

Abstract
The 1.1 Å, ultrahigh resolution neutron structure of hydrogen/deuterium (H/D) exchanged crambin is reported. Two hundred ninety-nine out of 315, or 94.9%, of the hydrogen atom positions in the protein have been experimentally derived and resolved through nuclear density maps. A number of unconventional interactions are clearly defined, including a potential O─H…π interaction between a water molecule and the aromatic ring of residue Y44, as well as a number of potential C─H…O hydrogen bonds. Hydrogen bonding networks that are ambiguous in the 0.85 Å ultrahigh resolution X-ray structure can be resolved by accurate orientation of water molecules. Furthermore, the high resolution of the reported structure has allowed for the anisotropic description of 36 deuterium atoms in the protein. The visibility of hydrogen and deuterium atoms in the nuclear density maps is discussed in relation to the resolution of the neutron data.
Keywords: hydrogen/deuterium exchange, neutron structure, PDB 4FC1, crystal, solvent
Approximately one-half of all atoms in a protein are hydrogen (H) atoms. H atoms play a variety of critical roles in proteins, including hydrogen bonding, electrostatic interactions, and catalysis. Unfortunately, H atoms are difficult to visualize in a three-dimensional context, and for the vast majority of crystal structures, their locations are inferred from the position of their neighboring heavy atom (C, N, O, S). These locations are based on atomic positions in databases of previously solved structures, general chemical knowledge, quantum mechanical calculations, or potential hydrogen bonding interactions. X-ray crystallography has the potential to reveal the positions of H atoms at ultrahigh resolution, typically at 1.0 Å resolution or better, from Fo-Fc difference electron density maps. In practice, however, even in the highest resolution macromolecular X-ray structures, only a limited number of H atom positions are experimentally determined, typically in well-ordered (i.e., low B factor) regions at the core of proteins (1).
Neutron crystallography, on the other hand, has the ability to resolve individual H atoms, even at medium resolution of around 2 Å due to the unique scattering properties of the H isotopes (2). The coherent neutron scattering length of deuterium (2H or D, +6.67 fm) is comparable to that of heavier atoms in proteins such as C, N, and O, whereas the light 1H isotope has a negative scattering length of -3.74 fm. The scattering properties of H and D allow for the accurate placement of these atoms in small molecule and macromolecular structures, in contrast to X-rays, where the single electron and proximity to heavier atoms make H atoms difficult to resolve. Moreover, the scattering characteristics of D have useful applications in neutron macromolecular crystallography, such as determining protonation states of active site residues in enzymes and establishing the identities and orientation of solvent molecules (3–15). Neutron diffraction has also been useful for determining orientations of methyl rotors and probing the energy barrier to rotation about a chemical bond (16, 17). By exchanging labile hydrogens in the amide backbone with deuterium, the relative populations of H and D in the main chain can be used as a sensitive probe of dynamics and breathing motion in proteins (18).
By analogy with high resolution X-ray crystallography, higher resolution neutron data can provide the positions of individual atoms, including Hs and Ds, that are determined to greater accuracy and precision than at lower resolutions. In addition to a more precise structure, the data-to-parameter ratio would be sufficient for refinement of anisotropic atomic displacement parameters (ADPs). Refining anisotropic ADPs potentially provides insight into asymmetric, vibrational, and librational motions of atoms, and has been useful in visualizing the dynamic properties of proteins in crystal structures. However, it is also important to take into account that anisotropic ADPs may also partially reflect the static disorder of atomic groups. Anisotropic ADP analysis is limited to the heavier atoms in proteins in X-ray structures, due to the weak scattering of X-rays by H atoms. The unique neutron scattering properties of D atoms make it possible to refine anisotropic ADPs for selected D atoms when high resolution neutron diffraction data are available.
Very few macromolecular crystals diffract X-rays beyond 1.0 Å resolution. Less than 0.7% of the X-ray structures in the Protein Data Bank (PDB; 493 structures out of 73,000 entries) are reported at ultrahigh resolution. We initially identified and screened a number of candidate proteins that could potentially provide crystals that diffract neutrons to ultrahigh resolution. After taking into account practical factors such as protein preparation, yield, and crystal symmetry, we chose to work with the small hydrophobic protein crambin (46 a.a., 4.7 kDa). The protein contains a mix of β-strand and α-helical regions (Fig. 1A) and forms exceptionally well-ordered crystals that diffract X-rays to 0.48 Å resolution using synchrotron radiation (19, 20). At that resolution, exquisitely detailed features relating to chemical bonding and deformation electron density can be seen, and it is possible to do charge density refinement (21). A lower resolution, 1.5 Å neutron dataset was collected on crambin in the early 1980s (22). We have measured neutron diffraction data from a crystal of crambin to ultrahigh resolution of 1.1 Å. This is the highest resolution neutron structure of a protein to date and allowed us to refine anisotropic ADPs of many atoms. Based on our refined structure we report here the anisotropic vibrational behavior of individual D atoms in a protein. The data quality has allowed us to visualize nearly all H and D atoms within the protein and to accurately orient a number of well-ordered solvent molecules in the first and second hydration shells. We can now take an inventory of all hydrogen bonding interactions within the protein and ascertain the categories and suggest strengths of H bonds in the system. We demonstrate the power of ultrahigh resolution neutron diffraction to resolve ambiguous hydrogen bonding networks and also to reveal unconventional structural features and interatomic interactions, such as apparent partial exchange of H atoms on Cα and a putative O─H…π interaction.
Fig. 1.

(A) Structure of crambin, with main chain amide H/D exchange pattern ranging from blue (unexchanged) to red (fully exchanged). (B) Side chains of residues R10, N14, N46, and T2 are depicted with anisotropic ellipsoids. D atoms are shown in green, with hydrogen bonds indicated.
Results and Discussion
Ultrahigh Resolution Neutron Structure of Crambin.
A neutron diffraction dataset of high completeness and ultrahigh resolution (1.1 Å) was collected on a single, 4 mm3 crystal of H/D exchanged crambin at the Protein Crystallography Station (PCS) at the Los Alamos Neutron Science Center (LANSCE). Phases from our previously reported 0.85 Å room temperature (RT) X-ray structure of H/D exchanged crambin were used as a starting model for the refinement (23, 24). After initial joint X-ray/neutron refinement in nCNS at 1.5 Å, the model was then refined in SHELX-97 against the full 1.1 Å neutron dataset alone, to a final Rfree/R1 of 0.254/0.211 for all data (0.217/0.170 for Fo > 4σ) (Table S1). The final model consisted of all 46 amino acids of the protein and 42 D2O molecules. There was additional weak nuclear density that could be interpreted as additional second shell, partially disordered solvent molecules, which were not modeled. The refined protein structure is virtually identical to the parallel 0.85 Å X-ray structure when the positions of the heavy atoms are compared. H atoms of functional groups on the side chains are observed to be exchanged with deuteriums and are clearly defined. In total, 299 out of 315, or 94.9%, of the H atoms in crambin are resolved at 1.5 sigma nuclear density, either in positive 2Fo-Fc maps as labile H exchanged with D or in negative 2Fo-Fc maps as unexchanged H. The complete set of H and D atoms can be resolved in 37 out of the 46 residues. The H atoms that are not visible are all on side chains, typically in terminal methyl groups.
Although the crystal of crambin used in this study was grown in 55% hydrogenated ethanol solution and sealed in a capillary containing perdeuterated ethanol in D2O, no obvious nuclear density was observed for the ethanol. Perdeuterated ethanol would be expected to scatter neutrons strongly due to the presence of six D atoms and has been observed clearly in previous neutron crystallographic studies of lysozyme, mainly at surface hydrophobic sites (25). Other smaller deuterated solvent molecules with less scattering power have also been observed in neutron macromolecular structures (26, 27). It is possible that perdeuterated ethanol does not have specific binding sites on the surface of crambin and is mostly present in the disordered solvent regions, lowering the free energy of crystal formation and helping produce a better diffracting crystal. Whereas ethanol or other polar organic solvents solubilize crambin, the crystal packing environment sequesters a hydrophobic patch on the protein, making the ethanol superfluous (22).
Anisotropic Behavior of H and D Atoms.
Refinement of anisotropic ADPs is normally applied to X-ray data extending to 1.2 Å resolution or better. Anisotropic ADPs are described by a tensor of six values, and their refinement can potentially lead to overfitting the data. This precludes the refinement of anisotropic ADPs at lower resolutions that have a limited number of observations. Because our neutron diffraction data extended to 1.1 Å, it was possible to refine the structure with anisotropic ADPs for a number of atoms, while still maintaining a reasonable data-to-parameter ratio (approximately 5 when refining anisotropic ADPs while restraining the protein molecule as a rigid body). The Rfree/R1 values dropped 0.6%/1.3% following refinement, and the difference between Rfree/R1 remained < 5%, indicating no overfitting of the structure.
Furthermore, as D atoms scatter neutrons strongly, we reasoned that anisotropic ADPs could also be refined for fully occupied D atoms in the structure. Anisotropic ADPs were refined for all 26 exchanged Ds on the side chains, seven fully exchanged backbone amide Ds, and the terminal 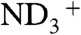 group (Fig. 1B). The reliability of the anisotropic ADP refinement with the neutron data can be gauged by comparing anisotropy values from neutron diffraction to those from X-ray diffraction. The anisotropy value is defined as the ratio of the minimum to maximum eigenvalues of the ANISOU tensor. A small value of anisotropy corresponds to a highly anisotropic atom, whereas a value close to 1.0 corresponds to a nearly isotropic atom. Of the 20 residues for which anisotropies of D atoms were refined, the anisotropy values of the individual atoms, as well as the trend in anisotropy, were generally consistent between the X-ray and neutron refinement (Fig. S1). Importantly, different refinement target functions and programs were used for refining the X-ray data (maximum likelihood, PHENIX) and the neutron data (conjugate gradient least squares, SHELX-97).
group (Fig. 1B). The reliability of the anisotropic ADP refinement with the neutron data can be gauged by comparing anisotropy values from neutron diffraction to those from X-ray diffraction. The anisotropy value is defined as the ratio of the minimum to maximum eigenvalues of the ANISOU tensor. A small value of anisotropy corresponds to a highly anisotropic atom, whereas a value close to 1.0 corresponds to a nearly isotropic atom. Of the 20 residues for which anisotropies of D atoms were refined, the anisotropy values of the individual atoms, as well as the trend in anisotropy, were generally consistent between the X-ray and neutron refinement (Fig. S1). Importantly, different refinement target functions and programs were used for refining the X-ray data (maximum likelihood, PHENIX) and the neutron data (conjugate gradient least squares, SHELX-97).
The anisotropy of each D atom was analyzed in the context of its bonded neighbor. On a qualitative level, out of 36 such pairs, 23 of the D atoms were more anisotropic (lower anisotropy value) than their bonded heavy atom neighbor, whereas 13 were less anisotropic (higher anisotropy value) (Fig. 2). Thus in about two-thirds of the cases, the light D atom tends to move or librate to a greater extent than its heavier neighbor, regardless of the chemical environment of the D atom. No correlation was observed between the principal axis of the D atom ADP ellipsoid and the direction of the H bond, although there are a limited number of such observations. These results should be interpreted with caution because the apparent atomic librations may also be due to static disorder. To quantitatively deconvolute static and dynamic disorder requires obtaining structures at different temperatures, whereas a translation, libration, screw tensor (TLS) analysis can also be performed to obtain a qualitative measure for a protein molecule. Nevertheless, the results raise the tantalizing possibility of further probing the vibrational behavior of individual H and D atoms in macromolecular structures, especially with perdeuterated samples and with ultrahigh resolution data.
Fig. 2.

Anisotropy of deuteriums and their bonded neighbors. Thirty-six pairs of atoms on 20 residues and their anisotropies are plotted.
Visibility of H Atoms at Different Resolution Cutoffs.
Empirically, difference electron density for H atoms in X-ray structures is visible at 1.0 Å and better. However, flexible (high B factor) regions in macromolecules and the fact that atom-H bonds can be highly polar tend to blur out the scattering contribution from a single electron, making H atoms effectively visible only in lower B factor regions of the protein molecule. To date, no protein crystal has diffracted neutrons to high enough resolution to experimentally ascertain the visibility of H atoms as a function of resolution. To better define the concept of atomic resolution in macromolecular neutron structures, nuclear density maps were calculated using phases from the final refined coordinates, truncating the neutron diffraction data at 1.1, 1.5, 2.0, and 2.5 Å resolution. “Cancellation” effects are observed in the nuclear density maps at carbon atoms of aliphatic side chains at 2.0 and 2.5 Å resolution. These effects arise from negative scattering of unexchanged H atoms bonded to neighboring carbon atoms, leading to nodes of absent nuclear density for CH2 and CH3 groups (Fig. 3A). At resolutions lower than 2.0 Å, side chains are poorly defined, whereas H/D exchanged groups at the termini of polar side chains are still characterized by strong nuclear density. Water molecule orientation is clear at 1.1 and 1.5 Å but becomes less straightforward at 2.0 Å. For resolutions around 2.5 Å, the placement and orientation of waters are dependent on the heavy atom positions and the resolution appears to be insufficient to accurately orient D2O molecules in hydrogen bonding networks (Fig. 3B). Nevertheless, exchanged functional groups remain relatively easy to identify at medium resolution, hence the utility of neutron crystallography for determining protonation states of key residues in enzyme active sites. In the case of unexchanged H atoms, their visibility is limited to 1.5 Å resolution or better (Fig. 3C). In the current structure, the resolution of the neutron data is high enough such that the positions of C and H atoms of aliphatic CH2 and CH3 groups are clearly resolved and do not show the cancellation effect seen in lower resolution nuclear density maps (7, 28). In this case, refinement against neutron diffraction data alone was sufficient for clear placement of almost all atoms in the model. For more typical neutron diffraction studies, with diffraction in the 1.8–2.5 Å range, joint X-ray/neutron refinement can “stabilize” the structure and the X-ray diffraction data can correctly place the side chain carbons, whereas the neutron data can correctly orient exchanged terminal groups on side chains.
Fig. 3.

Effect of resolution on visibility of atoms. (A) 2Fo-Fc nuclear density for L18, at 1.1, 1.5, 2.0, and 2.5 Å resolution. (B) Nuclear density for D2O molecules. (C) Nuclear density for Y44. Density maps are contoured at 1.5 sigma, with positive 2Fo-Fc density in green and negative 2Fo-Fc density in violet.
Ultrahigh Resolution and H/D Exchange.
H/D exchange of backbone amides is a powerful probe for assessing protein dynamics (18). The D occupancies of main chain amides were refined (Fig. 1A). Given the small size of crambin, only a limited number of residues can be characterized as interior residues, and relatively few residues are protected from H/D backbone exchange. Consistent with previous studies, backbone amides involved in hydrogen bonding between adjacent β strands remain unexchanged, whereas those facing the solvent are fully exchanged (7, 29). α-helical regions are generally unexchanged or partially exchanged, as the backbone amides are hydrogen bonded to the carbonyl oxygen in the adjacent helical turn.
The 1.1 Å data also revealed a number of unusual H/D exchange characteristics in the vicinity of residues T30 and G31, which lie in a 310 helical region at the end of the second helix (Fig. 4A). The carbonyl oxygen of C26 has two potential H bond donors in its proximity, the amide backbone hydrogen of residue T30 (H…O distance of 2.29 Å) and the side chain hydroxyl of T30 (D…O distance of 1.74 Å). Surprisingly, the hydroxyl group of T30 is fully exchanged, despite being solvent inaccessible and surrounded by mobile but unexchanged H atoms. The hydroxyl oxygen of T30 accepts an H bond from an unexchanged backbone amide H of C32. The H/D exchange of the T30 side chain may be explained by global protein breathing motions, the ability of water to interact with the hydroxyl group, or by periodic side chain rotation.
Fig. 4.

(A) H/D exchange pattern involving side chain of residue T30. The threonine side chain has exchanged for an OD, however neighboring backbone amide groups remain unexchanged, with no access to solvent for the T30 side chain. (B) Possible partial exchange of backbone hydrogen on G31. The 1.1 Å map shows that HA1 (left) on the α carbon may be partially exchanged for deuterium. Density maps are contoured at 1.5 sigma, with positive 2Fo-Fc density in green and negative 2Fo-Fc density in violet. Distances are indicated in Å.
Negative 2Fo-Fc nuclear density maps show that the α H atoms of G31 (and a lesser extent G37) appear to be nonequivalent, suggesting that one of them could be partially exchanged with D (Fig. 4B). The refined B factor of fully occupied HA1 was 13.3 Å2, compared to 7.6 Å2 for HA2, and 7.3 Å2 for the α carbon, with much weaker negative nuclear density than expected for a fully occupied H atom. This indicated that perhaps a stronger positive scatterer co-occupied the HA1 position, such as a D atom. We placed a partially occupied D at the HA1 position and then refined the relative H/D occupancies. The H/D occupancies of the site refined to around 80%/20%, with similar B factors, 7.4/8.5 Å2, respectively. This is consistent with no visible positive nuclear density that would be expected at a higher D occupancy. Because the glycine residues are fixed in conformation and should a priori have similar peaks and B factors for the H atoms, this unusual situation in G31 may be attributed to partial H/D exchange. Glycine, due to the lack of a side chain, is a considerably stronger carbon acid than the other amino acids (pKa for Cα approximately 22), and the elevated rate of H/D exchange could be attributed to the chemical environment of G31. The side chain hydroxyl groups on serine and threonine residues have a similar pKa (approximately 18–20) and are found to be fully exchanged in this structure, including nearby residue T30. This observation strengthens the theory that backbone α hydrogens can act as H bond donors in unconventional C─H…O interactions and participate in chemical exchange. Additional mass spectroscopy and NMR experiments will be necessary to thoroughly investigate and corroborate these observations.
We also note a set of potential Cα─H…O hydrogen bonds in the β strand region of the protein, between the carbonyl oxygen of T1 to the α H of I34 (2.51 Å), the oxygen of I33 to the α H of T2 (2.42 Å), and the oxygen of C3 to the α H of C32 (2.81 Å) (Fig. S2). This is in agreement with extensive computational evidence to the existence of and the role of the relatively weak Cα─H…O hydrogen bonds in stabilizing protein secondary and tertiary structure (30). Although it is difficult to verify this interaction experimentally, computational studies and analysis of structures in the PDB suggest that these H bonds contribute significantly to the repertoire of stabilizing interactions within a protein. Our observation of possible partial exchange in G31 also complements a number of earlier reports of partial C-H exchange in neutron structures (31, 32).
Hydrogen Bonding and Resolving Hydrogen Bond Networks.
Together with the hydrophobic effect, H bonds are one of the primary interactions contributing to the formation of secondary and tertiary structural motifs and are crucial for protein–ligand recognition. For almost all X-ray crystal structures, including those solved at atomic resolution, H bonds are normally inferred from the heavy atom positions of the donor–acceptor pair, and their distances. In our structure, H atom positions have been experimentally determined, and the high resolution of the neutron diffraction data and the accuracy of the model allow us to analyze the observed H bonds within the protein and the water network in more detail.
Out of the 124 conventional H bonds tabulated, 91 of them fall within the 1.5–2.2 Å range of D-to-acceptor distances for medium strength H bonds; the remaining 33 are 2.2 Å or longer, and are probably weak (Fig. S3). The two α helices in crambin have different hydrogen bonding characteristics; residues in the first helix (residues 6–18) have intrahelical H bond distances averaging 2.09 Å, whereas those in the second helix (residues 22–27) have somewhat longer H bonds averaging 2.19 Å. Therefore, even within a regular secondary structure such as an α helix, there are systematic differences that reflect on the relative stability of the structure. In the case of crambin, the second helix appears to be more mobile, with higher average B factors and residues showing a slightly greater degree of amide backbone H/D exchange. The donor-H… acceptor angles average 153.5° (Fig. S4), more linear than the average for intrachain H bonds in proteins, 151.7° (33). The average angle between the carboxyl/carbonyl group and the H bond donor is 134.9°, comparable to the average angle of 136.4° within proteins (33) (Fig. S5). For the Cα─H…O hydrogen bonds, the donor-H… acceptor angles average 135.9° (Fig. S4), and generally have longer H…O distances, averaging 2.80 Å, consistent with a weaker interaction (Fig. S3).
X-ray structures often give ambiguous information about the position and orientation of H atoms, especially for water molecules, even at ultrahigh resolution. In the crambin structure, an extensive network of water molecules contributes to a crystal contact near E23 and A24. The electron density maps and distances between the water oxygens suggest a variety of potential hydrogen bonding interactions for W1022, including symmetry-related W1007 (3.36 Å O…O distance), W1023 (2.79 Å), W1024 (3.00 Å), and W1026 (3.59 Å), in addition to the backbone amide H of E23 (3.04 Å) (Fig. 5A). The network of hydrogen bonding is clarified in the 1.1 Å nuclear density map, which shows that W1022 is oriented to donate an H bond to W1023, in addition to accepting an H bond from the backbone amide of E23 but does not interact with W1007, W1024, or W1026 (Fig. 5B).
Fig. 5.

Resolving a hydrogen bond network. (A) 0.85 Å X-ray electron density map showing potential H bond partners for W1022. (B) 1.1 Å nuclear density map showing that W1022 interacts with W1023, but not W1007, W1024, or W1026. (C) Hydration environment around Y44. A potential O─D…π interaction is highlighted involving W1023. Distances are indicated in Å.
Another ambiguous H bond network is seen around residue Y44, which lies in a special environment within the protein, acting as a buffer between hydrophobic and polar regions. One face of the phenolic group makes van der Waals interactions with the side chain of I33, whereas the other face is hydrated by several well-ordered water molecules. Although W1023 appears to be satisfied with the full complement of H bond donors and acceptors, the orientation of W1023 suggests that it can also participate in a weak O─H…π interaction with the aromatic ring of Y44. Atom D1 of W1023 points towards the aromatic ring and lies 3.20 Å from the center of the ring, with a 118.4° O─D…π angle (Fig. 5C). The oxygen of W1023 is 3.78 Å from the center of the ring, farther than the 3.5 Å distance suggested for an interaction between an O lone pair and a delocalized π system (34). The O─H…π interaction is important for a wide variety of molecular recognition events, such as drug–receptor interactions and hydration of nucleic acids. The orientation of W1023 is unambiguous in the 1.1 Å neutron maps but is not clear in the 0.85 Å electron density maps or the lower resolution neutron maps (Fig. S6).
Perspective on the Utility of Ultrahigh Resolution Neutron Crystallography.
The work presented here is a major technical achievement in neutron macromolecular crystallography, demonstrating the ability to collect data to ultrahigh resolution on the PCS within a period of 3 wk on a low symmetry (space group P21) crystal. Although medium resolution (approximately 2.0 Å) neutron data are sufficient to distinguish protonation states of amino acid residues and solvent molecules, one clear benefit of ultrahigh resolution neutron data is the accuracy of H and D atom locations and being able to more fully define the attributes of individual H atoms in proteins. Indeed, we have been able to describe the vibrational characteristics of all fully exchanged D atoms in the structure by anisotropic ADP refinement, which has not been possible until now. A more thorough, quantitative analysis of the exchanged D atoms on the protein, as well as the solvent molecules, should be possible with higher resolution data and/or perdeuterated sample. Another significant enhancement was our ability to resolve nearly all (95%) individual H and D atom positions from the 1.1 Å data. All missing H atoms were on side chains, generally in higher B factor regions of the molecule. Similarly, at 1.5 Å resolution, approximately 85% of H atoms could be individually resolved. Therefore, this gain in neutron resolution has a profound effect on the visibility of H atoms, as has been systematically examined with perdeuterated and H/D exchanged crystals (35). From our maps truncating the data, it appears that a resolution of 2.0 Å or better is necessary to unambiguously orient waters, and that 1.5 Å resolution or better can clearly resolve individual H atoms. In contrast, with the 0.85 Å RT X-ray data, less than 50% of H atoms in the protein, and none in the ordered solvent, were visible in Fo-Fc difference maps (24). Perdeuteration, when practical, increases the visibility of D atom positions at lower resolutions (35–37).
The refinement protocol used was also unique and at the same time has pointed out the need for continued improvements in the refinement of neutron and X-ray data. Initial refinement employed nCNS to a resolution of 1.5 Å by using the joint X-ray/neutron refinement strategy with the current neutron and published X-ray structure factors. Both datasets were collected on crystals of crambin at ambient temperature, where labile H atoms were exchanged against perdeuterated mother liquor (55% perdeuterated ethanol in D2O). The adjustment of water orientations lowered the Rfree considerably, with very few adjustments necessary in the protein chain. Refinement was then switched to SHELX-97, which was able to handle anisotropic ADP refinement for the heavy atoms as well as a number of well-defined D atoms. Refinement protocols introducing electrostatics can aid in the orientation of water molecules and placement of H atoms using neutron data at resolutions lower than 2.0 Å (38).
Through an ultrahigh resolution neutron structure, we have been able to more completely describe the position and behavior of the H and D atoms in crambin. Unlike techniques that can probe H atoms as an ensemble, neutron crystallography and NMR have the potential to define the attributes of individual H atoms. We expect that this work and future ultrahigh resolution neutron structures will help present a broader picture of the diverse roles of H atoms and elucidate their behavior and interactions in the context of a macromolecule. Future computational efforts addressing protein folding, dynamics, and protein-ligand interactions can also benefit from our more complete description of H atoms in proteins.
Materials and Methods
Neutron Data Collection.
Data were collected at the PCS at the spallation neutron source at LANSCE. Data collection statistics were reported previously and are summarized in Table S1 (24). Forty-four settings were used to collect neutron diffraction data from a single crystal of crambin (4 mm3) that were exchanged against perdeuterated synthetic mother liquor. Although diffraction was seen to beyond 1.05 Å, a conservative cutoff resolution of 1.1 Å was chosen, with high completeness and good merging statistics in the outer shell. The overall Rmerge of 23.1% is comparable with neutron datasets collected at spallation sources (39). Each image was processed using a version of d*TREK modified for wavelength-resolved Laue neutron protein crystallography (40, 41). Integrated reflections were wavelength normalized using LAUENORM, and then merged using SCALA in CCP4i (42, 43).
Refinement.
Initial refinement at moderate resolution (1.8 Å) was done in nCNS using joint neutron and X-ray refinement, utilizing the neutron dataset and the published RT X-ray data and coordinates (PDB ID 3U7T), excluding waters and with isotropic ADPs. Several rounds of rigid body, positional, and individual isotropic ADP refinement were performed. The resolution was extended to 1.5 Å, and solvent D2O molecules, including their D atoms, were oriented into the nuclear density maps, with a 5% drop in the Rwork due to the strong scattering from the D atoms in the water molecules. Refinement was completed in SHELX-97, using the neutron data alone to 1.1 Å resolution. Anisotropic ADP refinement was first carried out on all heavy atoms in the protein (C, N, O, S), with a reasonable drop in both the Rfree and R1, and then carried out on 36 fully exchanged D atoms on side chains and backbone amides. Solvent molecules were refined isotropically. Refinement statistics are reported in Table S1.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Mary Jo Waltman, Marat Mustyakimov, Blaine Schatz, and Pavel Afonine for their expert technical assistance and advice. J.C.-H.C. thanks Dr. Justin and Cynthia Chen for their support. The PCS is funded by the Department of Energy Office of Biological and Environmental Research (DOE-OBER). P.L. was partly supported by a grant from the National Institute of General Medical Science of the National Institutes of Health (R01GM071939). A.Y.K. was partly supported by an Laboratory Directed Research and Development grant from Los Alamos National Laboratory (20120256ER).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 4FC1).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1208341109/-/DCSupplemental.
References
- 1.Howard EI, et al. Ultrahigh resolution drug design I: Details of interactions in human aldose reductase-inhibitor complex at 0.66 Å. Proteins. 2004;55:792–804. doi: 10.1002/prot.20015. [DOI] [PubMed] [Google Scholar]
- 2.Blakeley MP. Neutron macromolecular crystallography. Crystallogr Rev. 2009;15:157–218. [Google Scholar]
- 3.Kossiakoff AA, Spencer SA. Direct determination of the protonation states of aspartic acid-102 and histidine-57 in the tetrahedral intermediate of the serine proteases: Neutron structure of trypsin. Biochemistry. 1981;20:6462–6474. doi: 10.1021/bi00525a027. [DOI] [PubMed] [Google Scholar]
- 4.Bennett B, et al. Complementary neutron and ultrahigh X-ray diffraction studies of Escherichia coli dihydrofolate reductase complexed with methotrexate. Proc Natl Acad Sci USA. 2006;103:18493–18498. doi: 10.1073/pnas.0604977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coates L, et al. The catalytic mechanism of an aspartic proteinase explored with neutron and X-ray diffraction. J Am Chem Soc. 2008;130:7235–7237. doi: 10.1021/ja801269x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kovalevsky AY, et al. Hydrogen location in stages of an enzyme-catalyzed reaction: Time-of-flight neutron structure of D-xylose isomerase with bound d-xylulose. Biochemistry. 2008;47:7595–7597. doi: 10.1021/bi8005434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blum M-M, et al. Rapid determination of hydrogen positions and protonation states of diisopropyl fluorophosphatase by joint neutron and X-ray diffraction refinement. Proc Natl Acad Sci USA. 2009;106:713–718. doi: 10.1073/pnas.0807842106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher Z, et al. Neutron structure of human carbonic anhydrase II: Implications for proton transfer. Biochemistry. 2010;49:415–421. doi: 10.1021/bi901995n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kovalevsky AY, et al. Metal ion roles and the movement of hydrogen during reaction catalyzed by D-xylose isomerase: A joint X-ray and neutron diffraction study. Structure. 2010;18:688–699. doi: 10.1016/j.str.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher Z, et al. Neutron structure of human carbonic anhydrase II: A hydrogen-bonded water network “switch” is observed between pH 7.8 and 10.0. Biochemistry. 2011;50:9421–9423. doi: 10.1021/bi201487b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howard EI, et al. Neutron structure of type-III antifreeze protein allows the reconstruction of AFP-ice interface. J Mol Recognit. 2011;24:724–732. doi: 10.1002/jmr.1130. [DOI] [PubMed] [Google Scholar]
- 12.Kawamura K, et al. X-ray and neutron protein crystallographic analysis of the trypsin-BPTI complex. Acta Crystallogr D. 2011;67:140–148. doi: 10.1107/S0907444910053382. [DOI] [PubMed] [Google Scholar]
- 13.Kovalevsky AY, et al. Identification of the elusive hydronium ion exchanging roles with a proton in an enzyme at lower pH values. Angew Chem Int Ed. 2011;50:7520–7523. doi: 10.1002/anie.201101753. [DOI] [PubMed] [Google Scholar]
- 14.Tomanicek S, et al. The active site protonation states of perdeuterated Toho-1 b-lactamase determined by neutron diffraction support a role for Glu166 as the general base in acylation. FEBS Lett. 2011;585:364–368. doi: 10.1016/j.febslet.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 15.Yokoyama T, et al. Hydrogen-bond network and pH sensitivity in transthyretin: Neutron crystal structure of human transthyretin. J Struct Biol. 2012;177:283–290. doi: 10.1016/j.jsb.2011.12.022. [DOI] [PubMed] [Google Scholar]
- 16.Kossiakoff AA, Shteyn S. Effect of protein packing structure on side-chain methyl rotor conformations. Nature. 1984;311:582–583. doi: 10.1038/311582a0. [DOI] [PubMed] [Google Scholar]
- 17.Wilson CC. Zero point motion of the librating methyl group in p-hydroxyacetanilide. Chem Phys Lett. 1997;280:531–534. [Google Scholar]
- 18.Kossiakoff AA. Protein dynamics investigated by the neutron diffraction-hydrogen exchange technique. Nature. 1982;296:713–721. doi: 10.1038/296713a0. [DOI] [PubMed] [Google Scholar]
- 19.Teeter MM, Roe SM, Heo NH. Atomic resolution (0.83 Å) crystal structure of the hydrophobic protein crambin at 130 K. J Mol Biol. 1993;230:292–311. doi: 10.1006/jmbi.1993.1143. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt A, Teeter M, Weckert E, Lamzin VS. Crystal structure of the small protein crambin at 0.48 Å resolution. Acta Crystallogr F. 2011;67:424–428. doi: 10.1107/S1744309110052607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jelsch C, et al. Accurate protein crystallography at ultra-high resolution: Valence electron distribution in crambin. Proc Natl Acad Sci USA. 2000;97:3171–3176. doi: 10.1073/pnas.97.7.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teeter MM, Kossiakoff AA. In: Neutrons in Biology. Schoenborn BP, editor. New York: Plenum Press; 1984. pp. 335–348. [Google Scholar]
- 23.Adams PD, Mustyakimov M, Afonine PV, Langan P. Generalized X-ray and neutron crystallographic analysis: More accurate and complete structures for biological macromolecules. Acta Crystallogr D. 2009;65:567–573. doi: 10.1107/S0907444909011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen JC-H, et al. Room temperature ultra-high resolution time-of-flight neutron and X-ray diffraction studies of H/D exchanged crambin. Acta Crystallogr F. 2012;68:119–123. doi: 10.1107/S1744309111051499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehmann MS, Mason SA, McIntyre GJ. Study of ethanol-lysozyme interactions using neutron diffraction. Biochemistry. 1985;24:5862–5869. doi: 10.1021/bi00342a026. [DOI] [PubMed] [Google Scholar]
- 26.Langan P, Jogl G, Lehmann MS, Wilkinson C, Kratky C. Laue neutron diffraction studies of coenzyme cob(II)alamin. Acta Crystallogr D. 1999;55:51–59. doi: 10.1107/S090744499800660X. [DOI] [PubMed] [Google Scholar]
- 27.Jogl G, et al. High-resolution neutron crystallographic studies of the hydration of the coenzyme cob(II)almin. Acta Crystallogr D. 2011;67:584–591. doi: 10.1107/S090744491101496X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Afonine P, et al. Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr D. 2010;66:1153–1163. doi: 10.1107/S0907444910026582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen JC-H, Mustyakimov M, Schoenborn BP, Langan P, Blum M-M. Neutron structure and mechanistic studies of diisopropyl fluorophosphatase (DFPase) Acta Crystallogr D. 2010;66:1131–1138. doi: 10.1107/S0907444910034013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheiner S, Kar T, Gu Y. Strength of the CαH--O hydrogen bond of amino acids. J Biol Chem. 2001;278:9832–9837. doi: 10.1074/jbc.M010770200. [DOI] [PubMed] [Google Scholar]
- 31.Chatake T, Tanaka I, Umino H, Arai S, Niimura N. The hydration structure of a Z-DNA hexameric duplex determined by a neutron diffraction technique. Acta Crystallogr D. 2005;61:1088–1098. doi: 10.1107/S0907444905015581. [DOI] [PubMed] [Google Scholar]
- 32.Niimura N, Chatake T, Ostermann A, Kurihara K, Tanaka I. High resolution neutron protein crystallography. Hydrogen and hydration in proteins. Z Krist. 2003;218:96–107. [Google Scholar]
- 33.Xu D, Tsai C-J, Nussinov R. Hydrogen bonds and salt bridges across protein–protein interfaces. Protein Eng. 1997;10:999–1012. doi: 10.1093/protein/10.9.999. [DOI] [PubMed] [Google Scholar]
- 34.Jain A, Ramanathan V, Sankararamakrishnan R. Lone pair…pi interactions between water oxygens and aromatic residues: Quantum chemical studies based on high-resolution protein structures and model compounds. Protein Sci. 2009;18:595–605. doi: 10.1002/pro.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gardberg AS, et al. Unambiguous determination of H-atom positions: Comparing results from neutron and high-resolution X-ray crystallography. Acta Crystallogr D. 2010;66:558–567. doi: 10.1107/S0907444910005494. [DOI] [PubMed] [Google Scholar]
- 36.Shu F, Ramakrishnan V, Schoenborn BP. Enhanced visibility of hydrogen atoms by neutron crystallography on fully deuterated myoglobin. Proc Natl Acad Sci USA. 2000;97:3872–3877. doi: 10.1073/pnas.060024697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munshi P, et al. Rapid visualization of hydrogen positions in protein neutron crystallographic structures. Acta Crystallogr D. 2012;68:35–41. doi: 10.1107/S0907444911048402. [DOI] [PubMed] [Google Scholar]
- 38.Fenn T, et al. Reintroducing electrostatics into macromolecular crystallographic refinement: Application to neutron crystallography and DNA hydration. Structure. 2011;19:523–533. doi: 10.1016/j.str.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanson BL, et al. A preliminary time-of-flight neutron diffraction study of Streptomyces rubiginosus D-xylose isomerase. Acta Crystallogr D. 2004;60:241–249. doi: 10.1107/S0907444903025873. [DOI] [PubMed] [Google Scholar]
- 40.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 41.Langan P, Greene G. Protein crystallography with spallation neutrons: Collecting and processing wavelength-resolved Laue protein data. J Appl Crystallogr. 2004;37:253–257. [Google Scholar]
- 42.Helliwell JR, et al. The recording and analysis of synchrotron X-radiation Laue diffraction photographs. J Appl Crystallogr. 1989;22:483–497. [Google Scholar]
- 43.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr D. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.