

Abstract
We describe a negative feedback autocrine regulatory circuit for glucose-stimulated insulin secretion in purified human islets in vitro. Using chronoamperometry and in vitro glucose-stimulated insulin secretion measurements, evidence is provided that dopamine (DA), which is loaded into insulin-containing secretory granules by vesicular monoamine transporter type 2 in human β-cells, is released in response to glucose stimulation. DA then acts as a negative regulator of insulin secretion via its action on D2R, which are also expressed on β-cells. We found that antagonism of receptors participating in islet DA signaling generally drive increased glucose-stimulated insulin secretion. These in vitro observations may represent correlates of the in vivo metabolic changes associated with the use of atypical antipsychotics, such as increased adiposity.
The β-cells of the islets of Langerhans integrate a variety of external signals into the timely release of physiologically appropriate amounts of insulin and thus accurately regulate blood glucose levels commensurate with metabolic demand. Some external signals act as amplifying agents that have little or no effect by themselves but enhance the sensitivity of the β-cell glucose-sensing apparatus (reviewed in Ref. 1). For example, certain amino acids synergize with d-glucose in promoting insulin secretion by β-cells. Net insulin production and glucose homeostasis is regulated by other small molecules as well, including several classical neurotransmitters (2, 3) that act directly on β-cells and indirectly through other tissues active in glucose homeostasis such as liver and skeletal muscle. Neurotransmitters participating in glucose homeostasis can be released from sympathetic and parasympathetic innervation, the adrenal medulla, or as we demonstrate in this report, directly from islets acting in an autocrine or paracrine manner to regulate islet insulin secretion.
Comparative microanatomy of human vs. rodent islets and islet innervation reveals important differences that may impact operant mechanisms of glucose homeostasis (4). Relative to the structure of mouse islets, human islets are sparsely innervated with few contacts to autonomic and cholinergic axons (5). Moreover, in human islets, sympathetic axons are associated with the smooth muscle cells of blood vessels located around and deep within the islet rather than directly contacting β-cells. To reconcile the apparent autonomy of human islets with the known effects of autonomic stimulation on rodent islet hormone secretion, it has been suggested that neurotransmitter spillover from innervation might be responsible for downstream effects on hormone secretion (6). However, an alternate possibility is autocrine and/or paracrine release of insulin secretory modulators.
Negative feedback regulation and paracrine or autocrine signaling are common control mechanisms within the central nervous system (CNS). For example, in mammalian brain, the nigrostriatal dopamine (DA) system is necessary for voluntary motor activity. It is well established that the activity of striatal neurons is regulated by autoregulatory negative feedback loops (reviewed in Ref. 7) where released DA acts on presynaptic DA type 2 receptors (D2R) to decrease DA synthesis and release (8), thereby reducing downstream signaling to postsynaptic neurons.
As in the CNS, gene expression studies reveal that human islet tissue expresses a variety of molecules associated with the biosynthesis, storage, degradation, and response to several neurotransmitters (9), including DA (10). β-Cells express vesicular monoamine type 2 transporters (VMAT2) (11), a molecule critical for the vesicular storage of DA (12), and DA type 1–5 receptors (13), and DA is present in rodent β-cell vesicles (14). In this report, we show evidence that DA is stored within human pancreatic islets, released in response to glucose stimulation, and acts on D2R (also expressed by human β-cells) resulting in the down-regulation of insulin secretion. The existence of a DA-mediated negative feedback regulatory circuit in human islets may be particularly relevant in the context of the association between the use of atypical antipsychotic drugs (ATA) and development of metabolic syndrome and type 2 diabetes (T2D). Given that the single unifying property of ATA is their D2R antagonist activity, the prediction is that D2R blockade would blunt the endogenous DA- and D2R-mediated negative feedback in glucose-stimulated insulin secretion (GSIS), and we provide evidence that this is indeed the case in human islets.
Materials and Methods
Drugs and reagents
GBR 12909 dihydrochloride (vanoxerine), benzothiophenylcyclohexylpiperidine (BTCP), α-methylparatyrosine (AMPT), haloperidol hydrochloride, serotonin (5-HT), sulpiride, DA hydrochloride, quinpirole hydrochloride, clozapine, and d-glucose were obtained from Sigma-Aldrich Corp. (St. Louis, MO). Tetrabenazine (TBZ) was obtained from Tocris Bioscience (Ellisville, MO). Dihydrotetrabenazine (DTBZ) was obtained from the National Institute of Mental Health's Chemical Synthesis and Drug Supply Program. Olanzapine was obtained from E. Lilly (Indianapolis, IN). [ring 2,5,5-3H]DA was obtained from American Radiolabeled Chemicals (St. Louis, MO). All other chemicals were of the highest commercial quality available.
Pancreas and islet procurement and islet culture
Whole human pancreata from donors without known history of diabetes and fixed in 10% neutral buffered formalin were procured from the National Disease Research Interchange (Philadelphia, PA). Human islets isolated from cadaveric nondiabetic donors were obtained from the Integrated Islet Distribution Program (City of Hope National Medical Center, Duarte, CA). The average purity of islets was 90 ± 5% (sem) as determined by dithizone staining, the average age of the donors (n = 36) was 42 ± 2 yr (sem). The average body mass index was 32 ± 1 (sem). The isolated human islets were normally cultured in supplemented CMRL-1066 medium for no longer than 2 d before being shipped. On arrival, islets were placed in CMRL-1066 medium containing 5.5 mm glucose, 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin and incubated at 37 C with 5% CO2. All cell culture media and supplements were obtained from Life Technologies (Grand Island, NY). Tissue culture plates were obtained from Falconware (Becton-Dickinson, Inc., Oxnard, CA). Islets used in these analyses were cultured for at least 24 h but for no longer than 5 d. All experiments regarding human islets and pancreata were approved by our Institutional Review Board.
Immunohistochemistry and colocalization microscopy
Fixed pancreas tissue was embedded in paraffin and 5-μm sections on glass slides prepared. Tissue was initially evaluated by hematoxylin and eosin staining; if autolysis was present, specimens were not evaluated in the study. Next, sections were stained for both insulin and D2R. The primary antibodies used, guinea pig antiinsulin (A0564; Dako, Carpinteria, CA) and a rabbit polyclonal anti-D2R (PA1-23584; Thermo Scientific, Rockford, IL), were chosen based on previous experience and published research (15). Primary antibodies were incubated on paraffin-embedded slides overnight at 4 C with the exception of insulin. Insulin was incubated for 2 h at room temperature. Secondary antibodies conjugated to fluorescein isothiocyanate or Cy5 were diluted to 1:100 (Jackson ImmunoResearch, West Grove, PA). All slides were coverslipped with Vectashield 4′,6-diamidino-2-phenylindole mounting medium (Vector Laboratories, Burlingame, CA), stored in the dark at 4 C, and analyzed within 1 d of staining. Slides were evaluated on a Nikon Eclipse 80i microscope using QCapture 51 to image insulin and D2R immunofluorescent colocalization.
Analysis of D2R, VMAT2, DAT, and LAT1 transcripts by RT-PCR
Human pancreas (catalog item 540023-41) and striatum (catalog item 540135-41) total RNA were from Agilent Technologies (MVP human normal adult tissue total RNA; Santa Clara, CA). Total RNA from islets or purified acinar tissue (9) was isolated using the RNA RNeasy Mini Kit (QIAGEN, Valencia, CA). cDNA was generated using the VILO cDNA synthesis kit (Life Technologies). All PCR assays were performed using the amount of cDNA obtained retro-transcribing 30 ng total RNA. Platinum PCR SuperMix High Fidelity (Life Technologies) was used for semiquantitative PCR assays at the conditions recommended by the manufacturer; in particular, 40 cycles of PCR amplification were performed with an annealing temperature of 56 C and an extension time of 45 sec. Accumulation of specific transcripts was measured by real-time PCR, using the SmartCycler System (Cepheid, Sunnyvale, CA). The QuantiTect SYBR Green PCR Kit (QIAGEN) was used to perform all the quantitative PCR assays with annealing temperatures of 55–60 C and extension times of 30–45 sec, depending on the couple of primers used. All the primers (Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org) were synthesized by Eurofins MWG Operon (Huntsville, AL), except for those specific for human β-actin (QuantiTect Primer Assay; QIAGEN). Quantitative RT-PCR reagent controls (reagents without any template or with 30 ng non-retro-transcribed RNA) were included in all the assays. Each assay was run in triplicate and independently repeated at least three times to verify the results; the mean copy number was used for analysis. The relative amount of specific transcripts was calculated by the comparative cycle threshold method given by Schmittgen and Livak (16). To correct for sample to sample variations in quantitative RT-PCR efficiency and errors in sample quantitation, the level of ACTB transcripts was tested for use in normalization of specific RNA levels.
Experimental animals
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee at Columbia University's College of Physicians and Surgeons. Male Sprague-Dawley (SD) and Zucker obese rats were obtained from Harlan Laboratories (Somerville, NJ). Rodents were housed under conditions of controlled humidity (55 ± 5%), temperature (23 ± 1 C), and lighting (lights on 0600–1800 h) with ad libitum access to standard laboratory rat chow and water.
Intraperitoneal glucose tolerance testing (IPGTT)
IPGTT was performed as previously described in 6-h-fasted unanesthetized rats as described previously (17). Sixty minutes before IPGTT, isoflurane anesthesia of male rats was induced and maintained in an oxygen mixture. The anesthetized rats were administered TBZ, GBR 12909, or vehicle alone at the indicated dose by iv injection at the penile vein. The animals were fully recovered for at least 30 min before receiving IPGTT, which was performed without anesthesia.
Insulin, DA, and DNA assays
Human insulin measurements were performed using the AlphaLISA human insulin research immunoassay (kit AL2904Cm; PerkinElmer, Waltham MA). Rat Insulin measurements were performed using ELISA (kit DOP31-K01; Eagle Bioscience, Nashua, NH). DA measurements were performed using ELISA (kit BA E-6300; Rocky Mountain Diagnostics, Colorado Springs, CO). All measurements were made following the kit manufacturer's protocols using a BioTek Synergy 2 reader with the appropriate filters (BioTek, Winooski, VT) DNA concentration measurements, used to normalize the mass of islets used in the static incubation experiments, were performed using the QuantiFluor double-stranded DNA system (catalog item E2670; Promega Corp., Madison, WI) according to manufacturer's recommendations.
Human islet insulin secretion and DA uptake assays
For the static incubation experiments, human islets were washed once in Krebs Ringers bicarbonate buffer (KRBB) (125 mm NaCl, 4.8 mm KCl, 2.6 mm CaCl2, 1.2 mm MgCl2, 25 mm NaHCO3, 10 mm HEPES (final pH 7.4) supplemented with 0.2% (wt/vol) serum albumin, preheated to 37 C, and bubbled with 95% O2 and 5% CO2] and then preincubated for 1 h in KRBB with 1.5 mm glucose (basal conditions).
In some experiments, this preincubation step was performed in the presence of 250 mg/liter AMPT. Islets were washed again in KRBB and seeded into 48-well tissue culture plates (BD Falcon 353230; BD Biosciences, Bedford, MA) at 100–500 islets/ml × 0.5 ml/well in KRBB. Solutions containing the indicated drug and/or glucose were prepared at twice the indicated concentration in KRBB (e.g. 30 mm glucose plus 100 nm DTBZ). These prewarmed (37 C) solutions (0.5 ml) were then added to wells with islets to yield the indicated final concentrations. Islets were then cultured for 1 h at 37 C with an atmosphere of 5% CO2/95% Air. At the end of each incubation period, a 0.5-ml aliquot of the culture medium was carefully collected to avoid aspiration of islets and frozen (−80 C) for analysis of insulin concentration and the remaining 0.5 ml set aside and frozen (−80 C) for later DNA measurements. The insulin concentrations measurements were normalized to the DNA content of the well to compensate for the variability of the well-seeding technique.
For the perfusion experiments, eight to 18 islets (diameters > 200 μm) were perfused in a temperature- and pH-controlled 100-μl chamber slide (Ibidi μslide I luer, 0.4 mm height; AutoMate Scientific, Inc., Berkeley, CA USA) and fractions collected for later analysis of insulin concentration. The perfusion apparatus consisted of 1) a temperature- and pH-controlled reservoir containing KRBB with constant aeration, connected by C-Flex tubing to 2) a peristaltic pump (dual-channel microperfusion pump; AutoMate Scientific), followed by 3) an inline solution heater and 4) the Ibidi chamber slide. The Ibidi chamber slide was mounted on a temperature-controlled transparent glass stage (TC-invivo; Bioscience Tools, San Diego, CA). Feedback control of the chamber temperature and perfusate was achieved with an inline temperature probe connected to a temperature controller (TC-TP; Bioscience Tools). The line out of the Ibidi chamber was covered with a 50-μm nylon mesh to prevent escape of islets, and the 100-μl chamber was partially filled with inert polystyrene beads (160–200 μm diameter; Solohill Engineering Inc., Ann Arbor, MI) to reduce chamber dead volume and aid in the enumeration and measurement of islets and their diameters. The chamber line out was connected by 0.031-in. tubing to a Gilson fraction collector.
Islets in KRBB were loaded into the Ibidi chambers, and the contents of the transparent chambers were examined and the number of islets and their size recorded. The filled chambers were placed in the apparatus and perfused at a rate of 600–700 μl/min with KRBB with 1.5 mm glucose for 30 min to equilibrate. At time zero, the collection of 0.5-min fractions began. Twenty minutes later, the glucose concentration was raised to 15 mm glucose in the presence and absence of TBZ and an additional 30 min of islet perfusion was performed. Collected fractions were stored at −80 C for later assay of insulin content by AlphaLISA. The average coefficient of variation for AlphaLISA-based insulin measurements was less than 12%. At the end of the experiment, the chamber was reexamined to confirm the islet count. In some experiments, where DA and insulin concentration measurements were both measured in the same fraction, we increased the number of islets per chamber to 1200 ± 200 to allow simultaneous detection by ELISA.
Measurements of tritiated DA uptake by islets were performed as follows: islets were seeded (about 500 per well) into BD Falcon cell culture inserts for 24-well plates (8.0-μm pores, translucent polyethylene terephthalate membrane) containing 0.5 ml KRBB with 1.5 or 8.0 mm glucose and 0.5% wt/wt BSA. To some wells, haloperidol was added to a final concentration of 10 μm. Islets were next incubated at 37 C in an atmosphere of 5% CO2/95% Air for 45 min. Next, some wells received GBR 12909 and/or BCTP to a final concentration of 10 μm. Islets were incubated for 30 min followed by the addition of 1 × 107 decays per minute (dpm)/well of [3H]DA. Islet cultures were incubated for 1 h and followed by washing and harvesting. Islets within the inserts were washed three times with 1 ml cold 4 C Ringers with 0.5% BSA using gentle suction to not disrupt the islets. After the third wash, 250 μl 0.1 n HCl was added to the insert and the contents frozen at −80 C until analysis. Upon thawing, 25 μl supernatant was removed for DNA assay and the remainder mixed with 5 ml Ecolite scintillation cocktail. Samples were counted in a calibrated liquid scintillation counter, and the background-corrected disintegrations per minute for each sample was calculated and then normalized to the well's DNA content.
Human islet glucose-stimulated DA release assays
Nafion-coated carbon fiber microelectrodes (nCFM) (diameter 30 μm × length 100 μm) were obtained from World Precision Instruments, Sarasota, FL (CFN30-100) or coated de novo as previously described (18). Chronoamperometric and voltammetry measurements were performed with a MicroC potentiostat (World Precision Instruments) using a combined reference/auxiliary electrode. The reference electrode was a dry-type Ag/AgCl cylinder (World Precision Instruments), encapsulated in an insulating tube, as supplied with the MicroC such that the active surface was a 2-mm-diameter Ag/AgCl disc. The time constant on the MicroC was set at 1 msec rise time. To drive the potentiostat, we used a Rigol DG1000 series function/arbitrary waveform generator (Rigol USA, Oakwood Village, OH). The MicroC potentiostat converts detected currents to a voltage signal that was collected using a 100-msec sampling period via a high-resolution USB data logger (ADC-20; Pico Technology, Cambridgeshire, UK) and the supplied PicoLog data acquisition software. The data signal was low-pass filtered (<50 Hz) to remove all noise at frequencies above 50 Hz from the signal before sampling and then stored in a personal computer in real time. Data reduction was performed using Excel software (Microsoft Corp., Redmond, WA) and macros created with the visual basic editor.
Before using the electrodes to measure DA release by islets in vitro, we characterized the in vitro response of the nCFM to both authentic DA and 5-HT. The nCFM were first preconditioned in PBS by applying a 70-Hz triangle wave (from 0 to +3.0 V relative to the Ag/AgCl reference electrode) for 20 sec, followed by holding the electrode in PBS at 1.5 V for 20 sec (19), followed by holding the electrode in 150 mm NaCl (pH 9.5) at 1.2 V for 5 min. Cyclic voltammograms for KRBB alone, KRBB with authentic DA added, and KRBB with authentic 5-HT added were collected using the following parameters: waveform, triangular; period, 8 sec; applied voltage maximum, 1000 mV; applied voltage minimum, −200 mV; scan rate, 350 V/sec, and duration, more than 12 cycles. The potentiostat gain used was 1 nA/mV to 100 pA/mV (see Supplemental Information).
In preliminary experiments, we found that using our instrumentation, the application of cyclic voltammetry to the measurement of DA release was insensitive to DA concentrations below 100 nm. Instead, we applied a related previously described square-wave or normal-pulse voltammetry/chronoamperometry technique (20–22). After the activation of the nCMF electrodes described above, square-wave pulses corresponding to the oxidation peak of DA were applied to the detection of DA released from islets in vitro. The parameters used were as follows: waveform, square; duty cycle, 50%; applied voltage maximum, +220 mV; applied voltage minimum, + 80 mV; and period, 8 sec. The potentiostat gain was set a 1 pA/mV. The nCFM and reference electrode were placed in the well of a 24-well tray filled with 1.0 ml Ringers buffer and the background current allowed to stabilize for 10 min under an applied voltage of +220 mV, after which the average background current was set to 0. The working electrode was situated 1–2 mm above the well floor. The tray and its contents were maintained at 37 C in a temperature-monitored micro-incubator. Next, the square waveform was applied to KRBB in the well, and the current vs. time profile generated by the applied voltage profile was collected for 20 min. No current was observed in KRBB supplemented with up to 20 mm glucose in the absence of added islets. During this time, human islets were washed in warm (37 C) KRBB and suspended at 1000 islets/ml in 0.5 ml KRBB. At time zero, the data collection was reinitiated, and 0.5 ml KRBB was removed from the well and replaced with the 0.5-ml suspension of human islets in KRBB. At 15 min, the glucose concentration was raised to 1.5 mm with a concentrated solution of glucose prepared in Ringers, at 30 min, the glucose concentration in the well was again raised to 15 mm glucose. At 1 h, data acquisition was stopped, and two thirds of culture supernatant was carefully removed to not disturb the islets and replaced with fresh warm KRBB four times, resulting in a dilution of glucose to a final concentration of less than 0.2 mm. Data acquisition was started again, and after 20 min, the glucose concentration was raised to 30 mm glucose. At 120 min, the potentiostat gain was reduced to 10 pA/mV from 1 pA/mV, and the islet-containing well was spiked with authentic DA to a final concentration of 500 nm. About 8 min later, an additional spike of authentic DA was added to bring the final concentration to 800 nm DA.
Statistical evaluation
Data are presented as the mean ± sem. The significance of comparisons of statistical means were calculated using the two-tailed Students t test. Statistically significant insulin pulse pulses were identified with the Cluster Analysis program, a computerized pulse analysis algorithm (23). Test cluster sizes for the nadir and peak widths were assigned to 4. The minimum t statistics was specified to 4.0 for both upstrokes and downstrokes. The settings detected peaks with less than 1% false-positive errors. Cluster analyses also provided information regarding peak height and periodicity. A sliding two-tailed Student's t test with a three-point window was also used to compare peak heights between DTBZ-treated and untreated islet perfusion profiles.
Results
D2R colocalize with insulin staining in human pancreas sections and are found predominantly in islets
Rubí et al. (13) showed colocalization of insulin and D2R in purified human islets. In sections of human pancreas, we examined the distribution of D2R and its relationship to insulin staining of islets in situ (Fig. 1). The staining pattern of D2R and its overlap with insulin staining is similar to that of VMAT2 (15) and suggests that D2R is present in insulin granules on resting β-cells. The fluorescent immunohistochemical labeling of sections also showed that within the pancreas, most of the D2R expression was limited to β-cells.
Fig. 1.

Human D2R and insulin staining in a representative pancreas section and islet from a healthy nondiabetic subject. Left panels, Immunofluorescent staining of a representative islet from a nondiabetic subject for D2R (top, green), insulin (middle, red), merged with 4′,6-diamidino-2-phenylindole staining (bottom); right panel, wide-field view of section showing islet and distribution of D2R and insulin staining. D2R staining was confined mainly to the insulin-positive islet.
D2R are highly expressed in the striatum, on dopaminergic presynaptic neurons as well as on medium spiny neurons and corticostriatal neurons. Alternative splicing of the D2R gene results in at least two transcript variants encoding different isoforms: the abundant long isoform (D2RL) including 29 amino acids derived from exon 6 and a short isoform (D2RS) missing these amino acids (24). To further understand the function of D2R in β-cells, we examined the expression of D2R at the transcript level in human total pancreas and purified cadaveric islets (Fig. 2). Using primers specific for both long and short isoforms, short isoform alone, long isoform alone, and total D2R (Supplemental Table 1), we amplified cDNA prepared from pancreas, purified islets, and as a control, human striatum. We found that, similar to striatum, D2RL was the predominantly expressed D2R isoform in human islets with D2RS not visible under the experimental conditions. Furthermore, the sum of the transcripts derived from all the known isoforms of D2R mRNA was higher in islets relative to total pancreas. The amount of D2R message in human islets was comparable to that found in human striatum.
Fig. 2.

Expression of D2R transcripts in human striatum, pancreas, and purified cadaveric islets. A and B, RT-PCR semiquantitative assay using the primer pairs 4hD2_F/3hD2_R (A) and 4hD2_F/4hD2_R (B), amplifying cDNA derived from both the long and short mRNA isoform of D2R. The 234-bp and 532-bp products expected from the amplification of D2RS cDNA are not visible in A and B at 40 cycles. When the reaction in A was extended to 55 cycles, the D2RS cDNA 234-bp produce became visible. C, RT-PCR semiquantitative assay using the primer pair Long_hD2_F/Long_hD2_R, specific for the long isoform of D2R. D, RT-PCR semiquantitative assay using the primer pair Short_hD2_F/3hD2R, specific for the short isoform of D2R. The 215-bp product expected from the amplification of D2RS cDNA is not visible. When the reaction in D was extended to 55 cycles, the D2RS cDNA 215-bp product became visible. E, RT-PCR semiquantitative assay using the primer pair 2hD2_F/2hD2R, amplifying cDNA derived from all the known mRNA isoforms of D2R. F, RT-PCR semiquantitative assay using the primer pair Hs_ACTB_2_SG, amplifying β-actin-specific cDNA. This figure shows the results of one of five similar experiments. All primer pairs and primer sequences are listed in Supplemental Table 1.
We used real-time PCR with the same set of primers to precisely quantitate levels of specific D2R isoforms (Fig. 3) in purified human islet, total human pancreas, and human striatum cDNA. We found that 1) in human islets, the abundance of D2RL and D2RS transcripts is approximately 5-fold greater than that found in human striatum and 50 times higher than that found in total pancreas; 2) the amount of D2RL-specific mRNA paralleled the amount of total D2R transcripts; and 3) the copy number of D2RS-specific mRNA, although detectable, was low and consistent with the semiquantitative results shown in Fig. 2 (data not shown).
Fig. 3.

Expression of D2R, insulin (INS), SLC7A5, SLC3A2, SLC18A2, and SLC6A3 mRNA in purified human cadaveric islets by quantitative RT-PCR. cDNA was amplified with the primer pair 2hD2_F/2hD2R, amplifying all D2R-specific cDNA (D2R), the primer pair Long_hD2_F/Long_hD2_R specific for the long isoform of D2R (D2RL). The amounts of transcripts in each tissue were normalized to ACTB transcript content in the respective samples and the results for D2R are reported as relative to the amount of specific cDNA found in striatum, which was arbitrarily set to 1. The results for insulin, SLC7A5, SLC3A2, SLC18A2, and SLC6A3 are reported as relative to the amount of specific cDNA found in whole pancreas, which was arbitrarily set to 1. Error bars show the sem. The differences in expression of D2R between striatum, pancreas, and islets were significant at the P < 0.05 level using Student's t test. *, Significant difference from whole pancreas or exocrine tissue at the P < 0.05 level using Student's t test.
In a separate set of experiments, we probed cDNA, prepared from whole pancreas, purified islets, and purified acinar exocrine tissue, for the expression of insulin (INS), as an islet purity control, and four other products of the SLC (solute carrier) gene superfamily, SLC7A5 (also known as LAT1 or CD98 light chain), SLC3A2 (also known as MDU1 or CD98), SLC18A2 (also known as VMAT2), and SLC6A3 (also known as DA reuptake transporter, DAT) relevant to a putative DA-mediated negative feedback regulatory circuit of insulin secretion. As expected, both insulin and VMAT2 gene expression was more than 30-fold enriched in islets relative to whole pancreas and more than 4 × 103-fold greater than the level measured in purified exocrine tissue. We also found that islets were enriched, relative to whole pancreas or pancreas exocrine tissue, for the expression of SLC7A5, SLC3A2, and SLC6A3.
Antagonism of monoamine transporters (MAT) and DA receptors enhances insulin secretion by human islets in vitro
The transport of monoamine neurotransmitters (e.g. DA) across cellular and vesicular membranes is mediated by members of a family of transporters known as MAT. In a previous report, we demonstrated that TBZ-mediated antagonism of VMAT2 resulted in enhanced GSIS of isolated islets (25). To determine whether a similar effect might be observed in cultures of human islets, we examined GSIS in the presence and absence of DTBZ (Fig. 4). Because DTBZ effects in this context are believed to be mediated through DA depletion (26, 27), we also tested GSIS in the presence of DA. As an additional control, we tested the effects of haloperidol, a first-generation antipsychotic and antagonist of DA action at D2R. We found that DTBZ enhanced GSIS at both high (15 mm) and low (8 mm) glucose concentrations but did not stimulate insulin secretion in the absence of glucose (Fig. 4). The amount of glucose-stimulated insulin release, normalized to islet DNA content in the sample, in these static incubation assays was similar to that detected previously (28). As reported for rodent islets (29), we found that DA (1.0 μm) significantly inhibited GSIS, whereas the antagonist of DA action at D2R, haloperidol, enhanced GSIS. Additional studies of GSIS using islets from different donors confirmed the generality of our findings with DTBZ, DA, haloperidol, and the tyrosine hydroxylase inhibitor AMPT (Fig. 5). As additional controls, we tested the effects of quinpirole (a selective D2 receptor agonist) and sulpiride (a selective D2 and D3 receptor antagonist) on human islet insulin secretion. As expected, agonism and antagonism at D2R, respectively, decreased and increased GSIS in human islets (Fig. 5).
Fig. 4.

Measurement of GSIS in islets from a single donor in the presence of glucose, TBZ, haloperidol, or DA. The islets were incubated in basal KRBB for 1 h and then seeded in wells and exposed to the indicated glucose and drug concentration for 1 h. The mean insulin concentration, normalized to the well DNA content, was then determined for sextuplicate wells. Error bars indicate the sem. *, Significantly different (P < 0.0.05) from the wells with 8 mm glucose alone; **, significantly different from wells with 15 mm glucose alone; ***, significantly different from wells with 3 mm glucose alone.
Fig. 5.

Measurement of GSIS in islets from multiple donors in the presence of glucose, TBZ, GBR 12909, DA, AMPT (pretreatment), 5-HT, and selected D2R agonists and antagonists. Static incubations were performed as indicated in Fig. 4. Each islet donor was tested against 1.5 mm glucose, 15 mm glucose, and one or more of the indicated compounds. Each bar represents the mean values obtained from three or more islet donors. For each donor and condition tested, the GSIS (nanograms insulin per nanogram DNA) was normalized to mean GSIS obtained in 1.5 mm glucose for that donor. Error bars indicate the sem. The mean GSIS for each compound tested was then compared with mean GSIS obtained in 15 mm glucose alone. *, Significantly different (P < 0.0.05) from the wells with 15 mm glucose alone; **, significantly different (P < 0.0.05) from the wells with 1.5 mm glucose alone.
In the CNS behind the blood-brain barrier (BBB), DA released at the synapse is recycled into vesicles by the action of the DAT. Here, DAT inhibitors increase extracellular DA concentrations because diffusion of hydrophilic molecules like DA is restricted by the BBB (30), but inactivation of DAT by knockout reduces tissue DA content (31). Because islets are not bordered by a BBB, yet are heavily vascularized and richly perfused, we hypothesized that inhibition of DAT might affect a net loss of DA signaling and inhibition of DAT by GBR 12909 might have similar effects on GSIS as did DA depletion by DTBZ. We found that GBR 12909 enhanced GSIS in human islets in vitro (Fig. 5), perhaps by blocking recycling and allowing DA to diffuse out of the cultured islet tissue. Supporting this idea was the additional finding that islet tissue stimulated with 8 mm glucose and treated with GBR 12909 contained less DA than control islet tissue stimulated with 8 mm glucose alone (Fig. 6A).
Fig. 6.

A, DA content of islet tissue and supernatants after GSIS. Static incubations were performed with 500 islets per well in KRBB with the indicated concentration of DTBZ or GBR 12909, with islets placed within the well inserts. Islets were obtained from two or more different donors as indicated by the value of n. At the end of the incubation, the supernatants were harvested and the DA and insulin content assayed. Islet tissue contained within the inserts were washed twice and then harvested in 1 ml buffer and assayed for insulin, DA, and DNA. DA and insulin concentration measurements were normalized to the DNA content of the well, and the mean value of at least triplicate wells was calculated. To compare groups, the amount of DA measured in the supernatant of each donor after stimulation with 8 mm glucose was set to represent 100%. All measurements in subsequent groups (supernatant vs. intracellular vs. treatment with GBR 12909) from that donor were expressed as the fraction of 8 mm glucose control supernatant value. B, DA release during perfusion experiments. Insulin and DA content were measured in selected fractions collected (flow rate = 750 μl/min at one fraction/30 sec) from approximately 1200 islets perfused in KRBB buffer with 8.0 mm glucose. Results are from a representative experiment in a series of two. *, Significant difference at the P < 0.05 level. C, DA uptake by cultured human islets. Islets were first cultured in 10 μm haloperidol to block [3H]DA binding to D2R. To these cultures 10 μm GBR 12909 or BCTP was then added to inhibit DAT function. [3H]DA was then added to the cultures for 1 h followed by washing and harvesting the islets. The amount of DA associated with the islets was then determined by scintillation counting, and the results are given as dpm normalized to the well DNA content. Each bar represents the average results obtained from two different islet donors, and the uptake for each condition was determined as the mean of triplicate wells. Error bars represent the sem. The results are given as background-corrected normalized dpm or as a percentage of control using the mean dpm value from cultures with 1.5 mm glucose and no haloperidol or DAT inhibitors added as the denominator. *, Significantly different (P < 0.0.05) from the wells with 1.5 mm glucose alone; **, significantly different (P < 0.0.05) from the wells containing 1.5 mm glucose and 10 μm haloperidol.
On the basis of these results, we formed the working hypothesis that DA stored in β-cell vesicles by action of VMAT2 is coreleased into the extracellular space with insulin upon glucose stimulation. DA acting at D2R in an autocrine manner or on nearby adjacent β-cells partially suppresses insulin secretion. Remaining DA molecules not internalized with D2R, or removed by the circulation, would be recycled via DAT.
Antagonism of VMAT2 by TBZ enhances insulin secretion in human islets in vitro by increasing secretion pulse height
To better understand how this DA circuit might be operating in islets, we examined the kinetics of GSIS under conditions of VMAT2 antagonism by DTBZ. The insulin concentration vs. time profile arising from perfusion of human islets with 1.5 mm glucose vs. 15 mm glucose with and without added DTBZ was measured and revealed insulin pulses (Fig. 7). After the perfusate glucose concentration was increased to 15 mm from 1.5 mm, the insulin concentration in collected fractions increased above basal levels. At 15 mm glucose, several significant peaks of insulin secretion (averaging about 7 × 102 amol/ml per islet) were observed with an average period of 5 ± 0.5 min (sem), consistent with previous reports (32, 33). In the presence of DTBZ, the amplitudes of the insulin pulses were significantly increased without a significant change in their periodicity. The mass of insulin secreted, as determined by the area under the curve during the high-glucose perfusion, was higher in the presence of DTBZ than in its absence, consistent with the static incubation experiments (Fig. 5).
Fig. 7.
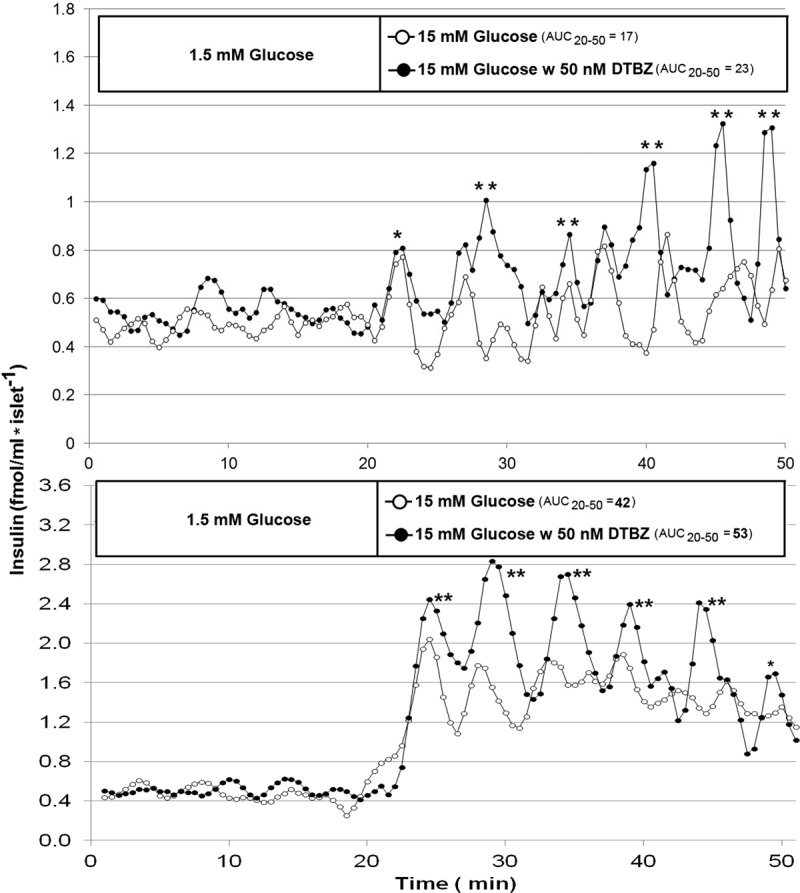
Effects of raising glucose from 1.5 to 15 mm in the presence and absence of DTBZ on the release of insulin from perfused human islets from two different donors. Upper panel shows results of 18 islets (donor a); bottom panel shows results of eight islets (donor b). Human islets were perfused (∼700 μl/min) in KRBB under basal glucose conditions for 20 min followed by raising the perfusate glucose concentration to 15 mm glucose with and without added DTBZ. Insulin concentrations were measured in duplicate or triplicate from 30-sec fractions of the perfusate. Cluster analysis (23) of the insulin concentration vs. time profile revealed the significant peaks of insulin secretion (indicated by asterisks). **, Peaks obtained during perfusion in 15 mm glucose with DTBZ that were significantly larger (P < 0.05) than those obtained without DTBZ as determined by a sliding t test. The area under the curve was determined using the trapezoidal rule. Two representative experiments from a series of three are shown.
Human islets in vitro release DA in a glucose-dependent manner
In investigating a possible β-cell autocrine or paracrine circuit involving DA-mediated regulation of GSIS and having shown that human islets and β-cells express D2R, and respond to DA and DA depletion in vitro, we next examined whether islets also released DA in response to glucose stimulation. Amperometry and voltammetry at microelectrodes have been demonstrated to possess sufficient sensitivity and temporal resolution to detect exocytosis of DA in the CNS (34). Similar techniques have been successfully applied to measurements of 5-HT and acetylcholine exocytosis from rodent and human islets (6, 35–37), but release of DA has not been characterized. To demonstrate release of DA, we first characterized the response of our detection system to DA and 5-HT (see Supplemental Fig. 1). Under our conditions, the nCFM was at least 50-fold more responsive to DA than to 5-HT.
Next, we probed the supernatant of islet cultures stimulated with glucose using an electrochemical technique and nCFM with preferential sensitivity to DA. The readout of this chronoamperometric technique is a current proportional to the DA concentration (Fig. 8C). Islets were first incubated in KRBB without glucose, followed by KRBB supplemented with glucose to 1.5 mm and then to 15 mm glucose. The average currents detected under these conditions were 21 ± 2, 23 ± 3, and 64 ± 3 pA (mean ± sem), respectively. Although the observed average current at 15 mm glucose was significantly greater (P < 0.001) than the current at no glucose and 1.5 mm glucose, the current detected at 1.5 mm glucose was not significantly different from the current observed at no glucose, although there was evidence of an increased number of current spikes, which may signify evidence of pulsatile DA release. After incubation in 15 mm glucose, the glucose concentration was reduced nearly 30-fold to 0.5 mm, and we observed a corresponding drop in the average current. Subsequently, we increased the glucose concentration to 30 mm glucose and observed a rise in the current. The average current rose significantly above the baseline, but this time, we also observed decay in the average current, possibly signaling DA depletion in the cultured islets. To provide evidence that the electrodes were still responsive to DA, at the end of the experiments, we added known concentrations of authentic exogenous DA to the islet cultures and measured the faradaic currents. On this basis, we estimate that DA release by islets was about 102 fmol/islet per current spike during stimulation by 15 mm glucose and similar to release of 5-HT as reported previously for mouse pancreatic β-cells (36).
Fig. 8.

Chronoamperometry of glucose-sensitive islet DA release. A, Input voltage waveform to the potentiostat (square wave pulses +0.08 to +0.220 V vs. Ag/AgCl with an 8-sec period and 50% duty cycle). B, Output current signals from a nCFM in KRBB. The initial voltage pulse was accompanied by a large increase in the double-layer current, which discharged well before the end of 50% duty cycle mark. C, The faradaic current (i.e. generated from the oxidation of DA) was measured during the last second of the +220-mV pulse (gray bar in A and B). The glucose concentration was varied as indicated, and the time vs. current data were plotted (C) and the average currents at each glucose concentration calculated. At the end of the experiment (time > 2 h) authentic DA was added to the well to obtain the corresponding faradaic current to confirm sensitivity to DA. Results are shown from a representative experiment in a series of three.
To confirm the findings of DA release by human islets in response to glucose stimulation, we measured the DA concentration in supernatants obtained from the static incubation experiments outlined above using the technique of ELISA. Here DA-specific antibodies are used to capture and detect DA from solution. The cross-reactivity of this assay with other endogenous monoamines (e.g. norepinephrine) was less than 1%. We found the amount of DA released by islets, normalized to the DNA content of the well, was significantly greater in 15 mm glucose relative to 1.5 mm glucose (mean concentration 0.28 ± 0.03 vs. 0.61 ± 0.07 ng DA/ng DNA) (Fig. 6A). We next measured both the DA and insulin content of the collected fraction from islet perfusion experiments performed under stimulation by 8.0 mm glucose (Fig. 6B). We found that peaks of DA immunoreactivity were largely, but not entirely, coincident with peaks of insulin immunoreactivity. Lastly, we measured tritiated DA uptake by cultured human islets. Total islet uptake of DA under the conditions tested was likely to reflect both binding and internalization by D2R and a component specifically mediated by DAT. To discriminate between these components, total uptake was measured in Ringers buffer with 1.5 mm glucose and the presence and absence of a D2R antagonist (haloperidol) and one or both of two specific DAT inhibitors [GBR 12909 (38) or BTCP (39)] (Fig. 6C). It was observed that addition of GBR 12909 and/or BCTP reduced the islet associated radioactivity to about 60% of the control level. Haloperidol blocked up to 30% of the total uptake of [3H] DA. When GBR 12909 or BCTP was added to the cultures containing haloperidol, the internalized counts dropped an additional 35% to about 45% of the total control radioactivity and were significantly (P < 0.05) less than internalized counts observed in the presence of haloperidol blocking alone. Similar results were found in cultures performed in 8.0 mm glucose.
Antagonism of VMAT2 or DAT reduces glucose excursions during IPGTT in the Zucker obese T2D rat model in vivo
Our in vitro experiments suggested that the monoamine transporters, DAT and VMAT2, were important control points in the regulation of GSIS. To determine whether this observation could be extended to in vivo models, we next examined the in vivo effects of TBZ and GBR 12909 in the Zucker fatty (ZF) rodent model of human pre-type 2 diabetes (Fig. 9, top and middle panel). As an additional control, we examined the effects of GBR 12909 on blood glucose levels during IPGTT in SD rats (Fig. 9, middle panel). In vivo inhibition of both VMAT2 and DAT during IPGTT reduced the glucose excursions after ip glucose challenge. The reduced glucose excursions measured after GBR 12909 administration and 30 min after glucose challenge were accompanied by increased serum insulin levels in both the SD and ZF rodent models (Fig. 9, bottom panel). Previously, we reported similar effects on insulin secretion when TBZ was administered before in vivo glucose challenge (i.e. increased serum insulin) (25).
Fig. 9.
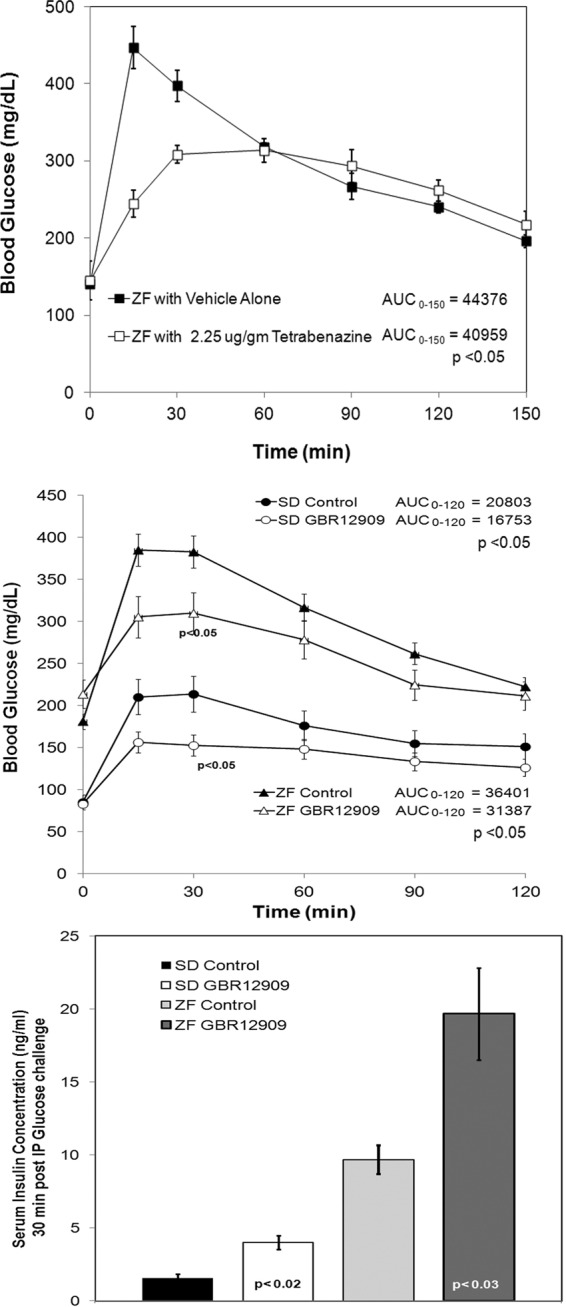
Top panel, TBZ reduces the blood glucose excursion during Intraperitoneal Glucose Tolerance Test (IPGTT) in male ZF (obese) rats. Mean blood glucose values during IPGTT of ZF rats treated with vehicle alone (■, n = 4) or with TBZ at 2.25 μg/g body weight (□, n = 5). Error bars represent the sem. The area under the curve (AUC) for each rodent's blood glucose vs. time profile was calculated using the trapezoid rule. The average AUC was calculated and compared between control and TBZ-treated groups. The P value of the significance of the comparison of AUC by t test is shown. Middle panel, GBR 12909 reduces the blood glucose excursions during IPGTT in male ZF (obese) and SD rats. Mean blood glucose values during IPGTT of rats treated with vehicle alone (▴ and ●, n = 5) or with GBR 12909 at 5.0 μg/g body weight (▵ and ○, n = 5). The comparison of AUC was performed as detailed above. Bottom panel, Sera collected at 30 min after IPGTT from the experiments shown above were assayed for insulin. The mean serum insulin concentrations for treated rats were compared with the values of untreated rats. The significance of comparison was determined by t test and is indicated within the bar.
ATA enhance in vitro insulin secretion by human islets
The ATA (e.g. clozapine and olanzapine), are characterized by potent antagonist activity at DA D2 and D3 and 5-HT2A receptors. In immunohistochemistry experiments, we demonstrated that D2R colocalized with insulin on β-cells and that D2RL was the predominant isoform expressed by β-cells. In static incubation experiments, we demonstrated that human islet GSIS was inhibited by 5-HT, DA (1 μm), and the D2R selective agonist quinpirole and enhanced by D2R antagonists such as haloperidol and sulpiride. On this basis, we predicted that ATA might also enhance human islet GSIS. We performed static incubation measurements of GSIS in the presence of a range of concentrations of either clozapine or olanzapine using islets obtained from several donors (Figs. 10 and 11). In general, we found that both clozapine and olanzapine significantly enhanced GSIS but that the effect differed in magnitude from donor to donor (Fig. 11).
Fig. 10.
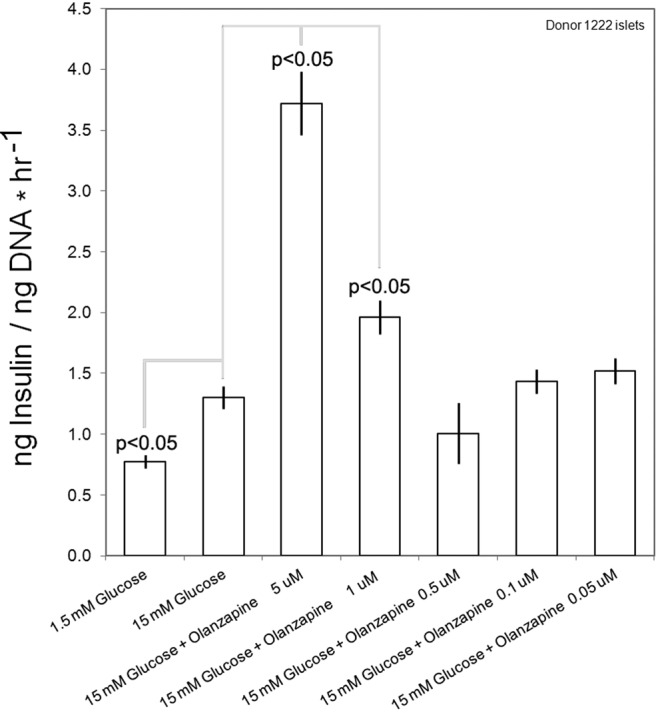
Measurement of GSIS in islets from a single donor in the presence of the ATA olanzapine. The islets were incubated in basal KRBB for 1 h and then seeded in wells and exposed to the indicated glucose and drug concentration for 1 h. The mean insulin concentration, normalized to the well DNA content, was then determined for sextuplicate wells. Mean insulin concentrations, normalized to the well DNA content, under different conditions were compared with the mean response to 15 mm glucose alone by t test. The significance of each comparison, where P < 0.05, is shown. Error bars indicate the sem.
Fig. 11.
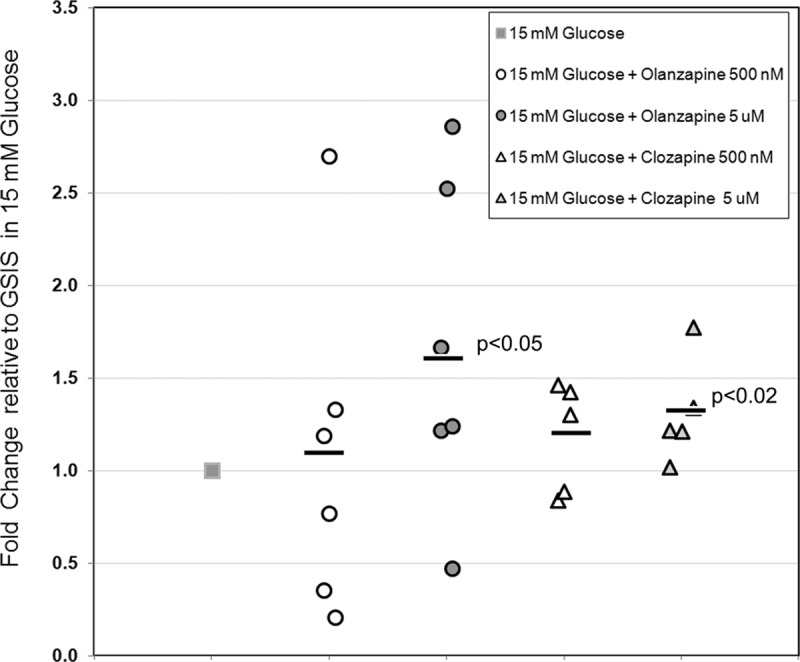
Measurement of GSIS in islets from multiple donors treated with olanzapine or clozapine. Static incubations were performed as indicated in Fig. 4. Each islet donor was tested against 15 mm glucose alone and 15 mm glucose plus the indicated compounds and dose. For each donor and condition tested, the GSIS (nanograms insulin per nanogram DNA) was normalized to mean GSIS obtained in 15 mm glucose alone for that donor to obtain the relative change. The mean relative change of GSIS among all donors tested for each compound tested (black bar) was then compared with mean GSIS obtained in 15 mm glucose alone. The P value obtained by Student's t test, where P < 0.05, is shown for each condition tested in the population.
Discussion
The regulatory role of intrinsic innervation on islet insulin secretion has been expertly summarized by Ahrén (3) and Thorens (40). Although past studies have mostly relied on rodent islets, more recent microanatomical studies in human islets suggest a diminished role for direct innervation of β-cells in regulation of insulin secretion (5). Our present study provides evidence of a novel DA-mediated regulatory autocrine circuit for insulin secretion by human islets in vitro. Such a circuit may partially support overall regulation of GSIS given the observed relative autonomy of human islets from sympathetic innervation (5).
An appreciation of the presence of monoamine neurotransmitters within islets and the role of these transmitters in regulation of glucose secretion began in the 1960s (41). During the following five decades, it became clear not only that rodent islets contain neurotransmitters (42) but also that β-cell vesicles contained insulin and 5-HT (43, 44) and/or DA as well. Next, it was found that β-cells probably express most if not all the enzymes needed for de novo synthesis of DA, its catabolism (13, 45, 46), and the VMAT that sequester cytosolic neurotransmitters into secretory storage granules (11). In the human pancreas, the expression of VMAT2 is largely confined to β-cell granules containing insulin (11, 15). Low levels of VMAT2 expression are present in exocrine pancreas (47) and endocrine PP cells, but α-cells (glucagon) and δ-cells (somatostatin) do not stain for VMAT2 protein (15). Similar to VMAT2, β-cells also express D2R, and as shown in this report, D2R expression in the pancreas is also largely confined to the β-cells. We found that in man, similar to what was observed in the rat (13), islets predominantly express the D2RL-specific isoform with D2RS-specific transcript representing a minority. In the CNS, neuronal DA release can be down-regulated using DA receptor agonists via DA autoreceptors (8). Knockout experiments in mice reveal that this autoreceptor's activity is most closely associated with the D2RS isoform (48). Additional study will be needed to understand the molecular physiology of islet DA receptor activity.
Based upon our earlier experiments, we hypothesized that DA, accumulated in β-cell vesicles by action of VMAT2, is released into the extracellular space along with insulin upon glucose stimulation. DA acting at D2R, newly exposed at the cell surface after a round of GSIS or expressed on vicinal β-cells, partially suppresses insulin secretion and residual DA, not internalized with D2R or removed by the circulation, would then be recycled by the action of DAT (49). To test this hypothesis, we first examined the expression of DAT in the pancreas. We observed that DAT was preferentially expressed in the islet tissue relative to whole pancreas (>6-fold) and at least 10-fold greater than that measured in exocrine tissue. We next studied the in vitro effects of TBZ's VMAT2 antagonism as well as its DA-depleting action on GSIS. ELISA-based measurements revealed that intra-islet DA content was reduced and GSIS was enhanced in the presence of DTBZ. In addition, we found that inhibition of l-3,4-dihydroxyphenylalanine (l-DOPA, the direct precursor of DA) synthesis by AMPT, an inhibitor of tyrosine hydroxylase, also enhanced islet GSIS. In this regard, it is interesting to note that islets also express the large neutral amino acid transporter system, composed of a heavy subunit encoded the SLC3A2 gene and a light subunit encoded by the SLC7A5 gene. This heterodimer preferentially transports branched-chain and aromatic (tryptophan and tyrosine) amino acids, including l-DOPA (50), and is highly expressed in brain capillary endothelial cells that form the BBB (51). As a consequence of the expression of DAT, the large neutral amino acid transporter system and VMAT2, islet β-cells appear to have the capacity to remove DA and its biosynthetic precursors (e.g. l-DOPA) (52) from the circulation and concentrate DA intravesicularly. As a direct consequence of expression of these molecules by dopaminergic neurons, the concentration of DA at release sites have been estimated to be about 1–100 μm, well within the upper limits of DA concentrations tested here. Nevertheless, it is not excluded that other intravesicular monoamines (e.g. norepinephrine) are active here. In addition to blocking DA transport at the level of insulin granules, we also tested the effects of antagonism of the β-cell's plasma membrane DAT. As in the case of VMAT2 inhibition, we found that antagonism of DAT by GBR 12909 enhanced islet GSIS. We speculated that inhibition of DAT in islets might result in loss of DA signaling, and although our in vitro and in vivo experiments support this conclusion, additional study and attention to possible off-target effects is needed.
We also found that human islet GSIS was enhanced in the presence of antagonists of D2R such as sulpiride and haloperidol. D2R agonists such as exogenous DA and quinpirole, as expected, diminished human islet GSIS. A study of the kinetics of islet GSIS in the presence of DTBZ revealed that enhanced insulin secretion was probably due to increases in the amplitude of pulsatile release rather than increases in the periodicity of insulin release. Lastly, we probed human islets in vitro for DA uptake mediated by DAT and glucose-dependent DA release. We found that islets could internalize exogenous [3H]DA through a GBR 12909- or BTCP-sensitive pathway. We also observed that the amount of DA released into culture supernatants increased with increasing ambient glucose concentrations and that DA release was concomitant with insulin release. The confirmation of glucose-dependent DA release by human islets and the additional above observations satisfy the conditions required by a DA-mediated autocrine feed-forward negative regulatory circuit.
We next examined whether this putative in vitro circuit could be manipulated to improve glucose clearance in vivo using a pre-type 2 diabetes rodent model. We found that antagonism of VMAT2 reduced the in vivo glucose excursions after glucose tolerance testing and that inhibition of DAT by GBR 12909 resulted in enhanced serum insulin concentrations after an ip glucose challenge as might be expected based on the results of our in vitro experiments, although it is not excluded that these drugs target sites beyond the endocrine pancreas. Although these results suggest that off-label use of existing VMAT2 inhibitors or D2R antagonists might be applied to enhance insulin secretion in response to glucose; in practice, however, placing too high a demand on insulin secretion could result in β-cell exhaustion (53). Consistent with this, the too-much-of-a-good-thing hypothesis (54) was proposed, in part, based on the results of the ADOPT study (55) showing that driving increased insulin secretion with sulfonylureas had worse long-term outcomes than pharmacotherapy with insulin sensitizers.
Because ATA have potent D2R antagonist activity (56), they have potential to act in a D2R-dependent manner at targets found within the islet autocrine DA circuit. In addition, the use of ATA has been associated with increased risk of developing T2D. In the past, increased risk of developing T2D associated with ATA use has been ascribed to increases in adiposity (and consequent insulin resistance) mediated via their CNS effects on food intake and physical activity (57, 58). We reasoned that an additional contributor to overall risk might be that, if certain ATA directly antagonize D2R on β-cells, an additional increase in insulin secretion might result through the DA/D2R-mediated autocrine feed-forward mechanism proposed above. We tested the effects of ATA clozapine and olanzapine on human islet GSIS in vitro and found that GSIS in the presence of these ATA was indeed enhanced. Similar experiments measuring GSIS in rodent islets treated with ATA have shown mixed results, ranging from no effect to effects only on basal insulin secretion (59–61), suggesting additional study is needed.
Because both drugs also have 5-HT receptor antagonist activity (including at 5-HT2A receptors), we also tested the effects of 5-HT on in vitro GSIS. As previously reported for rodent islets (62), we found that 5-HT inhibited islet GSIS, and thus it is reasonable to conclude that 5-HT receptor antagonism would parallel the effects of D2R antagonism. Thus, ATA via antagonism at D2R and 5-HT receptors could potentially promote β-cell loss by allowing β-cells to produce more insulin (63, 64) with resultant endoplasmic reticulum stress (65). Lastly, the efficacy of bromocriptine, a potent D2R agonist, in the management of T2D (66) might also be explained via its local effects on β-cell insulin secretion in addition to its central effects.
Although human β-cells may express the full complement of enzymes needed for de novo synthesis of DA, β-cells are probably not the only source of DA in the pancreas. Other DA sources might include, for example, spillover from the nerve terminals that innervate the pancreas (5, 67), cells of the exocrine pancreas, and/or reuptake from arterial blood, because the pancreas is fed by three interlocking arterial circles (47). Reliance on such sources of spillover DA would require that β-cells express DAT. In this report, we have shown indirect evidence that that DAT is expressed in islets and has a role in regulating GSIS in vitro and in vivo.
In this context, it is interesting to note that the brain is only a minor source of peripheral DA. As is the case for 5-HT, the major source of circulating DA is the gastrointestinal tract including the gastric mucosa (68). This fact, coupled with the findings that there are large postprandial spikes of serum DA in man (52) and that in vitro islets have a functional DAT, suggests that DA in its role of regulating β-cell insulin secretion represents an anti-incretin hypothesized to explain the beneficial effects of bariatric surgery on T2D (69). Here surgical procedures that possibly lower circulating DA levels (e.g. sleeve gastrectomy with duodenal switch) might be predicted to have enhanced insulin secretion. Driving increased β-cell function, particularly when there is weight loss failure, could result in the eventual postoperative reoccurrence of T2D as has been observed in a subpopulation of bariatric surgery patients (70).
Supplementary Material
Acknowledgments
This work was supported by the U.S. Public Health Service, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (5 R01 DK063567) and the Helmsley Charitable Trust (09PG-T1D020) (http://helmsleytrust.org). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure Summary: P.E.H. and A.M. are inventors on U.S. patent applications 20100204258 and 20110118300. N.S., M.F., S.B., Z.F., J.J., and R.L.L. have nothing to declare.
Footnotes
- AMPT
- α-Methylparatyrosine
- ATA
- atypical antipsychotic drugs
- BBB
- blood-brain barrier
- BTCP
- benzothiophenylcyclohexylpiperidine
- CNS
- central nervous system
- DA
- dopamine
- DAT
- DA transporter
- l-DOPA
- l-3,4-dihydroxyphenylalanine
- D2R
- DA type 2 receptor
- dpm
- decays per minute
- DTBZ
- dihydrotetrabenazine
- GSIS
- glucose-stimulated insulin secretion
- 5-HT
- serotonin
- IPGTT
- intraperitoneal glucose tolerance testing
- KRBB
- Krebs Ringers bicarbonate buffer
- MAT
- monoamine transporter
- nCFM
- Nafion-coated carbon fiber microelectrodes
- SD
- Sprague Dawley
- TBZ
- tetrabenazine
- T2D
- type 2 diabetes
- VMAT2
- vesicular monoamine type 2 transporter
- ZF
- Zucker fatty.
References
- 1. Henquin JC. 2000. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49:1751–1760 [DOI] [PubMed] [Google Scholar]
- 2. Brunicardi FC, Shavelle DM, Andersen DK. 1995. Neural regulation of the endocrine pancreas. Int J Pancreatol 18:177–195 [DOI] [PubMed] [Google Scholar]
- 3. Ahrén B. 2000. Autonomic regulation of islet hormone secretion–implications for health and disease. Diabetologia 43:393–410 [DOI] [PubMed] [Google Scholar]
- 4. Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A. 2006. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci USA 103:2334–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rodriguez-Diaz R, Abdulreda MH, Formoso AL, Gans I, Ricordi C, Berggren PO, Caicedo A. 2011. Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab 14:45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, Ricordi C, Roper SD, Berggren PO, Caicedo A. 2011. α-Cells secrete acetylcholine as a non-neuronal paracrine signal priming β-cell function in humans. Nat Med 17:888–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wolf ME, Roth RH. 1987. Dopamine neurons projecting to the medial prefrontal cortex possess release-modulating autoreceptors. Neuropharmacology 26:1053–1059 [DOI] [PubMed] [Google Scholar]
- 8. Westerink BH, de Vries JB. 1989. On the mechanism of neuroleptic induced increase in striatal dopamine release: brain dialysis provides direct evidence for mediation by autoreceptors localized on nerve terminals. Neurosci Lett 99:197–202 [DOI] [PubMed] [Google Scholar]
- 9. Maffei A, Liu Z, Witkowski P, Moschella F, Del Pozzo G, Liu E, Herold K, Winchester RJ, Hardy MA, Harris PE. 2004. Identification of tissue-restricted transcripts in human islets. Endocrinology 145:4513–4521 [DOI] [PubMed] [Google Scholar]
- 10. Kutlu B, Burdick D, Baxter D, Rasschaert J, Flamez D, Eizirik DL, Welsh N, Goodman N, Hood L. 2009. Detailed transcriptome atlas of the pancreatic β-cell. BMC Med Genomics 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anlauf M, Eissele R, Schäfer MK, Eiden LE, Arnold R, Pauser U, Klöppel G, Weihe E. 2003. Expression of the two isoforms of the vesicular monoamine transporter (VMAT1 and VMAT2) in the endocrine pancreas and pancreatic endocrine tumors. J Histochem Cytochem 51:1027–1040 [DOI] [PubMed] [Google Scholar]
- 12. Henry JP, Botton D, Sagne C, Isambert MF, Desnos C, Blanchard V, Raisman-Vozari R, Krejci E, Massoulie J, Gasnier B. 1994. Biochemistry and molecular biology of the vesicular monoamine transporter from chromaffin granules. J Exp Biol 196:251–262 [DOI] [PubMed] [Google Scholar]
- 13. Rubí B, Ljubicic S, Pournourmohammadi S, Carobbio S, Armanet M, Bartley C, Maechler P. 2005. Dopamine D2-like receptors are expressed in pancreatic β-cells and mediate inhibition of insulin secretion. J Biol Chem 280:36824–36832 [DOI] [PubMed] [Google Scholar]
- 14. Ericson LE, Håkanson R, Lundquist I. 1977. Accumulation of dopamine in mouse pancreatic B-cells following injection of l-DOPA. Localization to secretory granules and inhibition of insulin secretion. Diabetologia 13:117–124 [DOI] [PubMed] [Google Scholar]
- 15. Saisho Y, Harris PE, Butler AE, Galasso R, Gurlo T, Rizza RA, Butler PC. 2008. Relationship between pancreatic vesicular monoamine transporter 2 (VMAT2) and insulin expression in human pancreas. J Mol Histol 39:543–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 17. Weksler-Zangen S, Yagil C, Zangen DH, Ornoy A, Jacob HJ, Yagil Y. 2001. The newly inbred cohen diabetic rat: a nonobese normolipidemic genetic model of diet-induced type 2 diabetes expressing sex differences. Diabetes 50:2521–2529 [DOI] [PubMed] [Google Scholar]
- 18. Pennington JM, Millar J, L Jones CP, Owesson CA, McLaughlin DP, Stamford JA. 2004. Simultaneous real-time amperometric measurement of catecholamines and serotonin at carbon fibre ‘dident’ microelectrodes. J Neurosci Methods 140:5–13 [DOI] [PubMed] [Google Scholar]
- 19. Gonon FG, Fombarlet CM, Buda MJ, Pujol JF. 1981. Electrochemical treatment of pyrolytic carbon fiber electrodes. Anal Chem 53:1386–1389 [Google Scholar]
- 20. Michael AC, Justice JB., Jr 1987. Oxidation of dopamine and 4-methylcatechol at carbon fiber disk electrodes. Anal Chem 59:405–410 [DOI] [PubMed] [Google Scholar]
- 21. Ahn SS, Blaha CD, Alkire MT, Wood E, Gray-Allan P, Marrocco RT, Moore WS. 1991. Biphasic striatal dopamine release during transient ischemia and reperfusion in gerbils. Stroke 22:674–679 [DOI] [PubMed] [Google Scholar]
- 22. Crespi F, Cespuglio R, Jouvet M. 1983. Differential pulse voltammetry in brain tissue. III. Mapping of the rat serotoninergic raphe nuclei by electrochemical detection of 5-HIAA. Brain Res 270:45–54 [DOI] [PubMed] [Google Scholar]
- 23. Veldhuis JD, Johnson ML. 1986. Cluster analysis: a simple, versatile, and robust algorithm for endocrine pulse detection. Am J Physiol 250:E486–E493 [DOI] [PubMed] [Google Scholar]
- 24. Tan S, Hermann B, Borrelli E. 2003. Dopaminergic mouse mutants: investigating the roles of the different dopamine receptor subtypes and the dopamine transporter. Int Rev Neurobiol 54:145–197 [DOI] [PubMed] [Google Scholar]
- 25. Raffo A, Hancock K, Polito T, Xie Y, Andan G, Witkowski P, Hardy M, Barba P, Ferrara C, Maffei A, Freeby M, Goland R, Leibel RL, Sweet IR, Harris PE. 2008. Role of vesicular monoamine transporter type 2 in rodent insulin secretion and glucose metabolism revealed by its specific antagonist tetrabenazine. J Endocrinol 198:41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leysen JE, Wynants J, Eens A, Janssen PA. 1989. Ketanserin reduces a particular monoamine pool in peripheral tissues. Mol Pharmacol 35:375–380 [PubMed] [Google Scholar]
- 27. Reches A, Hassan MN, Jackson VR, Fahn S. 1983. Lithium attenuates dopamine depleting effects of reserpine and tetrabenazine but not that of α methyl-p-tyrosine. Life Sci 33:157–160 [DOI] [PubMed] [Google Scholar]
- 28. Kühtreiber WM, Ho LT, Kamireddy A, Yacoub JA, Scharp DW. 2010. Islet isolation from human pancreas with extended cold ischemia time. Transplant Proc 42:2027–2031 [DOI] [PubMed] [Google Scholar]
- 29. Shankar E, Santhosh KT, Paulose CS. 2006. Dopaminergic regulation of glucose-induced insulin secretion through dopamine D2 receptors in the pancreatic islets in vitro. IUBMB Life 58:157–163 [DOI] [PubMed] [Google Scholar]
- 30. Kristensen AS, Andersen J, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ, Strømgaard K, Gether U. 2011. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev 63:585–640 [DOI] [PubMed] [Google Scholar]
- 31. Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. 1996. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379:606–612 [DOI] [PubMed] [Google Scholar]
- 32. Hellman B, Salehi A, Gylfe E, Dansk H, Grapengiesser E. 2009. Glucose generates coincident insulin and somatostatin pulses and antisynchronous glucagon pulses from human pancreatic islets. Endocrinology 150:5334–5340 [DOI] [PubMed] [Google Scholar]
- 33. Song SH, Kjems L, Ritzel R, McIntyre SM, Johnson ML, Veldhuis JD, Butler PC. 2002. Pulsatile insulin secretion by human pancreatic islets. J Clin Endocrinol Metab 87:213–221 [DOI] [PubMed] [Google Scholar]
- 34. Crespi F. 1990. In vivo voltammetry with micro-biosensors for analysis of neurotransmitter release and metabolism. J Neurosci Methods 34:53–65 [DOI] [PubMed] [Google Scholar]
- 35. Aspinwall CA, Huang L, Lakey JR, Kennedy RT. 1999. Comparison of amperometric methods for detection of exocytosis from single pancreatic β-cells of different species. Anal Chem 71:5551–5556 [DOI] [PubMed] [Google Scholar]
- 36. Smith PA, Duchen MR, Ashcroft FM. 1995. A fluorimetric and amperometric study of calcium and secretion in isolated mouse pancreatic β-cells. Pflugers Arch 430:808–818 [DOI] [PubMed] [Google Scholar]
- 37. Barbosa RM, Silva AM, Tome AR, Stamford JA, Santos RM, Rosario LM. 1998. Control of pulsatile 5-HT/insulin secretion from single mouse pancreatic islets by intracellular calcium dynamics. J Physiol 510(Pt 1):135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matecka D, Rothman RB, Radesca L, de Costa BR, Dersch CM, Partilla JS, Pert A, Glowa JR, Wojnicki FH, Rice KC. 1996. Development of novel, potent, and selective dopamine reuptake inhibitors through alteration of the piperazine ring of 1-[2-(diphenylmethoxy)ethyl]-and 1-[2-[bis(4-fluorophenyl)methoxy] ethyl]-4-(3-phenylpropyl)piperazines (GBR 12935 and GBR 12909). J Med Chem 39:4704–4716 [DOI] [PubMed] [Google Scholar]
- 39. He X, Raymon LP, Mattson MV, Eldefrawi ME, de Costa BR. 1993. Further studies of the structure-activity relationships of 1-[1-(2-benzo[b]thienyl)cyclohexyl]piperidine. Synthesis and evaluation of 1-(2-benzo[b]thienyl)-N,N-dialkylcyclohexylamines at dopamine uptake and phencyclidine binding sites. J Med Chem 36:4075–4081 [DOI] [PubMed] [Google Scholar]
- 40. Thorens B. 2010. Central control of glucose homeostasis: the brain-endocrine pancreas axis. Diabetes Metab 36(Suppl 3):S45–S49 [DOI] [PubMed] [Google Scholar]
- 41. Falck B, Hellman B. 1964. A fluorescent reaction for monoamines in the insulin producing cells of the guinea-pig. Acta Endocrinol (Copenh) 45:133–138 [DOI] [PubMed] [Google Scholar]
- 42. Cegrell L, Falck B, Rosengren AM. 1967. Dopamine and 5-hydroxytryptamine in the guinea-pig pancreas. Life Sci 6:2483–2489 [DOI] [PubMed] [Google Scholar]
- 43. Jaim-Etcheverry G, Zieher LM. 1968. Electron microscopic cytochemistry of 5-hydroxytryptamine (5-HT) in the β-cells of guinea pig endocrine pancreas. Endocrinology 83:917–923 [DOI] [PubMed] [Google Scholar]
- 44. Zawalich WS, Tesz GJ, Zawalich KC. 2001. Are 5-hydroxytryptamine-preloaded β-cells an appropriate physiologic model system for establishing that insulin stimulates insulin secretion? J Biol Chem 276:37120–37123 [DOI] [PubMed] [Google Scholar]
- 45. Ito K, Hirose H, Maruyama H, Fukamachi S, Tashiro Y, Saruta T. 1995. Neurotransmitters partially restore glucose sensitivity of insulin and glucagon secretion from perfused streptozotocin-induced diabetic rat pancreas. Diabetologia 38:1276–1284 [DOI] [PubMed] [Google Scholar]
- 46. Iturriza FC, Thibault J. 1993. Immunohistochemical investigation of tyrosine-hydroxylase in the islets of Langerhans of adult mice, rats and guinea pigs. Neuroendocrinology 57:476–480 [DOI] [PubMed] [Google Scholar]
- 47. Mezey E, Eisenhofer G, Harta G, Hansson S, Gould L, Hunyady B, Hoffman BJ. 1996. A novel nonneuronal catecholaminergic system: exocrine pancreas synthesizes and releases dopamine. Proc Natl Acad Sci USA 93:10377–10382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Usiello A, Baik JH, Rougé-Pont F, Picetti R, Dierich A, LeMeur M, Piazza PV, Borrelli E. 2000. Distinct functions of the two isoforms of dopamine D2 receptors. Nature 408:199–203 [DOI] [PubMed] [Google Scholar]
- 49. Bannon MJ. 2005. The dopamine transporter: role in neurotoxicity and human disease. Toxicol Appl Pharmacol 204:355–360 [DOI] [PubMed] [Google Scholar]
- 50. Kageyama T, Nakamura M, Matsuo A, Yamasaki Y, Takakura Y, Hashida M, Kanai Y, Naito M, Tsuruo T, Minato N, Shimohama S. 2000. The 4F2hc/LAT1 complex transports l-DOPA across the blood-brain barrier. Brain Res 879:115–121 [DOI] [PubMed] [Google Scholar]
- 51. Boado RJ, Li JY, Nagaya M, Zhang C, Pardridge WM. 1999. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc Natl Acad Sci USA 96:12079–12084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goldstein DS, Swoboda KJ, Miles JM, Coppack SW, Aneman A, Holmes C, Lamensdorf I, Eisenhofer G. 1999. Sources and physiological significance of plasma dopamine sulfate. J Clin Endocrinol Metab 84:2523–2531 [DOI] [PubMed] [Google Scholar]
- 53. Hansen JB, Arkhammar PO, Bodvarsdottir TB, Wahl P. 2004. Inhibition of insulin secretion as a new drug target in the treatment of metabolic disorders. Current medicinal chemistry 11:1595–1615 [DOI] [PubMed] [Google Scholar]
- 54. Aston-Mourney K, Proietto J, Morahan G, Andrikopoulos S. 2008. Too much of a good thing: why it is bad to stimulate the β-cell to secrete insulin. Diabetologia 51:540–545 [DOI] [PubMed] [Google Scholar]
- 55. Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O'Neill MC, Zinman B, Viberti G. 2006. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 355:2427–2443 [DOI] [PubMed] [Google Scholar]
- 56. Nord M, Farde L. 2011. Antipsychotic occupancy of dopamine receptors in schizophrenia. CNS Neurosci Ther 17:97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Elias AN, Hofflich H. 2008. Abnormalities in glucose metabolism in patients with schizophrenia treated with atypical antipsychotic medications. Am J Med 121:98–104 [DOI] [PubMed] [Google Scholar]
- 58. Holt RI, Peveler RC. 2006. Antipsychotic drugs and diabetes: an application of the Austin Bradford Hill criteria. Diabetologia 49:1467–1476 [DOI] [PubMed] [Google Scholar]
- 59. Johnson DE, Yamazaki H, Ward KM, Schmidt AW, Lebel WS, Treadway JL, Gibbs EM, Zawalich WS, Rollema H. 2005. Inhibitory effects of antipsychotics on carbachol-enhanced insulin secretion from perifused rat islets: role of muscarinic antagonism in antipsychotic-induced diabetes and hyperglycemia. Diabetes 54:1552–1558 [DOI] [PubMed] [Google Scholar]
- 60. Melkersson K. 2004. Clozapine and olanzapine, but not conventional antipsychotics, increase insulin release in vitro. Eur Neuropsychopharmacol 14:115–119 [DOI] [PubMed] [Google Scholar]
- 61. Melkersson K, Khan A, Hilding A, Hulting AL. 2001. Different effects of antipsychotic drugs on insulin release in vitro. Eur Neuropsychopharmacol 11:327–332 [DOI] [PubMed] [Google Scholar]
- 62. Lindström P, Sehlin J. 1983. Opposite effects of 5-hydroxytryptophan and 5-hydroxytryptamine on the function of microdissected ob/ob-mouse pancreatic islets. Diabetologia 24:52–57 [DOI] [PubMed] [Google Scholar]
- 63. Grill V, Björklund A. 2009. Impact of metabolic abnormalities for β-cell function: clinical significance and underlying mechanisms. Mol Cell Endocrinol 297:86–92 [DOI] [PubMed] [Google Scholar]
- 64. Chang-Chen KJ, Mullur R, Bernal-Mizrachi E. 2008. β-Cell failure as a complication of diabetes. Rev Endocr Metab Disord 9:329–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Qian L, Zhang S, Xu L, Peng Y. 2008. Endoplasmic reticulum stress in β-cells: latent mechanism of secondary sulfonylurea failure in type 2 diabetes? Med Hypotheses 71:889–891 [DOI] [PubMed] [Google Scholar]
- 66. Meier AH, Cincotta AH, Lovell WC. 1992. Timed bromocriptine administration reduces body fat stores in obese subjects and hyperglycemia in type II diabetics. Experientia 48:248–253 [DOI] [PubMed] [Google Scholar]
- 67. Kirchgessner AL, Gershon MD. 1990. Innervation of the pancreas by neurons in the gut. J Neurosci 10:1626–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Eisenhofer G, Aneman A, Friberg P, Hooper D, Fåndriks L, Lonroth H, Hunyady B, Mezey E. 1997. Substantial production of dopamine in the human gastrointestinal tract. J Clin Endocrinol Metab 82:3864–3871 [DOI] [PubMed] [Google Scholar]
- 69. Rubino F, R'bibo SL, del Genio F, Mazumdar M, McGraw TE. 2010. Metabolic surgery: the role of the gastrointestinal tract in diabetes mellitus. Nature reviews Endocrinology 6:102–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. DiGiorgi M, Rosen DJ, Choi JJ, Milone L, Schrope B, Olivero-Rivera L, Restuccia N, Yuen S, Fisk M, Inabnet WB, Bessler M. 2010. Re-emergence of diabetes after gastric bypass in patients with mid- to long-term follow-up. Surg Obes Relat Dis 6:249–253 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.