

Abstract
Transcriptional profiling has been widely used as a tool for unveiling the coregulations of genes in response to genetic and environmental perturbations. These coregulations have been used, in a few instances, to infer global transcriptional regulatory models. Here, using the large amount of transcriptomic information available for the bacterium Escherichia coli, we seek to understand the design principles determining the regulation of its transcriptome. Combining transcriptomic and signaling data, we develop an evolutionary computational procedure that allows obtaining alternative genomic transcriptional regulatory network (GTRN) that still maintains its adaptability to dynamic environments. We apply our methodology to an E. coli GTRN and show that it could be rewired to simpler transcriptional regulatory structures. These rewired GTRNs still maintain the global physiological response to fluctuating environments. Rewired GTRNs contain 73% fewer regulated operons. Genes with similar functions and coordinated patterns of expression across environments are clustered into longer regulated operons. These synthetic GTRNs are more sensitive and show a more robust response to challenging environments. This result illustrates that the natural configuration of E. coli GTRN does not necessarily result from selection for robustness to environmental perturbations, but that evolutionary contingencies may have been important as well. We also discuss the limitations of our methodology in the context of the demand theory. Our procedure will be useful as a novel way to analyze global transcription regulation networks and in synthetic biology for the de novo design of genomes.
Keywords: automated design, synthetic genomics, genome refactoring, evolutionary computation
Organisms have evolved mechanisms for regulating transcription to better adapt to changing environments. Could such regulation be engineered in a different way (1, 2)? Recent experiments investigating the evolvability of bacterial transcriptional regulatory networks (TRNs) have shown that the massive addition of new links to the network does not significantly alter cell growth. Isalan et al. (3) added transcriptional fusions of promoters with different master transcriptional regulators and showed that Escherichia coli (E. coli) tolerated almost all rewired networks; however, growth was perturbed by as much as 5% (3). This inherent predisposition of E. coli networks to dampen extreme changes in their circuitry enables the possibility of conducting genome-wide rewiring (4). Global transcription regulation could also be analyzed by comparing the regulatory models from distant organisms, provided they show a similar response to the set of studied environments. In this way, they could provide alternative regulatory models, although the lack of knowledge of species-specific selective pressures may blur the conclusions. We will propose here an alternative evolution experiment, which will be conducted computationally thanks to the availability of a quantitative model for the genomic transcriptional regulatory network (GTRN) of E. coli.
Global models of transcription regulation are essential to understand the function of an organism in alternative environments. The analysis of the structure of GTRN has unveiled many design principles, such as the identification of local patterns of regulation with defined function (5). How predictable should a model be in order to be able to evolve a global TRN? The relationship between network structure and function is best described by models based on ordinary differential equations (ODEs) that implement instances of the regulatory network. Monitoring of gene expression at a genome-wide scale allows assigning parameter values to global models of transcription regulation (6). If it were possible to create an ODE model for the global transcriptional regulation and signaling of a given genome, then we would be able to predict the function of a network even after rewiring it in silico, allowing the generation of alternative models with similar behavior. We will show that this can be done by adapting an existing ODE model for the TRN of E. coli (7) to include the required signal transduction. The evolutionary computational methodology here proposed is general, and it could be used with other ODE models for TRNs (8–13).
The computational design of small TRNs was first proposed by using computational evolution with a system of ODEs describing the TRN (14), although no nucleotide sequence was generated for the evolved TRN. Recently, the use of a modular approach based on the assembly of biological part models has allowed the assignation of nucleotide sequences to the evolved TRN (15), which opened the door to the automatic design of genomic-sized sequences. For genomic-scale TRNs, we could take advantage of the available high-throughput functional genomics data to infer the required ODE models (7). Evolutionary TRN optimization requires defining a fitness function. A simple fitness function could be defined based on the expression levels of some selected genes. Alternatively, a more complex fitness function could be defined by linking gene expression to cell growth, which would allow evolving whole genome TRNs. We call this a GTRN, defined as a TRN (including signaling) together with a fitness function accounting for cell growth. It has recently been shown that the transcriptomic expression profiles are good predictors for instantaneous cell growth in Saccharomyces cerevisiae (16). Assuming that this relationship is true for other organisms, it can be hypothesized that the expression profile of a given system determines cell growth. This can also be rationalized by arguing that natural selection results in nearly optimal biomass production by favoring regulatory pathways that confer optimal levels of gene expression in a given environment. In this line, Tagkopoulos et al. (17) used Pearson correlations between the abundance of cell resources and the response of gene expression as a fitness function to computationally evolve the biochemical network of E. coli in variable environments. In this work, we propose to use the similarity of the expression profile of a GTRN and the wild-type (WT) as fitness function. Therefore, if we evolve a GTRN by only rewiring the transcription regulation yet keeping the same expression profile, we would expect that the solutions still have optimal growth.
Here, we analyze the transcriptional complexity required for robust growth under changing environments by developing a mathematical framework to evolve GTRNs (Fig. 1). We start by summarizing the proposed methodology for the computational evolution of GTRNs. Afterwards, we choose an organism, E. coli, for which an ODE for its TRN is known, and we analyze its predictability once we construct the GTRN. Next, we show that it is accurate enough to make predictions even if its topology is locally modified. Afterwards, we will analyze the resulting TRN after computational evolution under changing levels of oxygen, carbon, and nitrogen. Finally, we discuss the implications of our rewired TRN on the design principles of regulatory networks. We conclude that our methodology for rewiring genomic TRN is a useful tool to explore the design principles of transcription regulation and signaling. Our methodology will also be useful for the future re-engineering of genomes.
Fig. 1.

Our approach for the computational evolution of a GTRN. Each step of our methodology (blue, green, and red arrows) was validated in Fig. 2.
Computational Methods
We need to have a suitable GTRN, which we construct here by using a genome-wide model of E. coli gene transcription in response to selected external signals able to predict changes in cell growth after transcriptional modifications (Materials and Methods). The model is used to estimate kinetic parameters from experimental steady-state data (18). Given a GTRN described by a set of ODE for the concentrations of each gene product in a given genome, we propose to evolve it by an iterative procedure involving cycles of generalized mutations and selection. As generalized mutations, we consider modifications in the ODEs that could implement the move of a gene to a different operon or the addition of synthetic promoters (Fig. S1). For the selection step, we use as fitness function the similarity to a WT transcriptional profile, providing in this way the variation of cell growth. The fitness function is used in a Monte Carlo procedure to select or discard the suggested mutations (Materials and Methods).
Results
Environmental Adaptation of the WT GTRN.
To construct the GTRN, we extended our ODE model for the TRN of E. coli (7) to sense environmental changes at the molecular level. We evaluated the model by quantifying how the expression of a given transcription factor (TF) changes upon the perturbation of a specific uptake factor(s) (Fig. 1 and Dataset S1). Next, we investigated how the model responds to environmental changes. We evaluated a distance, Sexp, between the optimal expression profile (defined as the expression profile measured for E. coli growing at the maximum rate for a given environmental condition) and the expression profile of the model in each environment. As it is not clear which genes will be most relevant to cell growth during our evolution, we explored six sets of genes to define Sexp (physiological adaptation genes, defense pathway genes, a combination of genes related to these two functions, genes that protect against abiotic stresses, genes encoding central metabolism enzymes, and all genes). Fig. S2A shows the optimality degree, defined as the relative growth that E. coli exhibits in environments that are optimal except in the concentration of a single component, such as oxygen or glucose (Materials and Methods) (19). Fig. S2B shows calculations of Sexp based on our model from the expression profiles predicted under 100 different environmental conditions. The largest variations of the expression score and optimality degree were obtained when selecting a gene set related to defense functions, and the smallest variation was obtained after considering genes related to enzymatic activity. This difference is expected, because the defense responses are highly inducible and specific to given environmental stimuli, whereas metabolism is able to buffer external stimulus through a critical set of metabolic pathways.
Predictability of GTRN upon Genetic and Environmental Changes.
We sought to determine whether a GTRN model able to assign parameters to promoters and TF sequences predict the transcriptome of E. coli under different environmental conditions and/or after genetic modifications. To test our inferred model, we perform a K-fold cross-validation to ensure that gene expression profiles predicted from experimental measures of TF expression do not depend on the selection of the testing set (Fig. 2A and Fig. S3). We also evaluated the performance of the GTRN in predicting responses to environmental stresses and genetic changes by introducing such modifications in the model (Dataset S1). For illustrative purposes, Fig. 2B shows the predicted versus experimental profiles for two examples of master regulator knockouts (fnr and soxS) under aerobic and anaerobic conditions and for two environmental perturbations in which glucose, oxygen, and glycerol sources were changed. To validate Sexp, we compared the predicted fitness values to data from E. coli experimental evolution. Recently, Conrad et al. (20) characterized all acquired adaptive mutations of E. coli strains from a short-term laboratory evolution in minimal lactate medium. Fig. 2C shows a significant correlation (Pearson r = 0.82, 6 df, p < 0.05) between observed and predicted fitnesses when considering only TFs were considered in the computation of Sexp, thus validating our choice of the fitness function (Fig. S4E). Furthermore, we also attempted to predict the phenotypic response of E. coli after adding new regulations in its TRN (3). Fig. 2D show a significant correlation (r = 0.65, p < 0.0001) between growth rate and predicted fitness when only the contributions of TFs to Sexp was considered, corroborating that our fitness function is able to capture large changes in the TRN (SI Materials and Methods).
Fig. 2.

(A) Histogram of Pearson correlations among predicted and experimental gene expressions in the 380 experimental conditions of M3D by using models trained obtained from different subsets (10-fold cross-validation) or all set of conditions (white and black bars, respectively) (Fig. S3). (B) Prediction of expression profiles of E. coli upon genetic changes (knockout of fnr and soxS), environmental perturbations (modification of oxygen and carbon availability), or both (fnr and soxS knockout under anaerobic conditions). Each dot in the scatter plots represents a value obtained from a different hybridization experiment plotted against the algorithm prediction. The red line represents the exact prediction. (C) Correlation between predicted fitness considering only TFs (Fig. S4E) and the growth rates of four strains and their intermediaries evolved in the laboratory under minimal lactate media. The GTRN of such strains was modeled by optimizing the unknown expression parameters for the mutated genes (SI Text). The fitness values (Sexp) of the WT GTRN under the different environments selected are shown in Fig. S4 (SI Text). (D) Correlation between predicted fitness considering only TFs and the growth rate of 37 strains with a rewired TRN.
Rewiring the E. coli GTRN by Computational Evolution.
In addition to fitness expressed as growth, Sexp, we needed another objective function that is related to the expected GTRN arrangement, Smod (Materials and Methods). Fig. 3A illustrates the trajectories of the Sexp and Smod functions and their weighted sums, which defines the fitness function to be used during the in silico evolution (SI Materials and Methods). First, this was done for different environments by maintaining the optimal gene expression levels only for metabolic enzymes (Fig. S4 A and B). The fitness function achieved similar values during the last steps of the evolution process for all simulated replicates of the rewired GTRNs. Interestingly, we observed a significant reduction in the complexity of the rewired TRN with respect to the WT. We computed the ratio between the number of regulatory interactions (Ξ < 0.31, p < 0.001) and the number of operons (Θ < 0.27, p < 0.001) for the rewired and WT GTRNs, which do not appear to depend on the environment. These GTRNs were optimized under the imposed constrain that only central metabolism enzymes expression must remain close to the optimal level. How does the reduction of TRN complexity depend on the selection of critical genes involved in the fitness function? To address this question (Figs. S5 and S6), we also explored the possibility that limiting the expression of only those genes related to defense and adaptation would allow larger reductions in complexity (Fig. S5D; Ξ < 0.25, p < 0.001; Θ < 0.23, p < 0.001). The smallest reductions in complexity were obtained when the entire genome was restricted (Fig. S5J; Ξ < 0.38, p < 0.001; Θ < 0.33, p < 0.001). Thus, high reductions in TRN complexity were obtained independently of the set of genes selected as critical predictors of transcriptomic fitness.
Fig. 3.

(A) Modularity score and transcriptomic similarity for rewired GTRNs in the evolutionary steps under permissive and challenging environments (blue and red lines, respectively). Random optimizations produced significantly lower biobjective function values than those of the WT GTRN. Error bars represent standard deviations of scores obtained from 10 evolutionary processes. (B) Functional similarity, depending on operon size, of rewired and random GTRNs (blue and red points, respectively) that have evolved in a neutral environment. Error bars show the minimum and maximum value of functional similarity of all operons with a given size. Note that selective pressure, Sexp, was computed scoring only genes relating to central metabolism (see Fig. S5 for selective pressures based on stress genes or all genome). (C and D) Optimality degree (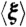 and
and 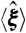 , respectively) as a measurement of adaptive behavior of rewired E. coli GTRNs evolved under selective pressures affecting either genes coding for enzymatic activity, genes related to adaptation and defense functions, or to the entire genome. The behavior of the designed GTRNs was measured by applying single environmental perturbations (C) that modified external fluxes of oxygen or carbon sources (blue and red bars, respectively) or simultaneous changes in the oxygen, carbon, and nitrogen sources (D). Adaptation was predicted by using cell fitness constraints depending on the critical genes selected in the in silico evolution process. Error bars represent standard deviations of the scores obtained from 10 evolutionary processes.
, respectively) as a measurement of adaptive behavior of rewired E. coli GTRNs evolved under selective pressures affecting either genes coding for enzymatic activity, genes related to adaptation and defense functions, or to the entire genome. The behavior of the designed GTRNs was measured by applying single environmental perturbations (C) that modified external fluxes of oxygen or carbon sources (blue and red bars, respectively) or simultaneous changes in the oxygen, carbon, and nitrogen sources (D). Adaptation was predicted by using cell fitness constraints depending on the critical genes selected in the in silico evolution process. Error bars represent standard deviations of the scores obtained from 10 evolutionary processes.
Next, we investigated whether genes with high functional similarity were grouped into the same operons or network modules; for example, we computed the functional similarity of all operons containing more than one gene in the rewired and random operon-organization GTRNs. Fig. 3B shows the highly statistically significant functional similarity of genes rewired into the same operon with respect to random evolutions (Kolmogorov-Smirnov test, p < 0.001; Mann-Whitney test, p < 0.001). It is especially interesting that the rewired GTRNs were characterized by operons containing genes of similar functions, a property that was not imposed during the evolutionary process. Specifically, the number of rewired operons with degrees of functional similarity < 0.8 considerably exceeded the number of those with random organization.
Analysis of Biochemical Adaptation in Rewired GTRNs.
Many signaling systems can adapt their expression programs in response to novel stimuli. Fig. S2 shows that a single, strong environmental perturbation induced WT TRN to reduce cell fitness to a minimal, but stable, value. This motivated us to investigate whether rewired systems acquired the ability to adapt to environmental changes more quickly than WT systems. Fig. 4
A and C shows an example of rewired GTRN showing the operons that contain rearranged TFs regulated by three environmental factors (EFs). Interestingly, those rewired operons are controlled by a set of new regulations that highly differ from the WT operons (Fig. 4
A and C), yet maintain the original transcriptomic behavior. We explored single environmental perturbations by simulating two sets of environments. We then used the optimality degree to assess the adaptation of rewired GTRN to the environments, considering three types of selection pressure in the expression score: selecting only genes coding for enzymes involved in central metabolism, stress-related genes, or the entire genome (Fig. 3C). Using the first two criteria for the evolutionary process, the average of the optimality degrees 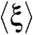 around the set of environmental perturbations was negative (i.e., cell fitness exceeded the optimal value for all re-engineered GTRNs (
around the set of environmental perturbations was negative (i.e., cell fitness exceeded the optimal value for all re-engineered GTRNs ( and -0.023, respectively). On the contrary, GTRNs rewired based on the third criterion achieved positive optimality degrees (
and -0.023, respectively). On the contrary, GTRNs rewired based on the third criterion achieved positive optimality degrees (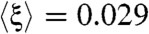 ). Defining the fragility of a GTRN as its optimality degree in different environments, rewired GTRNs were more fragile; anticipatory behavior disappeared (
). Defining the fragility of a GTRN as its optimality degree in different environments, rewired GTRNs were more fragile; anticipatory behavior disappeared ( , 0, and 0.025 for the three evolutionary criteria mentioned, respectively) when cell fitness was computed using an expression score from a set of critical genes different from those used during the design phase. It should be noted that the optimality degree under single perturbations did not significantly depend on alterations in metabolic uptake factors.
, 0, and 0.025 for the three evolutionary criteria mentioned, respectively) when cell fitness was computed using an expression score from a set of critical genes different from those used during the design phase. It should be noted that the optimality degree under single perturbations did not significantly depend on alterations in metabolic uptake factors.
Fig. 4.

Examples of the process of gene rearrangement (A) and rewiring (B and C) of a GTRN. Only WT or synthetic operons regulated by EFs are plotted. (A) WT and rewired E. coli genome maps showing operons regulated by the EFs [nitrogen (blue) and carbon (orange) sources, and oxygen (green)]. (B) Number of TFs regulating the promoters (in the WT or rewired GTRNs) of critical enzymes with rate limiting in the cellular respiration, glycolysis pathway, and nitrogen metabolism. (C) Operons containing TFs affected by some EFs in the WT (Top) and rewired (Lower; see also Fig. S7A) GTRNs. The dashed boxes surrounding genes exemplify the notation.
Next, we studied systems that were re-engineered under simultaneous multiple perturbations (Fig. 3D). We predicted GTRN optimality by altering oxygen and carbon source uptake factors in the same range defined by single perturbations, and we added a third sensing component related to the nitrogen source by adding nitrate to the environment. As before, rewired GTRNs achieved negative or zero degrees of normalized optimality with the two first evolutionary criteria (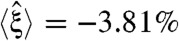 ), but for the third criterion, the average normalized optimality (
), but for the third criterion, the average normalized optimality (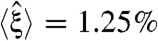 ) indicated that new systems retained the fitness of the optimal system.
) indicated that new systems retained the fitness of the optimal system.
Discussion
Design Principles of Genomic Adaptation to Environmental Changes.
One important implication of our results is inference of some genome design principles (2, 4). In particular, we studied the rewired TRNs that had achieved over-optimality or lost optimality. Our rewired GTRN were more susceptible to environmental perturbations when optimality was computed using transcriptomic fitness based on a different set of genes than those selected for the computational evolution. Recent work has shown that biochemical networks have evolved to capture the multidimensional structure of diverse environments and thus form internal representations (through regulatory networks) that allow the prediction of environmental changes. For example, Tagkopoulos et al. (17) provided evidence of anticipatory behavior of E. coli to changes in temperature and oxygen levels that occurred over evolutionary time scales (21, 22). We examined the anticipatory ability of our rewired GTRNs by computing their optimality using transcriptomic fitness with the same set of genes used in the in silico evolutionary process. Interestingly, we found that rewired GTRNs achieved greater optimality degrees than those of WT GTRNs for both single and multiple environmental perturbations. This suggests that natural selection may be “shortsighted” (i.e., it does not anticipate large changes over the long term) and that actual TRNs have thus evolved for optimal responses to regimes of small fluctuations.
We could induct some design principles by analyzing the genes and operons involved with the uptake of oxygen, nitrate, and glucose in the WT and rewired GTRNs (Fig. 4C, Upper and Lower). It is insightful to recall Savageau’s Demand theory (23), which in our context states that enzymes catabolizing the chemical inducers present more (less) often should be regulated by a positive (negative) transcription regulation mode. This rule is thought to provide robustness under mutational drift (5). We can check in Fig. 4B that the WT GTRN follows this rule by inspecting the regulation mode of the rate-limiting enzymes (genes in green boxes) associated to our chemical inducers (in high demand). As our methodology does not consider evolution in the context of a population, we do not expect it could comply with the propositions of such theory. Surprisingly, we can still see that the rewired GTRN also follows Savageau’s rules for the corresponding key enzymes. Probably this is due to our choice of fitness function that forces the rewired GTRN to have similar gene expression profiles than the WT under changing environments. In addition, as our GTRN evolution only relocates genes, it is difficult to change the mode of regulation in a single evolution step. We should notice that regulatory circuits designed in synthetic biology often lack robustness to mutational drift (24). Sometimes this lack of evolutionary robustness may be desired for biosafety reasons. Further work could consider incorporating such robustness into our fitness function.
Implications of Rewired GTRNs to Genome Organization.
Our results demonstrate that it is possible to rewire the GTRN of E. coli, achieving up to 69% reduction in the number of regulatory interactions and a 73% reduction in the number of operons, while maintaining its ability to physiologically respond to environmental perturbations. One limitation of the rewired GTRNs evolved under a single constant environment is that they will not behave like the WT GTRN under alternative environments. Fig. 4C, Lower, illustrates this point, where the arcA synthetic operon also contains the galS enzyme involved in the galactose metabolism. Fig. S7 shows how the transcriptomic fitness under galactose variation differs from the WT. In addition, we found that the rewired GTRNs contain operons that encompass several genes with similar functionality. This is an important result, given that the fitness function imposed to evaluate GTRNs performance did not consider gene function. This agrees with the experimental observation that genes within an operon have similar functions (25). Moreover, these GTRNs acquired the ability to adapt more rapidly to environmental changes, probably as a direct consequence of the reduced number of regulatory elements. Our methodology could also be applied to the de novo genome design problem if we had a perfect model of the WT GTRN. As the WT GTRN is still poorly known to aim for a faithful biological matching, it is not yet reasonable to seek any biological implementation of the rewired GTRNs, even if it would not be hard to assign a genomic nucleotide sequence for it. The de novo design of cells with synthetic genomes that are viable in a well-defined environment might require only the constitutive expression of the minimal set of genes required for life (26), but design of genomes adapted to various environments requires incorporating computational methodologies evolving GTRNs. We expect that the improvement of GTRNs and the rapid development of technologies allowing the synthesis of novel genomes and their introduction into hosts (27–29) will allow the construction of simplified genomes.
Materials and Methods
Mathematical Genome-Scale Model.
We used transcriptomic data to infer a continuous model for the transcription of all E. coli genes, which we then used to assign appropriate parameters to promoter and TF coding sequences. By assuming that these parameters do not depend on genomic context in most cases, we proposed our first methodology for the automatic evolution of rewired GTRNs under changing environments. Specifically, we constructed a GTRN for the WT genome that was able to predict gene regulation at the transcriptional and environmental levels (SI Text). For this, we adopted a linear model based on differential equations describing the time dynamics of each mRNA (7, 12) to infer kinetic parameters for promoter and TF sequences. Thus, the mRNA dynamics from the ith gene, yi, is given by dyi/dt = ai + Σjβijyj + ΣkγikΔvk - δiyi, where αi represents its constitutive transcription rate, βij represents the regulatory effect that gene j has on gene i, γik represents the effect that environmental factor (EF),that is, the metabolic uptake factor k, has on the expression of gene i; 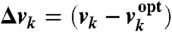 is the difference between the uptake factor measured under a given environmental condition, vk, and the uptake factor measured in the optimal environmental condition,
is the difference between the uptake factor measured under a given environmental condition, vk, and the uptake factor measured in the optimal environmental condition, 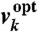 ; and δi represents the degradation and dilution rate constant.
; and δi represents the degradation and dilution rate constant.
Computational Evolution of GTRNs.
The main variables required for automatic evolution of GTRNs are the same as those required for any evolutionary algorithm: (i) an initial GTRN, (ii) evolutionary steps represented by changes in the genome (Fig. S1), and (iii) a fitness function that evaluates the performance of each mutant GTRN (SI Text). For the first step, we used the GTRN of the model bacterium E. coli. The second step was achieved by dissecting the bacterial GTRN into elementary modules (transcriptional model of the E. coli WT GTRN, http://repository.issb.genopole.fr/frontal/Technology/Tools/Carrera_SupDat2.xml/at_download/file), to which evolutionary rules were applied.
One design approach that we used involved the computational evolution of the GTRN, where we pursued two goals simultaneously (SI Materials and Methods): (i) simplifying the internal structure of the E. coli GTRN, and (ii) maintaining the external system function. To maximize the modularity of the system and thus simplify the TRN, we defined a measure based on the entropy of the TRN, 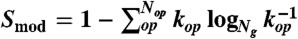 , where
, where 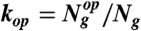 .
. 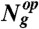 represents the number of genes in the operon op, Ng is the number of nonconstitutive genes in the WT GTRN, and Nop is the updated number of operons contained in the rewired GTRN. We also aimed to maximize the similarity of the expression profiles of the WT (yopt) and rewired (yenv) GTRN for a set of extreme environments (Nenv) and for a set of critical genes that guarantee the functionality of the rewired GTRN,
represents the number of genes in the operon op, Ng is the number of nonconstitutive genes in the WT GTRN, and Nop is the updated number of operons contained in the rewired GTRN. We also aimed to maximize the similarity of the expression profiles of the WT (yopt) and rewired (yenv) GTRN for a set of extreme environments (Nenv) and for a set of critical genes that guarantee the functionality of the rewired GTRN, 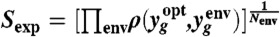 , where g denotes genes included in a set of critical genes that guarantee the optimal growth of the cell. We used the TRN model integrated with signal transduction to measure that similarity. Considering these two aims, we developed an optimization algorithm based on the mutation rules described in Fig. S2 to rewire the WT E. coli GTRN (SI Materials and Methods). Genes that are controlled by constitutive promoters were not involved in the computational evolution. These genes could always be regrouped in a straightforward way by assuming that they could be collapsed into large operons regulated by a gradient of different expression levels.
, where g denotes genes included in a set of critical genes that guarantee the optimal growth of the cell. We used the TRN model integrated with signal transduction to measure that similarity. Considering these two aims, we developed an optimization algorithm based on the mutation rules described in Fig. S2 to rewire the WT E. coli GTRN (SI Materials and Methods). Genes that are controlled by constitutive promoters were not involved in the computational evolution. These genes could always be regrouped in a straightforward way by assuming that they could be collapsed into large operons regulated by a gradient of different expression levels.
GTRN Optimality Degree.
We assumed that cell fitness could be estimated in terms of the Sexp objective function. This allowed the study of GTRN adaptation under changing environments in one (Δvk=i ≠ 0 and Δvk≠i = 0) or multiple (Δvk ≠ 0∀k) directions (14). To do this, we defined the optimality degree, ξΔvk, in a target environment characterized by  and different from the optimal environment as the difference between Sexp evaluated in an environment containing Δvk = 0 (i.e., fitness in the optimal condition) and that evaluated in the target environment containing
and different from the optimal environment as the difference between Sexp evaluated in an environment containing Δvk = 0 (i.e., fitness in the optimal condition) and that evaluated in the target environment containing  . Hence, we distinguished between positive and negative error adaptation corresponding to environmental states where cell fitness achieved sub- or over-optimal growth, respectively.
. Hence, we distinguished between positive and negative error adaptation corresponding to environmental states where cell fitness achieved sub- or over-optimal growth, respectively.
Functional Analysis of GTRNs.
Genes contained in the operons of all rewired GTRNs were functionally identified using 184 biological functions in GO (30). We defined the degree of functional similarity (ϕop) of a given operon, op, as the ratio between the maximum number of genes with the same functionality and the operon size. We imposed ϕop = 0 for those operons containing only one gene because more than one gene was needed to assess functional similarity.
Supplementary Material
ACKNOWLEDGMENTS.
This work was supported by FP7-ICT-043338 (Bacterial Computing with Engineered Populations), ATIGE-Genopole, TIN2006-12860 (Ministry of Science and Innovation [MICINN]), and the Fondation pour la Recherche Medicale grants (to A.J.). S.F.E. is supported by grant BFU2009-06993 (MICINN). We thank B. Palsson, T. Conrad, and M. Isalan for providing us with experimental data from their recent publications, J. Forment for help with computer resources; R. Estrela, G. Rodrigo, for discussions; J. Sardanyés, T. Landrain, L. Janniere, I. Junier, M. P. Zwart, and F. Kepes for critical reading of the manuscript; and the comments provided by anonymous reviewers.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1200030109/-/DCSupplemental.
References
- 1.Khalil AS, Collins JJ. Synthetic biology: Applications come of age. Nat Rev Genet. 2010;11:367–379. doi: 10.1038/nrg2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhardwaj N, Kim PM, Gerstein MB. Rewiring of transcriptional regulatory networks: Hierarchy, rather than connectivity, better reflects the importance of regulators. Sci Signaling. 2010;3:ra79. doi: 10.1126/scisignal.2001014. [DOI] [PubMed] [Google Scholar]
- 3.Isalan M, et al. Evolvability and hierarchy in rewired bacterial gene networks. Nature. 2008;452:840–845. doi: 10.1038/nature06847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bashor CJ, et al. Rewiring cells: Synthetic Biology as a tool to interrogate the organizational principles of living sytems. Annu Rev Biophys. 2010;39:515–537. doi: 10.1146/annurev.biophys.050708.133652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alon U. An introduction to systems biology: Design principles of biological systems. London: Chapman & Hall/CRC; 2007. [Google Scholar]
- 6.Ronen M, Rosenberg R, Shraiman BI, Alon U. Assigning numbers to the arrows: parameterizing a gene regulation network by using accurate expression kinetics. Proc Natl Acad Sci USA. 2002;99:10555–10560. doi: 10.1073/pnas.152046799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrera J, Rodrigo G, Jaramillo A. Model-based redesign of global transcription regulation. Nucleic Acids Res. 2009;37:e38. doi: 10.1093/nar/gkp022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrera J, Rodrigo G, Jaramillo A. Towards the automated engineering of a synthetic genome. Mol Biosyst. 2009;5:733–743. doi: 10.1039/b904400k. [DOI] [PubMed] [Google Scholar]
- 9.Chan LY, Kosuri S, Endy D. Refactoring bacteriophage T7. Mol Syst Biol. 2005;1:2005.0018. doi: 10.1038/msb4100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonneau R. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–1365. doi: 10.1016/j.cell.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 11.Covert MW, et al. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:92–96. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- 12.Gardner TS, et al. Inferring genetic networks and identifying compound mode of action via expression profiling. Science. 2003;301:102–105. doi: 10.1126/science.1081900. [DOI] [PubMed] [Google Scholar]
- 13.Carrera J, Rodrigo G, Jaramillo A, Elena SF. Reverse-engineering the Arabidopsis thaliana transcriptional network under changing environmental conditions. Genome Biol. 2009;10:R96. doi: 10.1186/gb-2009-10-9-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francois P, Hakim V. Design of genetic networks with specified functions by evolution in silico. Proc Natl Acad Sci USA. 2004;101:580–585. doi: 10.1073/pnas.0304532101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodrigo G, Carrera J, Jaramillo A. Computational design of synthetic regulatory networks from a genetic library to characterize the designability of dynamical behaviors. Nucleic Acids Res. 2011;39:e138. doi: 10.1093/nar/gkr616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Airoldi EM, et al. Predicting cellular growth from gene expression signatures. PLoS Comput Biol. 2009;5:e1000257. doi: 10.1371/journal.pcbi.1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tagkopoulos I, Liu YC, Tavazoie S. Predictive behavior within microbial genetic networks. Science. 2008;320:1313–1317. doi: 10.1126/science.1154456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faith JJ, et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma W, et al. Defining network topologies that can achieve biochemical adaptation. Cell. 2009;138:760–773. doi: 10.1016/j.cell.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conrad TM, et al. Whole-genome resequencing of Escherichia coli K-12 MG1655 undergoing short-term laboratory evolution in lactate minimal media reveals flexible selection of adaptive mutations. Genome Biol. 2009;9:R118. doi: 10.1186/gb-2009-10-10-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perkins TJ, Swain PS. Strategies for cellular decision-making. Mol Syst Biol. 2009;5:326. doi: 10.1038/msb.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koide T, Pang WL, Baliga NS. The role of predictive modeling in rationally re-engineering biological systems. Nat Rev Microbiol. 2009;7:297–305. doi: 10.1038/nrmicro2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savageau MA. Demand theory of gene regulation. I. Quantitative development of the theory. Genetics. 1998;149:1665–1676. doi: 10.1093/genetics/149.4.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sleight SC, Bartley BA, Lieviant JA, Sauro HM. Designing and engineering evolutionary robust genetic circuits. J Biol Eng. 2010;4:12. doi: 10.1186/1754-1611-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keseler IM, et al. EcoCyc: A comprehensive database of Escherichia coli biology. Nucleic Acids Res. 2011:39, D583–D590. doi: 10.1093/nar/gkq1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forster AC, Church GM. Towards synthesis of a minimal cell. Mol Syst Biol. 2006;2:45. doi: 10.1038/msb4100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lartigue C, et al. Genome transplantation in bacteria: Changing one species to another. Science. 2007;317:632–638. doi: 10.1126/science.1144622. [DOI] [PubMed] [Google Scholar]
- 28.Dymond JS, et al. Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature. 2011;477:471–476. doi: 10.1038/nature10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Temmea K, Zhaob D, Voigt CA. Refactoring the nitrogen fixation gene cluster from Klebsiella oxytoca. Proc Natl Acad Sci USA. 2012;109:7085–7090. doi: 10.1073/pnas.1120788109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ashburner M, et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.