

Abstract
The p110α catalytic subunit (PIK3CA) is one of the most frequently mutated genes in cancer. We have examined the activation of the wild-type p110α/p85α and a spectrum of oncogenic mutants using hydrogen/deuterium exchange mass spectrometry (HDX-MS). We find that for the wild-type enzyme, the natural transition from an inactive cytosolic conformation to an activated form on membranes entails four distinct events. Analysis of oncogenic mutations shows that all up-regulate the enzyme by enhancing one or more of these dynamic events. We provide the first insight into the activation mechanism by mutations in the linker between the adapter-binding domain (ABD) and the Ras-binding domain (RBD) (G106V and G118D). These mutations, which are common in endometrial cancers, enhance two of the natural activation events: movement of the ABD and ABD–RBD linker relative to the rest of the catalytic subunit and breaking the C2–iSH2 interface on binding membranes. C2 domain mutants (N345K and C420R) also mimic these events, even in the absence of membranes. A third event is breaking the nSH2–helical domain contact caused by phosphotyrosine-containing peptides binding to the enzyme, which is mimicked by a helical domain mutation (E545K). Interaction of the C lobe of the kinase domain with membranes is the fourth activation event, and is potentiated by kinase domain mutations (e.g., H1047R). All mutations increased lipid binding and basal activity, even mutants distant from the membrane surface. Our results elucidate a unifying mechanism in which diverse PIK3CA mutations stimulate lipid kinase activity by facilitating allosteric motions required for catalysis on membranes.
Keywords: H/D exchange, PI3 kinase, signaling
The oncogenic potential of phosphoinositide 3-kinases (PI3Ks) was first established when it was shown that an activated form of the PI3K isoform p110α (v-P3K) was encoded by an avian sarcoma virus and that this viral gene and its cellular counterpart can cause cell transformation (1). The advent of large-scale sequencing established that cancer-linked somatic mutations of p110α are among the most common mutations associated with human tumors (2–4). Tumor-associated mutations appear throughout the sequence of p110α (Fig. 1A). The majority of these are in hotspots in the helical (E545K) or kinase (H1047R) domains. The synergy of these two mutations in activating p110α suggests they act independently, and both structural and functional studies indicate they work by distinct mechanisms (5–7). The presence of both of these mutations in colorectal tumors, compared with the singly mutated form, strongly correlates with decreased survival (8). However, it is not clear how other mutations throughout the sequence activate p110α, nor is it clear whether there is any unifying principle governing all of the oncogenic mutations. It is becoming increasingly apparent that the p110α mutations do not act alone in tumor development. Mice with the most common p110α mutation (H1047R) expressed in ovaries develop hyperplasias, but not tumors, whereas coupled with the loss of the lipid phosphatase PTEN results in rapid ovarian tumor development (9).
Fig. 1.

Distribution and activity of cancer-associated p110α/p85α mutants. (A) Frequency of somatic mutations in p110α versus residue number, with domains of p110α colored. Bars for mutations analyzed in this study are highlighted in red. (B) Locations of cancer-linked mutants analyzed in this study are shown as spheres mapped on the crystal structure of p110α/niSH2-p85α (Protein DataBank, pdb: 3HHM). Schematics of the crystal structure with catalytic subunit in gray and p85 in green simplify the views presented. The p85 domains missing from the crystal structure are outlined in green. (C) Lipid kinase assays of somatic mutants. Assays measured 32P-PIP3 production. Assays were performed in duplicate and repeated twice.
The activity of p110α is stringently regulated by a tightly associated p85 regulatory subunit, which stabilizes the enzyme, inhibits lipid kinase activity, and allows activation downstream of receptor tyrosine kinases (RTKs) (10, 11). The N-terminal SH2 domain (nSH2) and the inter-SH2 domain (iSH2) of p85 are the minimal fragment of the regulatory subunit required for full inhibition of p110α lipid kinase activity (11). The binding of phosphorylated YXXM motifs in activated RTKs and adapter proteins disrupts the inhibitory contact between the nSH2 and catalytic subunit (12, 13). The p85 subunit is also somatically mutated in several tumor types (14, 15), with frequent mutations in primary endometrial tumors (14). These p85 mutations lead to stimulation of the associated catalytic subunit (16).
The p110α catalytic subunit is composed of an adapter-binding domain (ABD), a Ras-binding domain (RBD), a C2 domain, helical domain, and a kinase domain (17). Gain-of-function mutations resulting in increased oncogenic transformation are associated with all of these domains (18, 19). The mechanism of activation of some oncogenic mutants, in particular mutants in the ABD domain and the linker between the ABD and RBD domains, has been unclear, because these mutations are distant from both the lipid-binding surface as well as the kinase domain active site. However, these mutations are frequent in primary endometrial cancers (18). It has been proposed that mutations in the ABD domain could disrupt interactions with the rest of the catalytic subunit (12, 20); however, no clear mechanism by which this conformational shift would activate lipid kinase activity has been revealed. The E545K mutation increases activity by disrupting the nSH2–helical domain interface, similar to binding of RTKs (12, 13). The structure of the H1047R mutant showed a conformational change in the proposed lipid-binding region of the kinase domain (13). Compared with the WT, H1047R has higher affinity for lipids and increased soluble RTK phosphopeptide (pY)-stimulated activity (5, 7). The N345K mutant is located at the interface of the iSH2 and C2 domains, and it causes a disruption of this interface, similar to the N564D mutant in the iSH2 domain (19, 21).
Hydrogen/deuterium exchange mass spectrometry (HDX-MS) has become a powerful tool for understanding the dynamics of complex protein systems (22) and it is particularly useful for understanding protein–lipid interactions (23–27). The rate of exchange of amide hydrogens with deuterium is strongly dependent on secondary structure and solvent accessibility, and this can be used to examine dynamic structural perturbations caused by different stimuli. We have used HDX-MS to determine conformational changes that occur in the full-length WT p110α/p85α complex during activation by pY and when binding to membranes. Our results show that four unique events are required for maximal catalysis. Furthermore, we show that cancer-linked mutations located in different domains of p110α either mimic or enhance the same conformational changes induced in the wild-type enzyme by pY binding or lipid interaction.
Results
Lipid Kinase Activity of Cancer-Linked Mutations.
We characterized 11 mutants covering almost every domain of the p110α catalytic subunit, including the ABD domain (R88Q, R93W, and G106V), the ABD–RBD linker (K111E and G118D), the C2 domain (N345K, C420R, and E453K), helical domain (E545K), and kinase domain (H1047R) (Fig. 1 A and B). We also characterized one p85α mutant in the iSH2 domain (N564D). Lipid kinase assays were carried out with 5% PIP2-containing vesicles (25). All cancer-linked mutations, including mutants in the ABD and ABD–RBD linker, increased basal lipid kinase activity compared with WT, with H1047R acting as the most potent activator, followed by N345K and E545K (Fig. 1C).
The WT enzyme’s lipid kinase activity was stimulated by a soluble phosphopeptide (PDGFR residues 735–767, with pY740 and pY751, referred to hereafter as pY). When looking into the stimulation of cancer mutants by pY, we find that almost all mutants had a higher pY-activated lipid kinase activity compared with the pY-activated WT. This indicates that these p110α somatic mutants do not simply mimic the pY-activated form of the enzyme. This was especially surprising for the ABD and ABD–RBD linker mutants, because they are very distant from both the active site and the lipid-binding site. In contrast, the E545K helical domain mutant as well as the p85α engineered oncogenic mutant K379E in the nSH2 (a residue in contact with E545 in the structure of the p110/p85 heterodimer) were not stimulated to a greater level than the pY-activated WT p110α and were insensitive to pY activation (<1.5-fold activation).
HDX-MS of WT p110α/p85α in the Presence of pY and/or Membranes.
For both the WT and cancer-linked mutants, we characterized three distinct states: basal, pY activated, and membrane-bound, using HDX-MS. Amide hydrogen to deuterium exchange (HDX) was initiated by exposing the protein to D2O for various times (3, 30, and 300 s), followed by quenching. Deuterium incorporation was quantified on pepsin-digested peptides separated on a reversed-phase ultra performance liquid chromatography (UPLC), as described previously (25). The selected peptic peptides used for both p110α and p85α analysis are shown in Fig. S1. Full details of HDX methods are in Fig. S2, and the full list of 128 peptides in p110α and 82 peptides in p85α with their deuterium incorporation data are in Fig. S3. All peptides that showed differences in HDX levels either upon mutation and/or binding of either pY or membranes are shown in a structural representation in Fig. S4, and the percent differences in HDX at each time point are shown in Fig. S5 for p110α and Fig. S6 for p85α.
Dynamic changes in WT p110α/p85α upon pY activation.
Upon addition of pY, we observed differences in HDX both in expected regions located at the nSH2–helical interface, as well as in unexpected regions distant from this interface (Fig. 2 A and B and Figs. S5B and S6B). The presence of pY caused exposure of regions in both the helical and C2 domains, which are in contact with the nSH2 (Fig. 2A), validating that pY disrupts these contacts. However, pY binding also increased HDX in both the ABD–RBD linker and a region in the C2 domain that forms an interface with the iSH2. This reveals that pY binding triggers conformational shifts between the ABD and the rest of the catalytic subunit as well as a disruption of the iSH2–C2 interface. Regions in both the nSH2 and cSH2 domains of p85α showed decreases in HDX due to pY binding (Fig. S6), similar to previous results with p110δ/p85α (25).
Fig. 2.

Changes in HDX in full-length WT p110α/p85α in the presence of pY and PIP2 vesicles. (A) Time course of HDX incorporation for representative peptides that showed differences in HDX upon addition of pY, ±lipid vesicles. (B) Peptides in p110α and the iSH2 of p85α that showed differences in HDX both greater than 0.5 Da and 6% between the basal and pY-activated WT or (C) between the pY-activated WT with and without membranes, are highlighted on both the structure of p110α/iSH2-p85α (pdb: 3HHM) and on a schematic. Peptides with no differences are shown in gray for p110α and green for p85α. All further HDX figures use this same mapping scheme. The full set of HDX incorporation plots for peptides with differences in HDX are shown in Fig. S8, and differential HDX-MS data are in Figs. S5 and S6.
Dynamic changes in WT p110α/p85α upon binding membranes.
We next examined HDX differences in the pY-activated WT p110α/p85α upon binding to 5% PIP2 membranes. The ABD–RBD linker, which was partially exposed upon pY binding, was further exposed upon binding membranes (Fig. 2 A and C and Fig. S5). Regions in p85 at or near the iSH2–C2 interface were also further exposed upon membrane binding, similar to the ABD–RBD linker. This indicates that the conformational changes in the C2/iSH2 interface and the ABD/RBD linker may be mechanistically linked. Interestingly, we observed decreased HDX upon membrane binding in a region of the kinase domain that is located near the ABD–kinase domain interface (Fig. 2 A and C). Movement of the ABD domain may be required to allow for proper orientation of the kinase domain at the membrane.
Dynamic Changes in p110α Cancer-Linked Mutants.
For HDX-MS experiments, we selected the most activating mutations in each of the p110α domains: G106V in the ABD, N345K in the C2 domain, E545K in the helical domain, and H1047R in the kinase domain. Three time points (3, 30, and 300 s) under three conditions (basal, pY activated, and membrane bound) were tested. To determine whether mutations in the same domain acted by a similar mechanism, we also carried out experiments on G118D (ABD–RBD linker) and C420R (C2 domain), at the 3-s time point.
Effect of mutations in the ABD and ABD–RBD: G106V and G118D.
The G106V mutation, located at the end of ABD domain, caused increased HDX in the ABD–RBD linker compared with the WT (Fig. 3 A and B). Upon pY activation of G106V, the ABD–RBD linker was even further exposed. Interestingly, this same region was exposed in the WT upon pY binding and further exposed upon membrane binding. Surprisingly, the major difference when comparing the pY-activated forms of G106V and the WT is that G106V shows greater exposure upon pY binding in the iSH2 region located at the iSH2–C2 interface (Fig. 3 A and C). The HDX level seen in this region in G106V upon pY binding is nearly the same as the WT upon membrane binding (Fig. 3A), suggesting that G106V, even in the absence of membranes, is able to undergo conformational changes that are normally caused by membrane binding. The G106V mutation is spatially distant from the iSH2–C2 interface, and this further substantiates a mechanistic link between the ABD–RBD linker and the iSH2–C2 interface. The rest of the protein in G106V showed similar changes in HDX with pY as observed in the WT (Fig. 3C and Figs. S5B and S6B).
Fig. 3.

Changes in HDX levels in the G106V and N345K cancer mutants either basally or upon pY activation. HDX differences caused by membrane binding for these mutants are in Fig. S4. (A) HDX curves for representative peptides with differences in HDX upon mutation or pY activation. Dotted lines across plots indicate the deuteration levels of the basal WT state. A more complete set of HDX curves is shown in Fig. S9, and the HDX differential data are in Figs. S5 and S6. (B) Peptides with differences in HDX between the WT and the G106V mutant, (C) upon pY binding of the G106V mutant, (D) between the WT and the N345K mutant, and (E) upon pY binding of the N345K mutant are highlighted on the structure of p110α/iSH2-p85α and on a schematic. Locations of cancer mutations are indicated with yellow spheres on the crystal structure and yellow stars on the schematic.
Consistent with tighter membrane association of G106V versus WT (see below), membranes caused a greater decrease in HDX in the kinase domain of G106V compared with WT (Figs. S4, S5C, and S6C), in regions either predicted to be part of lipid-binding surface (loop from 864 to 875) (13) or shown to be critical for membrane interaction (very C terminus of p110α) (5). The G118D mutant exhibited similar HDX behavior to G106V, indicating that these mutations work by a similar mechanism (Figs. S5 and S6).
Effect of mutations in the C2 domain: N345K and C420R.
The N345K mutation in the C2 domain increased HDX compared with the WT at the iSH2–C2 interface as expected; however, it also showed increased HDX in the spatially distant ABD–RBD linker (Fig. 3D and Figs. S5 and S6). The increase in HDX in N345K in the C2 domain and p85–iSH2 domain is consistent with this mutation disrupting the iSH2–C2 interface. This was also true for C420R, which showed increased HDX in the C2 domain compared with the WT (Fig. S5). Upon pY binding, the N345K mutant showed increased HDX in the ABD–RBD linker to a level normally caused by membrane binding (Fig. 3 A and E). This unexpected increase in HDX in the ABD–RBD linker caused by N345K also substantiates a coupling of conformational dynamics of the ABD–RBD linker and the iSH2–C2 interface. The rest of the protein in N345K showed similar changes in HDX with pY as observed in the WT (Fig. 3E and Figs. S5 and S6). Interestingly, N345K and G106V showed similar changes in HDX in p110α in the presence of membranes (Figs. S4, S5C, and S6C). One interesting exception was decreased HDX for N345K in the C2 domain, which has been postulated to interact with membranes (5, 28). HDX-MS experiments on N345K provide direct evidence for this.
Effect of mutation in the helical domain: E545K.
Compared with WT, the E545K mutation caused increased HDX in p110α in the ABD–RBD linker and C2 and helical domains as well as in the nSH2 and iSH2 domains of p85α (Fig. 4 A and B and Figs. S5A and S6A). All changes seen in the catalytic subunit were of the same magnitude and location as seen in WT upon pY activation, indicating that E545K mimics the pY activated state. Regions of the p85–nSH2 domain that make up the interface with the helical domain also showed increased HDX (Fig. S6A). These changes are consistent with breaking the interaction between the catalytic subunit and the nSH2 domain. Upon pY addition to E545K there were no further detectable changes in exchange in the catalytic subunit (Figs. S4, S5B, and S6B), which is consistent with the lack of activation upon pY binding. Upon binding to vesicles, E545K showed a pattern of decreased HDX in regions similar to the membrane-bound WT (Figs. S4, S5C, and S6C).
Fig. 4.

Changes in HDX levels in the E545K and H1047R cancer mutants either basally or upon membrane binding. HDX differences caused by pY binding for these mutants are in Fig. S4. (A) HDX curves for representative peptides with differences in HDX upon mutation, pY activation, or membrane binding. HDX rates for the 532–551 peptide from WT and H1047R are similar, whereas the E545K mutant differs. A more complete set is shown in Fig. S9, and the HDX differential data are in Figs. S5 and S6. (B) Peptides with differences in HDX between the WT and the E545K mutant, (C) between the WT and the H1047R mutant, and (D) upon membrane binding of the H1047R mutant are highlighted on the structure of p110α/iSH2-p85α and on a schematic.
Effect of mutation in the kinase domain: H1047R.
The H1047R mutation in the basal state caused increased HDX compared with WT in many regions in the C lobe of the kinase domain (Fig. 4C and Fig. S5A). All of these peptides are structurally linked through a network of interactions (13), explaining their simultaneous exposure. One of the regions exposed contained the C-terminal end of the activation loop, which is responsible for binding to lipid substrate, and is directly underneath the H1047R mutation. Upon pY activation, H1047R showed HDX differences similar to WT (Figs. S4, S5B, and S6B). Interestingly, all of the peptides that were exposed basally in H1047R were then protected upon binding to membranes (Fig. 4D and Fig. S4C), indicating that this mutation changes the dynamics of the membrane-binding surface.
Lipid Affinity of Cancer-Linked Mutations.
We performed protein–lipid FRET experiments to measure lipid binding (29). Experiments were done in the presence and absence of pY for all p110α/p85α constructs. We have previously shown for both p110δ (25) and p110α (5) that pY is required for binding to membranes. Protein–lipid FRET showed that binding of all mutants to vesicles, except E545K, was stimulated by pY and was tighter than the WT (Fig. 5B and Fig. S7). Surprisingly, we found that pY-activated mutants in the ABD and ABD–RBD linker showed increased membrane affinity compared with the pY-activated WT. Because these mutants are distant from the proposed membrane-binding surface, they appear to increase membrane affinity through an indirect mechanism.
Fig. 5.

Lipid binding of cancer mutants. (A) Protein–lipid FRET binding curves for a selection of mutants ±5 μM pY. Full lipid-binding curves used to calculate Ka values are in Fig. S7. (B) Protein–lipid FRET assays were performed with 5% PIP2 vesicles, and Ka was calculated for both ±5 μM pY. All experiments were performed in triplicate. In the absence of pY, the majority of proteins had FRET levels that were too low for reliable curve fitting and are indicated by IB (insufficient binding), and no Ka value is reported.
Discussion
Understanding the mechanisms by which somatic mutations activate the oncogenic potential of p110α is an important goal toward designing effective cancer therapeutics. Crystal structures of p110α have suggested how some cancer mutants activate lipid kinase activity (5, 13, 20), but the mechanism of activation for many rare mutants, including ones in the ABD and ABD–RBD linker frequently mutated in endothelial cancers (18), has remained unclear. Recent studies have identified novel aspects of PI3K regulation, for example, by phosphorylation of both the nSH2 and cSH2 domains of p85 downstream of PKC (30) and the cSH2 domain by IKK (31), leading to decreased affinity for tyrosine-phosphorylated activators. Phosphorylation of SH2 domains could also affect the basal activity by disruption of inhibitory contacts; however, this has not been tested. Furthermore, the p85 regulatory subunit directly interacts with and regulates the lipid phosphatase PTEN (32, 33). There might be yet undiscovered regulatory partners of class IA PI3Ks. However, all of the mutants we have examined activate p110α/p85 directly, even in the absence of binding partners, allowing us to directly probe dynamic changes associated with activation of both the WT and mutant complexes.
We find that all cancer mutants tested activated basal lipid kinase activity, and all mutants except E545K had increased affinity for membranes in the presence of soluble RTK–pY compared with WT p110α/p85. This shows that membrane binding can be increased, even by mutations not at the binding surface, through an indirect mechanism.
HDX-MS experiments carried out on both the WT and mutant p110α/p85α in three different states: basal, pY activated, and membrane bound, have identified four distinct conformational events in the PI3Kα catalytic cycle (Fig. 6). These four events are: (i) breaking the nSH2–helical interface, (ii) disrupting the iSH2–C2 interface, (iii) movement of the ABD domain relative to the kinase domain, and (iv) interaction of the kinase domain with lipid. All four of these events are detected in the activation of WT p110α/p85α, and cancer mutants affect these conformational events in distinct ways. Our results do not allow us to establish an order for these events. A summary of the effect of cancer mutants on these events is shown in Fig. 6.
Fig. 6.
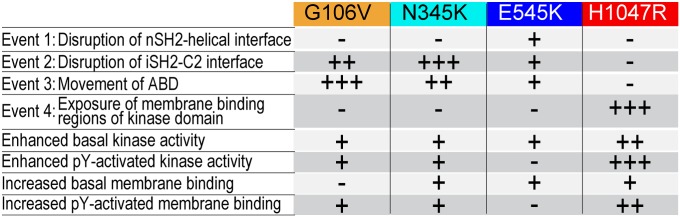
Conformational changes in the catalytic cycle of p110α/p85α. Summary of the effects of selected cancer mutants on conformational events, lipid kinase activity, and lipid binding.
The nSH2 domain of p85 inhibits p110α, whereas soluble RTK–pY releases this inhibition (11, 12). Our HDX-MS data are consistent with p110α having an nSH2-mediated inhibitory contact, and no contact with the cSH2. This is in contrast to p110β and p110δ, which have inhibitory contacts with both the nSH2 and cSH2 (25, 34). Breaking the nSH2–helical interface (event 1) also weakens the interaction between the C2 and iSH2 domains, as well as the interaction between the ABD domain and the rest of the catalytic subunit. This indicates that nSH2 contact not only inhibits the enzyme by interacting with the C lobe of the kinase domain (13), but that it also plays an important scaffolding role in rigidifying the enzyme and preventing interdomain movements required for membrane binding. All of our assays (kinase activity, lipid binding and HDX-MS) indicate that nSH2–helical mutants (E545K in p110α and K379E in p85α) mimic the RTK-activated form of the WT (Fig. 6), as expected (7, 12, 13, 35). Although E545K is insensitive to soluble RTK–pY, it does not mean that it would be insensitive to tyrosine phosphorylated proteins that are either directly or indirectly tethered to membranes, because E545K would have both the nSH2 and cSH2 free for binding to these pY-containing targets. Indeed, it has been shown that E545K is preferentially recruited to IRS1 compared with the WT enzyme (36), and in a cellular context E545K may be more sensitive to RTK signaling.
For the WT enzyme there was a further disruption of both the iSH2–C2 interface and the ABD–RBD linker upon binding to membranes. These conformational changes may be required for correct orientation of p110α on the membrane surface. Previous work showing a gain of function in mutants at the iSH2–C2 interface have left open the question of whether this interface is disrupted in the process of activating the wild-type enzyme (21). Our results indicate that this interface is indeed disrupted upon activation of the WT enzyme (event 2). Mutations at the iSH2–C2 interface (N345K and C420R), nSH2–helical interface (E545K), along with mutations at the ABD or ABD–RBD linker (G106V and G118D) caused disruption in the iSH2–C2 interface either basally or upon pY activation.
Mutations in the ABD and ABD–RBD linker are gain-of-function mutations (18, 19); however, the mechanisms of activation for mutants not directly at the ABD–kinase domain interface (R93W, G106V, K111E, and G118D) have been difficult to interpret from structural data (5, 13, 20). Our results suggest that mutations in the ABD, ABD–RBD, or in the C2 domain all cause movement of the ABD relative to the rest of the enzyme (event 3), either basally or upon pY activation. This implies a mechanistic link between the iSH2–C2 interface and the ABD–kinase domain interface (i.e., between events 2 and 3).
The final event that accompanies maximal lipid kinase activity was interaction of the kinase domain with lipid (event 4). It has been shown previously that H1047R rearranges part of the proposed lipid-binding surface (13) and increases the affinity for membranes (5, 13). Our HDX-MS results confirmed these previous studies; however, they showed a much broader area of dynamic changes in the lipid-binding region of the kinase domain upon mutation then expected from the crystal structure, and this was accompanied by an enhanced binding to membranes.
Our study has revealed a unifying principle by which oncogenic mutants in PI3Kα activate both lipid-binding and lipid kinase activity by mimicking or enhancing motions that occur in the catalytic cycle of the WT enzyme. The combinatorial effect of mutations that facilitate distinct conformational events could lead to more potent activation than the single mutations. For example, our studies show that H1047R is basally activated but still undergoes the same conformational changes upon pY binding and membrane binding as the WT. Therefore, H1047R combined with a mutation either in the ABD/RBD linker, the C2/iSH2 interface or in the helical domain, should show enhanced activity compared with single mutations. Indeed, a previous study showed that combined mutations in the helical and kinase domain (E545K and H1047R) greatly synergize in cell transformation (6). Similar enhancement could be happening with mutations in the ABD or ABD–RBD linker that co-occur with mutations in the kinase domain in endometrial tumors (18), because HDX-MS data indicate that these mutations facilitate distinct events. Resistance that develops to PI3K inhibitors has not been shown to involve mutations in the active site. Instead, mutations elsewhere in the pathway that bypass inhibition are common (37). The synergy of p110α mutants could form the basis for a new class of resistant mutations.
The increased affinity of the oncogenic mutants for lipid membranes and the HDX-MS observations are consistent with the mutations destabilizing a closed, cytosolic form of the enzyme. It may be that small-molecule inhibitors could be selected that would stabilize the closed form of the enzyme.
Methods
HDX Measurements.
HDX reactions were initiated by the addition of proteins (1 μM final) to either pY peptide solution (15 μM final) or blank followed by addition of 88% D2O solution, to a final concentration of 69% D2O. For HDX-MS membrane interaction experiments lipid vesicles were mixed with the D2O solution followed by addition of the pY-activated p110α/p85α. Three time points of “on exchange” (3, 30 and 300 s) at 23 °C were followed by addition of a quench buffer. Samples were frozen in liquid nitrogen and stored for no more than 1 wk at −80 °C until mass analysis.
Full experimental procedures are described in SI Methods.
Supplementary Material
Acknowledgments
We thank Mark Skehel, Sew Yeu Peak-Chew, and Farida Bergum for help with the HDX-MS setup, and Robin Irvine for critical reading of the manuscript. J.E.B. was supported by a European Molecular Biology Organization long-term fellowship (ALTF268-2009) and the British Heart Foundation (PG11/109/29247). O.V. was supported by a Swiss National Science Foundation fellowship (Grant PA00P3_134202). This work was funded by the Medical Research Council (file reference U105184308).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.A.E. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1205508109/-/DCSupplemental.
References
- 1.Chang HW, et al. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 1997;276:1848–1850. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- 2.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 3.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal. 2011;4:re2. doi: 10.1126/scisignal.2002165. [DOI] [PubMed] [Google Scholar]
- 5.Hon WC, Berndt A, Williams RL. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene. 2012;31:3655–3666. doi: 10.1038/onc.2011.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci USA. 2008;105:2652–2657. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carson JD, et al. Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem J. 2008;409:519–524. doi: 10.1042/BJ20070681. [DOI] [PubMed] [Google Scholar]
- 8.Liao X, et al. Prognostic role of PIK3CA mutation in colorectal cancer: Cohort study and literature review. Clin Cancer Res. 2012;18:2257–2268. doi: 10.1158/1078-0432.CCR-11-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinross KM, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122:553–557. doi: 10.1172/JCI59309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu J, et al. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18:1379–1387. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu J, Wjasow C, Backer JM. Regulation of the p85/p110alpha phosphatidylinositol 3′-kinase. Distinct roles for the N-terminal and C-terminal SH2 domains. J Biol Chem. 1998;273:30199–30203. doi: 10.1074/jbc.273.46.30199. [DOI] [PubMed] [Google Scholar]
- 12.Miled N, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239–242. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- 13.Mandelker D, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci USA. 2009;106:16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urick ME, et al. PIK3R1 (p85α) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 2011;71:4061–4067. doi: 10.1158/0008-5472.CAN-11-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaiswal BS, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16:463–474. doi: 10.1016/j.ccr.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun M, Hillmann P, Hofmann BT, Hart JR, Vogt PK. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc Natl Acad Sci USA. 2010;107:15547–15552. doi: 10.1073/pnas.1009652107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- 18.Rudd ML, et al. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin Cancer Res. 2011;17:1331–1340. doi: 10.1158/1078-0432.CCR-10-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci USA. 2007;104:5569–5574. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang CH, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–1748. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 21.Wu H, et al. Regulation of Class IA PI 3-kinases: C2 domain-iSH2 domain contacts inhibit p85/p110alpha and are disrupted in oncogenic p85 mutants. Proc Natl Acad Sci USA. 2009;106:20258–20263. doi: 10.1073/pnas.0902369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engen JR. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal Chem. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burke JE, et al. A phospholipid substrate molecule residing in the membrane surface mediates opening of the lid region in group IVA cytosolic phospholipase A2. J Biol Chem. 2008;283:31227–31236. doi: 10.1074/jbc.M804492200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burke JE, et al. Interaction of group IA phospholipase A2 with metal ions and phospholipid vesicles probed with deuterium exchange mass spectrometry. Biochemistry. 2008;47:6451–6459. doi: 10.1021/bi8000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burke JE, et al. Dynamics of the phosphoinositide 3-kinase p110δ interaction with p85α and membranes reveals aspects of regulation distinct from p110α. Structure. 2011;19:1127–1137. doi: 10.1016/j.str.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu YH, Burke JE, Li S, Woods VL, Jr, Dennis EA. Localizing the membrane binding region of Group VIA Ca2+-independent phospholipase A2 using peptide amide hydrogen/deuterium exchange mass spectrometry. J Biol Chem. 2009;284:23652–23661. doi: 10.1074/jbc.M109.021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Man P, et al. Accessibility changes within diphtheria toxin T domain upon membrane penetration probed by hydrogen exchange and mass spectrometry. J Mol Biol. 2011;414:123–134. doi: 10.1016/j.jmb.2011.09.042. [DOI] [PubMed] [Google Scholar]
- 28.Denley A, Kang S, Karst U, Vogt PK. Oncogenic signaling of class I PI3K isoforms. Oncogene. 2008;27:2561–2574. doi: 10.1038/sj.onc.1210918. [DOI] [PubMed] [Google Scholar]
- 29.Nalefski EA, Slazas MM, Falke JJ. Ca2+-signaling cycle of a membrane-docking C2 domain. Biochemistry. 1997;36:12011–12018. doi: 10.1021/bi9717340. [DOI] [PubMed] [Google Scholar]
- 30.Lee JY, Chiu YH, Asara J, Cantley LC. Inhibition of PI3K binding to activators by serine phosphorylation of PI3K regulatory subunit p85alpha Src homology-2 domains. Proc Natl Acad Sci USA. 2011;108:14157–14162. doi: 10.1073/pnas.1107747108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Comb WC, Hutti JE, Cogswell P, Cantley LC, Baldwin AS. p85α SH2 domain phosphorylation by IKK promotes feedback inhibition of PI3K and Akt in response to cellular starvation. Mol Cell. 2012;45:719–730. doi: 10.1016/j.molcel.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chagpar RB, et al. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 2010;107:5471–5476. doi: 10.1073/pnas.0908899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheung LW, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011;1:170–185. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, et al. Structure of lipid kinase p110β/p85β elucidates an unusual SH2-domain-mediated inhibitory mechanism. Mol Cell. 2011;41:567–578. doi: 10.1016/j.molcel.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaussade C, Cho K, Mawson C, Rewcastle GW, Shepherd PR. Functional differences between two classes of oncogenic mutation in the PIK3CA gene. Biochem Biophys Res Commun. 2009;381:577–581. doi: 10.1016/j.bbrc.2009.02.081. [DOI] [PubMed] [Google Scholar]
- 36.Yang X, et al. Using tandem mass spectrometry in targeted mode to identify activators of class IA PI3K in cancer. Cancer Res. 2011;71:5965–5975. doi: 10.1158/0008-5472.CAN-11-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci USA. 2011;108:E699–E708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.