins may not be attributed to c-Abl kinase alone, as both A549 and H1299 cells have activated EGFR (Zhang et al., 2004; Das et al., 2006).
Figure 2. Detection of activated c-Abl tyrosine kinase in two NSCLC cell lines deficient of FUS1.
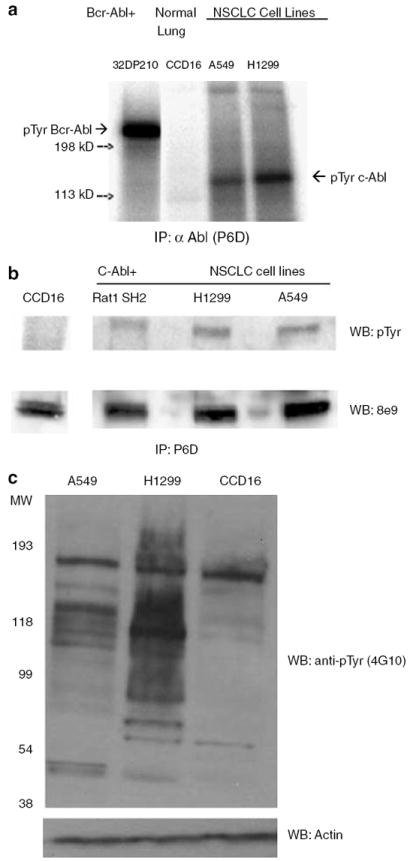
(a) NSCLC cell lines A549 and H1299 contain an active c-Abl tyrosine kinase, as measured in an in vitro kinase assay. Cells were lysed and incubated with the monoclonal anti-Abl antibody P6D, raised against c-Abl 51–64 residues (Liu et al., 1993). The immune complex was pulled down using protein A beads and subjected to either kinase assays or western blotting. 32D cells expressing Bcr-Abl were used as a positive control. Normal human lung fibroblast, CCD16 and 32D cells expressing Bcr–Abl were used as negative and positive controls, respectively. (b) Anti-phosphotyrosine western blotting demonstrates that NSCLC cell lines A549 and H1299 have an active c-Abl tyrosine kinase while normal human lung fibroblast CCD16 lack an active c-Abl tyrosine kinase. Phosphotyrosine c-Abl was detected by anti-phosphotyrosine antibody 4G10 and c-Abl was detected by monoclonal anti-Abl antibody 8e9. Transformed Rat 1cells (Rat1 SH2) that express activated c-Abl was used a positive control (Ling et al., 2003). (c) NSCLC cell lines have increased levels of phosphotyrosine-containing proteins compared to normal lung fibroblast cells (CCD16) as measured by 4G10.
Co-expression of FUS1 and c-ABL in COS1 cells inhibits c-Abl tyrosine kinase activity
To measure the ability of Fus1 to inhibit c-Abl tyrosine kinase, we coexpressed c-ABL cDNA with the cDNAs of wild-type full-length FUS1 [1–110], a mutant truncated form of FUS1 [1–80] or empty vector in COS1 cells and assessed the level of phosphorylation on tyrosine 412 (Figure 3a and b). Phosphorylation of tyrosine 412 of c-Abl is a measure of c-Abl tyrosine kinase activation (Brasher and Van Etten, 2000). Only expression of wild-type full-length FUS1 with c-ABL significantly inhibited the phosphorylation of tyrosine 412 on c-Abl (Figure 3a and b). In contrast, coexpression of truncated FUS1 [1–80] had little inhibitory effects on the phosphorylation of tyrosine 412 of c-Abl (Figure 3a and b). It has been reported that myristoylation of Fus1 is required for its tumor suppression activity in NSCLC (Uno et al., 2004). Coexpression of c-ABL with a myristoylation-deficient mutant of FUS1 in COS1 cell resulted in a minimal decrease in phosphotyrosine c-Abl when compared to coexpression of wild-type full-length FUS1 and c-ABL (Figure 3c). Of note, immunoprecipitation of Fus1 with an N-terminal Fus1 antibody co-precipitated c-Abl and immunoprecipitation of c-Abl antibody co-precipitated Fus1 suggesting that Fus1 is associated with c-Abl (Figure 3a and d).
Figure 3. Wild-type full-length Fus1 down regulates c-Abl tyrosine kinase.
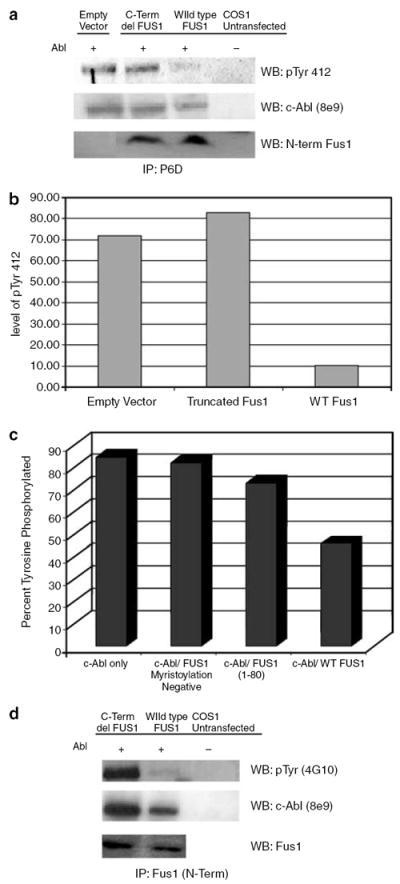
(a) c-ABL was co-transfected with either wild-type full-length FUS1, truncated FUS1, or empty vector in COS1 cells. At 48 h post-transfection, COS1 cells were harvested and lysate was incubated with the monoclonal anti-Abl antibody P6D. The immune complex was pulled down using protein A beads and subjected to western blotting. Antibody specific for pTyr412 of Abl (Abcam) was used to show kinase active c-Abl. The same blot was stripped and re-probed with monoclonal anti-Abl antibody 8e9. N-terminal Fus1 antibody was used to detect Fus1. (b) The intensities of the c-Abl pTyr 412 bands were normalized against c-Abl expression. (c) C-ABL was co-transfected with either wild-type full-length FUS1, truncated FUS1 or a non-myristoylated mutant FUS1 (Gly2Ala) in COS1 cells. At 48 h post-transfection, COS1 cells were harvested and the lysate was incubated with the monoclonal anti-Abl antibody P6D. The immune complex was pulled down using protein A beads and subjected to western blotting. Phosphotyrosine c-Abl was detected by anti-phosphotyrosine antibody 4G10 and the same blot was stripped and re-probed with monoclonal anti-Abl antibody 8e9 . The band intensities were normalized for c-Abl expression. (d) C-ABL was co-transfected with either wild-type full-length FUS1 or truncated FUS1 in COS1 cells. At 48 h post-transfection, COS1 cells were harvested and lysate was incubated with a polyclonal N-terminal Fus1 antibody. The immune complex was pulled down using protein A beads and subjected to western blotting. Phosphotyrosine c-Abl was detected by anti-phosphotyrosine antibody 4G10 and the same blot was stripped and re-probed with monoclonal anti-Abl antibody 8e9.
Imatinib inhibits colony formation of NSCLC cells
Imatinib mesylate (Gleevec) is widely known for its ability to inhibit the tyrosine kinase activity of c-Abl and Bcr-Abl (Buchdunger et al., 1996). A previous study has shown that imatinib inhibited the growth of NSCLC cell line A549 in vitro (Zhang et al., 2003). We therefore tested the effects of imatinib on colony formation of H1299 cells in soft agar (Figure 4). Imatinib treatment (concentrations 1–5 μM) strongly inhibited the colony-forming ability of H1299 cells; both the number and size of colonies were inhibited (Figure 4). We note that imatinib had little effect on the proliferation of the H1299 cells in cell culture. So the effects of imatinib were confined to the oncogenic behavior of H1299 cells, as measured by formation of growth of the cells in soft agar.
Figure 4. Imatinib decreases colony formation of NSCLC cell line H1299 in soft agar.
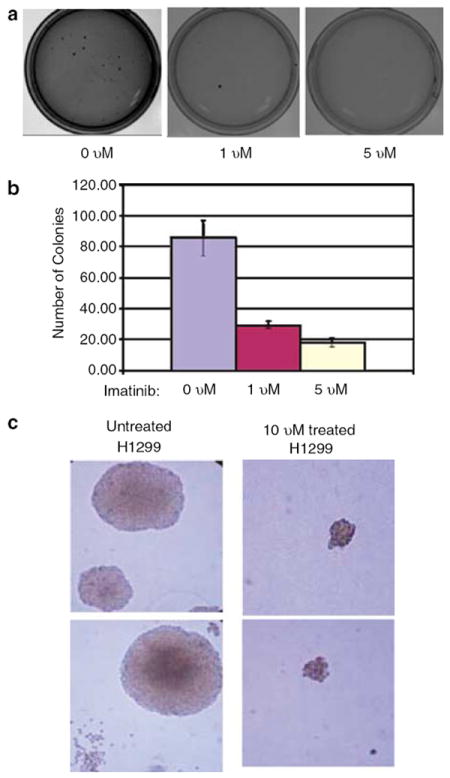
(a) A total of 1000 H1299 cells were plated in 0.35% soft agar containing either 0, 1, or 5 μM imatinib. Agar cultures were grown for 18 days changing the media every 2 days containing the respective concentrations of imatinib. (b) Dose-dependent inhibition of H1299 colony formation in soft agar by imatinib. (c) A total of 1000 H1299 cells were plated in 0.35% soft agar either containing 10 μM imatinib or untreated. Cells were grown for 18 days changing the media every 2 days containing the respective concentrations of imatinib.
H1299 cell expressing FUS1 leads to cell death and decrease kinase active c-Abl
Stable clones of human NSCLC H1299 cells were established by transfection with plasmid vectors containing wild-type FUS1 (wt-FUS1) or myristoylation-deficient mutant FUS1 (mt-FUS1, G2A) genes under the control of a ponasterone A (PA)-inducible promoter. After 48 h of induction with PA, H1299 cells expressing wt-FUS1 exhibit a steep increase in cell death, measured by transferase-mediated dUTP nick-end labeling (TUNEL) staining, whereas the empty vector (EV) and mt-FUS1 show little increase in cell death (Figure 5a). The tyrosine kinase activity of c-Abl was measured in these H1299 cells after 24 and 36 h of induction with PA. Tyrosine 245 of c-Abl is located in the linker domain between the SH2 domain and kinase domain. It is well known that phosphorylation of tyrosine 245 is critical to the activation of c-Abl kinase (Brasher and Van Etten, 2000). Not only is there a decrease in the phosphorylation of tyrosine 245 of c-Abl after FUS1 induction (Figure 5b, top panel) but induction of FUS1 leads to a decrease in the amount of c-Ab1 protein as well (Figure 5b, second panel). The total phosphotyrosine protein pattern of H1299 cells was also decreased upon FUS1 induction (Figure 5c), indicating that FUS1 expression decreased not only c-Abl activation (Figure 5b) but possibly targets of activated c-Abl as well. This is of interest since these cells have activated EGF receptor, which will most likely activate some of these targets as a result of tyrosine phosphorylation. Treatment of immune complexes of wild-type c-Abl with the stearate Fus1 peptide further supports that the Fus1–Abl interaction maylead to c-Abl degradation, as with increasing doses of stearate Fus1 peptide there is a decreasing amount of Abl protein (Figure 5d). In contrast, non-stearate Fus1 peptide and a stearate Bcr peptide did not induce degradation of c-Abl. Of interest, coexpression of wt-FUS1 with c-ABL in COS1 cells also reduced the level of the c-Abl protein (Figure 3a and d), suggesting the Fus1 interaction with c-Abl leads to its degradation in lung cells.
Figure 5. Induction of Fus1 in NSCLC H1299 cells induces cell death and decreases c-Abl kinase activity.
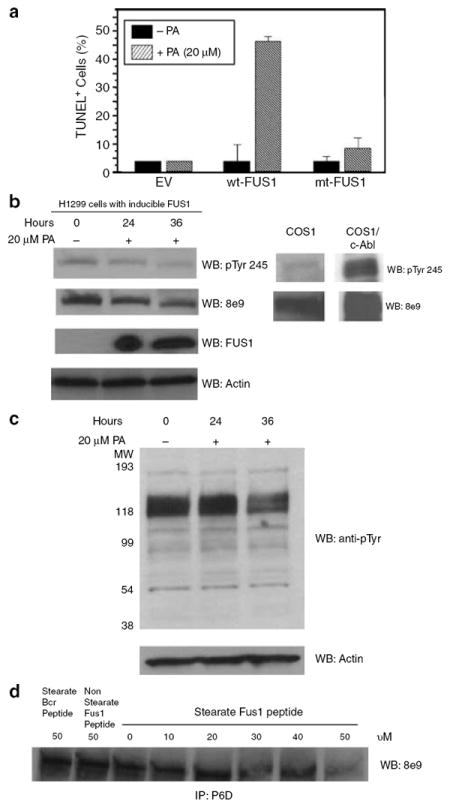
(a) Induction of FUS1 expression in H1299 NSCLC cells induces apoptosis. Stable clones of human NSCLC H1299 that express an inducible wild-type FUS1 (wt-FUS1) or myristoylation mutant FUS1 (mt-FUS1) were treated with of 20 μM of PA for 48 h. Apoptosis was measured by flow cytometry using a terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)-based fluorescence-activated cell sorter (FACS) analysis. DNA fragmentation was analysed by FACS. The relative apoptotic cells were calculated as the percentage of the TUNEL-positive cells in the total cell populations. (b) Induction of FUS1 expression inactivated c-Abl tyrosine kinase and reduced the level of the c-Abl protein. H1299 cells with inducible FUS1 were treated with PA for 24 and 36 h. Whole cell lysate was subjected to western blot and the membrane was probed with c-Abl pTyr 245 antibody (Abcam), c-Abl antibody 8e9 and N-terminal Fus1 antibody. COS1 cells transfected with c-Abl were used as a positive control. (c) FUS1 expression in H1299 cells reduces the level of phosphotyrosine-containing proteins. (d) Fus1 peptide decreases the level of the c-Abl protein in immune complexes of c-Abl from COS1. Immune complexes were treated with indicated amount of Fus1 peptide for 20 min at 30°C.
Discussion
Previous studies have demonstrated that FUS1 has antitumor activity (Kondo et al., 2001; Ji et al., 2002), but targets of the FUS1 gene have not been identified. In our studies, c-Abl was found to be negatively regulated by Fus1. Using a stearate Fus1 peptide derived from sequences lacking in the mutant FUS1 (1–80) (Kondo et al., 2001) seen in some NSCLC patients, we were able to inhibit the recombinant Abl kinase’s ability to phosphorylate Crk, a normal target of c-Abl (Feller et al., 1994) (Figure 1). Of note, increased expression of c-Crk is associated with aggressive phenotype in lung adenocarcinoma (Miller et al., 2003). Our results also show that only the stearic acid-modified form of the Fus1 peptide had inhibitory effects on purified c-Abl kinase (Figure 1). It has been shown that the myristoylation of FUS1 is required to produce the tumor suppression effects of FUS1 (Uno et al., 2004). Coincidentally, the N terminus of c-Abl (isoform 1b) also contains a myristate residue, which has been implicated in the autoinhibition of c-Abl (Hantschel et al., 2003).
Functional interaction of wild-type Fus1 and c-Abl was shown in cells by co-expression assays in COS1 cells (Figure 3). Phosphorylation of tyrosine 412 is a critical step that leads to the activation of c-Abl kinase (Brasher and Van Etten, 2000). Co-expression of wild-type FUS1 and c-ABL led to a significant decrease in both the total phosphotyrosine level of c-Abl and the level phosphotyrosine 412 content compared to only a very small decrease in either signal when c-Abl is co-expressed with either a truncated FUS1 [1–80], found in some NSCLC cell lines (Kondo et al., 2001), or the EV (Figure 3). This C-terminal deletion form of Fus1 lacks the peptide sequences, which are shown to inhibit c-Abl in kinase assays (Figure 1). In addition, it appears that Fus1 and c-Abl are in complex, as Fus1 and c-Abl co-precipitate with each other in IP assays (Figure 3a and d). Of note, the C-terminal truncated Fus1 still binds to c-Abl, but does not decrease phosphotyrosine-labeled c-Abl. The C-terminus of Fus1, the sequences found in the inhibitory Fus1 peptide, must contain a critical sequence that leads to the decrease of c-Abl kinase activation. These findings provide strong evidence for direct inhibition of c-Abl tyrosine kinase by the Fus1 protein.
Our studies have also demonstrated that NSCLC cell lines express the tyrosine kinase-active c-Abl (Figure 2). This result is interesting considering that c-Abl is a tightly regulated tyrosine kinase and its oncogenicity is usually associated with oligomeric sequences in a chimeric protein (for example, Bcr-Abl and Tel-Abl) in leukemia cells. It is also important to note that c-Abl is not overexpressed in these human NSCLC cell lines. In support of our findings showing activation of c-Abl in NSCLC lines, treatment of a NSCLC cell line with imatinib strongly inhibited colony formation in soft agar (Figure 4). Therefore, our findings identify c-Abl as a possible target of the TSG product FUS1 and indicate that c-Abl maybe a significant contributor to the oncogenicity of some forms of NCSLC, particularly those patients who are defective in FUS1 expression.
In previous studies, reduced expression of FUS1 or expression of defective mutants of FUS1, such as C-terminal deletion mutant of Fus1 (Kondo et al., 2001) (which lacks the c-Abl inhibitory sequence (Figure 1)) or expression of the myristoylated mutant (G2A) form of Fus1 (Uno et al., 2004) (the fatty acid stearic acid is required for the c-Abl inhibitory activity of the Fus1 inhibitory peptide (Figure 1)), are suggestive of an important role of Fus1 in controlling c-Abl and the oncogenic behavior of NSCLC cells. In support of the role of Fus1 in the regulation of c-Abl, we showed that forced expression of a full-length and functional Fus1 protein in H1299 cells induced apoptosis and inhibited the tyrosine kinase activity of c-Abl (Figure 5b). Of interest, not only was the c-Abl tyrosine kinase activity inhibited by forced Fus1 expression, but Fus1 expression in H1299 cells led to the reduction of the c-Abl protein in these cells (Figure 5b). Treatment of c-Abl immune complexes with the inhibitory stearate Fus1 peptide also reduced the level of c-Abl in these immune complexes in a dose-dependent manner (Figure 5d). Moreover, the non-stearate form of the Fus1 peptide and the stearate Bcr peptide had no effects on the level of c-Abl protein. On the basis of our previous studies with anti-Abl immunoprecipitates from Bcr-Abl + cells (Samanta et al., 2006), the degradative forces responsible for reduction of c-Abl protein in the immune complex are in the putative c-Abl network of proteins, or contaminating degradation enzymes in the immunoprecipitate are degrading the c-Abl in the c-Abl immune complex treated with stearate/Fus1 peptide. In summary, these findings suggest that Fus1 may negatively regulate the stability of c-Abl in lung cells.
Further, the facts that c-Abl tyrosine kinase can be activated downstream of EGFR via Src kinase (Plattner et al., 1999) and c-Abl plays a role in breast cancers overexpressing EGFR (Srinivasan and Plattner, 2006) strengthen the possibility of c-Abl contributing to the oncogenicity of NSCLC, especially since our studies show that the deficiency of expression of the functional FUS1 TSG contributes to increased active c-Abl kinase. Further, a recent study has shown that activated Abl kinase has a novel role in the regulation of EGFR endocytosis. The study shows that active Abl kinase phosphorylates EGFR and impairs EGFR internalization (Tanos and Pendergast, 2006). Thus, it seems likely that the well-known c-Abl inhibitor, imatinib, may have potential for the successful treatment of some forms of NSCLC.
Materials and methods
Cell lines and reagents
The human NSCLC cell lines, A549 and H1299, were obtained from Dr John Minna and colleagues. A549, H1299 and 32D P210 cells were cultured in RPMI (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS) and penicillin/streptomycin (Cambrex, Walkersville, MD, USA). Normal human lung fibroblast cell line, CCD16, was obtained from Dr Ray Meyn and were cultured in Alpha MEM (Invitrogen) with 10% FBS, 1mM non-essential amino acid mixture (Cambrex), MEM essential vitamin mixture (100 ×) (Cambrex), and penicillin/streptomycin. COS1 cells and Rat 1 SH2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) with 10% FBS and penicillin/streptomycin. Imatinib mesylate was purchased from the MD Anderson pharmacy and dissolved in sterilized Millipore water. PA was purchased from Invitrogen.
Kinase assays
Cell-free kinase assays using a bacterially purified, constitutively activated, commercial truncated 45 kDa Abl containing only the SH2 and kinase domain (New England Biolabs, Ipswich, MA, USA) were performed according to the manufacturer’s instruction using γ-32P ATP mixed with 100 μM cold ATP. GST–Crk was prepared as described and used as a substrate for the Abl tyrosine kinase. Reactions were run on an 8% SDS-PAGE gel and exposed to phosphoimager. Cell-free kinase assays, using an insect–purified, human, full-length active Abl (Upstate, Lake Placid, NY, USA), were performed as described by the manufacturer’s instructions, using the suggested Abl target peptide, EAIYAAPFAKKK. Immune complex kinase assays used procedures as described previously (Liu et al., 1996). The Abl target peptide absorbs to Whatman P81 filters and the filters in triplicate are counted in a liquid scintillation spectrometer.
Immunoprecipitation and western blotting
These procedures were described previously (Liu et al., 1996). Antibodies used were Abl antibody P6D, raised against c-Abl 51–64 residues (Liu et al., 1993), anti-Abl SH2 domain antibody, 8e9, anti-phosphotyrosine antibody 4G10 (Upstate, Charlottesville, VA, USA), anti-Abl-phosphotyrosine 245 (Abcam, Cambridge, MA, USA), anti-Abl-phosphotyrosine 412 (Abcam), and Fus1 antibodies were obtained from Dr Lin Ji.
Co-transfection assays
c-ABL was co-transfected with either wild-type full-length FUS1 (1–110), truncated FUS1 (1–80), or EV in COS1 cells. Fugene 6 (Roche, Indianapolis, IN, USA) was used for the co-transfection assay.
Generation of stable expressing FUS1 H1299 cell line
Stable clones of human NSCLC H1299 cells were established by transfection with plasmid vectors containing wt-FUS1 or myristoylation-deficient mutant FUS1 (mt-FUS1) genes under the control of a PA-inducible promoter and with a Neomycin-resistant gene as a selective marker. The cell clone containing EV without the gene insert was used as a negative control.
Colony formation in soft agar assays and foci formation
H1299 cells were seeded on to dishes in 0.35% agar; cells were maintained until visible colonies were observed. Plates were stained with crystal violet.
TUNEL assay
Apoptosis was measured by flow cytometry using a terminal deoxynucleotidyl TUNEL-based fluorescence-activated cell sorter (FACS) analysis. Cells were fixed in 1% paraformaldehyde, permeabilized with 70% ethanol, washed with PBS and stained with propidium iodide (PI) solution containing 40 g/ml of PI and 10 g/ml DNase-free RNase A. DNA fragmentation was analysed by FACS.
Acknowledgments
This work was funded in part by the Lung SPORE Grant P50CA70907. We thank Professor Ray Meyn for supplying the CCD16 normal lung fibroblasts.
References
- Brasher B, Van Etten R. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J Biol Chem. 2000;275:631–637. doi: 10.1074/jbc.M005401200. [DOI] [PubMed] [Google Scholar]
- Buchdunger E, Zimmerman J, Mett H, Meyer T, Muller M, Druker B, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–104. [PubMed] [Google Scholar]
- Das A, Sato M, Story M, Peyton M, Graves R, Redpath S, et al. Non-small cell lung cancers with kinase domain mutations in the epidermal growth factor receptor are sensitive to ionizing radiation. Cancer Res. 2006;66:9601–9608. doi: 10.1158/0008-5472.CAN-06-2627. [DOI] [PubMed] [Google Scholar]
- Dowell J, Minna J. Chasing mutations in the epidermal growth factor in lung cancer. N Eng J Med. 2005;352:830–832. doi: 10.1056/NEJMe058033. [DOI] [PubMed] [Google Scholar]
- Feller SM, Ren R, Hanafusa H, Baltimore D. SH2 and SH3 domains as molecular adhesives: the interactions of Crk and Abl. Trends Biochem Sci. 1994;19:453–458. doi: 10.1016/0968-0004(94)90129-5. [DOI] [PubMed] [Google Scholar]
- Girard L, Zochbauer-Muller S, Virmani A, Gazdar A, Minna J. Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer Res. 2000;60:4894–4906. [PubMed] [Google Scholar]
- Hantschel O, Nagar B, Guettler S, Kretzschmar J, Dorey K, Kuryian J, et al. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell. 2003;112:845–857. doi: 10.1016/s0092-8674(03)00191-0. [DOI] [PubMed] [Google Scholar]
- Ito I, Ji L, Tanaka F, Saito Y, Gopalan B, Branch C, et al. Liposomal vector mediated delivery of the 3p FUS1 gene demonstrates potent antitumor activity against human lung cancer in vivo. Cancer Gene Ther. 2004;11:733–739. 1–7. doi: 10.1038/sj.cgt.7700756. [DOI] [PubMed] [Google Scholar]
- Ji L, Nishizaki M, Gao B, Burbee D, Kondo M, Kamibayashi C, et al. Expression of several genes in the human chromosome 3p21.3 homozygous deletion region by an adenovirus vector results in tumor suppressor activities in vitro and in vivo. Cancer Res. 2002;62:2715–2720. [PMC free article] [PubMed] [Google Scholar]
- Kondo M, Ji L, Kamibayashi C, Tomizawa Y, Randle D, Sekido Y, et al. Overexpression of candidate tumor suppressor gene FUS1 isolated from the 3p21.3 homozygous deletion region leads to G1 arrest and growth inhibition of lung cancer cells. Oncogene. 2001;20:6258–6262. doi: 10.1038/sj.onc.1204832. [DOI] [PubMed] [Google Scholar]
- Ling X, Ma G, Sun T, Liu J, Arlinghaus R. Bcr and Abl interaction: oncogenic activation of c-Abl by sequestering Bcr. Cancer Res. 2003;63:298–303. [PubMed] [Google Scholar]
- Liu J, Wu Y, Arlinghaus R. Sequences within the first exon of BCR inhibit the activated tyrosine kinases of c-Abl and the Bcr-Abl oncoprotein. Cancer Res. 1996;56:5120–5124. [PubMed] [Google Scholar]
- Liu J, Campbell M, Guo J, Lu D, Xian Y, Andersson B, et al. BCR-ABL tyrosine kinase is autophosphorylated or transphosphorylates P160 BCR on tyrosine predominantly within the first BCR exon. Oncogene. 1993;8:101–109. [PubMed] [Google Scholar]
- Miller C, Chen G, Gharib T, Wang H, Thomas D, Misek D, et al. Increased C-CRK proto-oncogene expression is associated with an aggressive phenotype in lung adenocarcinomas. Oncogene. 2003;22:7950–7957. doi: 10.1038/sj.onc.1206529. [DOI] [PubMed] [Google Scholar]
- Pendergast A. The Abl family kinases: mechanisms of regulation and signaling. Adv Cancer Res. 2002:51–100. doi: 10.1016/s0065-230x(02)85003-5. [DOI] [PubMed] [Google Scholar]
- Plattner R, Kadlec L, DeMali K, Kazlauskas A, Pendergast A. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13:2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluk H, Dorey K, Superti-Furga G. Autoinhibition of c-Abl. Cell. 2002;108:247–259. doi: 10.1016/s0092-8674(02)00623-2. [DOI] [PubMed] [Google Scholar]
- Samanta A, Lin H, Wang Y, Kantarjian H, Arlinghaus R. Janus kinase 2: a critical target in chronic myelogenous leukemia. Can Res. 2006;66:6468–6472. doi: 10.1158/0008-5472.CAN-06-0025. [DOI] [PubMed] [Google Scholar]
- Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66:5648–5655. doi: 10.1158/0008-5472.CAN-06-0734. [DOI] [PubMed] [Google Scholar]
- Taagepera S, McDonald D, Loeb J, Whitaker L, McElroy A, Wang J, et al. Nuclear-cytoplasmic shuttling of C-ABL tyrosine kinase. Proc Natl Acad Sci. 1998;95:7457–7462. doi: 10.1073/pnas.95.13.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanos B, Pendergast AM. Abl tyrosine kinase regulates endocytosis of the epidermal growth factor receptor. J Biol Chem. 2006;28:32714–32723. doi: 10.1074/jbc.M603126200. [DOI] [PubMed] [Google Scholar]
- Uno F, Sasaki J, Nishizaki M, Carboni G, Xu K, Atkinson E, et al. Myristoylation of the fus1 protein is required for tumor suppression in human lung cancer cells. Cancer Res. 2004;64:2969–2976. doi: 10.1158/0008-5472.can-03-3702. [DOI] [PubMed] [Google Scholar]
- US Cancer Statistics Working Group. US Cancer Statistics: 2001 Incidence and Mortality. US Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; Atlanta, GA: 2004. [Google Scholar]
- Wang J. Regulation of cell death by the Abl tyrosine kinase. Oncogene. 2000;19:5643–5650. doi: 10.1038/sj.onc.1203878. [DOI] [PubMed] [Google Scholar]
- Wen S, Van Etten R. The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 1997;11:2456–2467. doi: 10.1101/gad.11.19.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodring P, Hunter T, Wang J. Regulation of F-actin-dependent processes by the Abl family of tyrosine kinases. J Cell Sci. 2003;116(PT 13):2613–2626. doi: 10.1242/jcs.00622. [DOI] [PubMed] [Google Scholar]
- Zabarovsky E, Lerman M, Minna J. Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene. 2002;21:6915–6935. doi: 10.1038/sj.onc.1205835. [DOI] [PubMed] [Google Scholar]
- Zhang P, Gao W, Turner S, Ducatman B. Gleevec (STI-571) inhibits lung cancer cell growth (A549) and potentiates the cisplatin effect in vitro. Mol Cancer. 2003;2:1. doi: 10.1186/1476-4598-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhang X, Bai C, Chen J, Wei M. Inhibition of epidermal growth factor receptor expression by RNA interference in A549 cells. Acta Pharmacol Sin. 2004;25:61–67. [PubMed] [Google Scholar]